

Abstract
Our understanding of the biological principles of mycobacterial tolerance to antibiotics is crucial for developing shorter anti-tuberculosis regimens. Various in vitro approaches have been developed to identify the conditions that promote mycobacterial persistence against antibiotics. In our laboratories, we have developed a detergent-free in vitro growth model, in which mycobacteria spontaneously grow at the air–medium interface as self-organized multicellular structures, called biofilms. Mycobacterial biofilms harbor a sub-population of drug tolerant persisters at a greater frequency than their planktonic counterpart. Importantly, development of these structures is genetically programmed, and defective biofilms of isogenic mutants harbor fewer persisters. Thus, genetic analysis of mycobacterial biofilms in vitro could potentially be a powerful tool to unravel the biology of drug tolerance in mycobacteria. In this chapter we describe a method for screening biofilm-defective mutants of mycobacteria in a 96-well format, which readily yields a clonally pure mutant for further studies.
Keywords: Biofilm, Genetic mutant, Mycobacteria, Drug tolerance
1 Introduction
Many, if not most, microbes grow as biofilms—self-organized multicellular structures encapsulated within a matrix of extracellular polymeric substance. There are numerous diverse forms, ranging from microcolonies on submerged substrata to pellicles at air–medium interfaces [1–4]. biofilms develop through genetically programmed pathways and harbor phenotypically heterogeneous but genetically clonal populations of constituent cells [5–16]. Microbial biofilms display a variety of behaviors that are not associated with dispersed planktonic growth, including tolerance to environmental and immunological stresses and antibiotic resistance. biofilms represent an important persistence strategy of pathogenic microbes in chronic infections [17–19].
Mycobacterium tuberculosis is the causative agent of human tuberculosis, although most (>90–95 %) infections of immuno-competent individuals do not lead to active disease. Instead, a latent infection is established which can subsequently activate into disease when the immune system is compromised [20–22]. Treatment of active tuberculosis requires at least 6 months with multiple antibiotics [23], and drug resistance is an emerging problem in controlling the disease. Together, these characteristics of M. tuberculosis infections underscore the extraordinary ability of the pathogen to persist in the face of chemical and immunological challenges. Although it is unclear as to how and where in the host the persistent M. tuberculosis bacilli survive against the host-derived stresses and antibiotics, it is plausible that the mechanisms underlying in vivo persistence overlap with the intrinsic stress tolerance displayed by the bacilli when grown in vitro.
In detergent-free liquid cultures in vitro, most mycobacterial species including M. tuberculosis form macroscopic structures leading to the development of pellicles at the air–medium interface [5– 7, 16, 24, 25]. Over the last several years it has become apparent that mycobacterial pellicles develop through distinct stages with specific genetic requirements, and that these pellicles harbor bacilli that are phenotypically tolerant to high concentrations of antibiotics [5, 7, 16, 24, 25]. Thus, these mycobacterial pellicles represent a genetically programmed developmental process, in common with the many other microbes that form similar biofilms. Further genetic studies of these developmental processes will likely contribute towards identifying novel targets against recalcitrant infections, and facilitate an improved understanding of host–pathogen interactions. These information can subsequently be exploited for drug discovery and vaccine design against TB.
In this chapter we describe detailed methods for growing and investigating mycobacterial biofilms. Broadly, the methods involve four steps; (1) Establishing a biofilm assay in 96-well format for high-throughput screening, (2) making a high-density transposon library of mycobacteria, (3) screening the library using the 96-well format of the biofilm assay, and (4) mapping the sites of transposon insertion in the biofilm-defective mutants. While biofilm assays and construction of transposon libraries of M. smegmatis and M. tuberculosis mutants have been independently published elsewhere [7, 16, 24, 26, 27], we integrate these methods in this chapter to provide a composite workflow for studying the genetics of mycobacterial biofilms.
2 Materials
2.1 Biofilm Assay in a 96-Well Format
A mycobacterial strain—either Mycobacterium smegmatis mc2 155, ATCC 700084; Mycobacterium tuberculosis H37Rv, ATCC # 25618; or mc2 7000 (see Note 1).
10 % w/v d -pantothenate : Dissolve 1 g of d -pantothenic acid hemicalcium salt in 9 mL of deionized water. Filter-sterilize with 0.22 μm filter, and store at room temperature (see Note 2).
7H9ADCTw : Dissolve 4.7 g of Middlebrook 7H9 base in 890 mL of deionized water. Add 5 mL of 100 % glycerol. Adjust the volume to 900 mL. Dispense 90 mL aliquots in bottles, autoclave, and cool to room temperature. To each 90 mL of aliquot, add 10 mL of ADC enrichment (albumen/dextrose/catalase supplement—Becton Dickinson), and 250 μL of 20 % v/v Tween 80 (final concentration of 0.05 % v/v) (see Note 2).
7H10ADC : Dissolve 19 g of Middlebrook 7H10 agar base in 890 mL of deionized water. Add 5 mL of 100 % glycerol, and adjust the volume to 900 mL with deionized water. Heat with stirring to dissolve the agar. Autoclave and cool to 50 °C. Add 100 mL of either 10× ADC enrichment, and pour 25 mL for each 85 mm petri dish. For 7H10ADCTw, add 2.5 mL of 20 % Tween 80 per liter of the media (see Note 2).
Complete Sauton's medium: Dissolve 0.5 g KH2 PO4, 0.5 g MgSO4, 4 g l-asparagine monohydrate, 2 g citric acid, 0.05 g ferric ammonium citrate to 900 mL deionized water. Add 60 mL glycerol. Adjust pH to 7.0 with 1 M NaOH. Autoclave and store at room temperature. Just before inoculating the cells, add 0.1 mL of sterile 1 % ZnSO4 per liter of medium. If culturing planktonic cells, add 2.5 mL of 20 % Tween 80 per liter of medium (see Note 2).
Complete biofilm medium : Dissolve 13.6 g of KH2 PO4 in 900 mL of water. Add 2 g of (NH4)2 SO4. Adjust pH to 7.2 with 10 M NaOH. Add 0.5 mg of FeSO4 ·7H2 O and 5 g of casamino acids. Bring up the volume to 1,000 mL with water. Autoclave and store at room temperature. Just before inoculating the cells, aseptically add 5 mL of 40 % glucose, 1.0 mL of 0.1 M CaCl2, and 0.1 mL of 1 M MgSO4 to 94 mL of the sterile biofilm base medium.
2.2 Construction and Screening of Transposon Library
A mycobacterial strain (see Subheading 2.1, item 1).
A shuttle phasmid, phAE781, carrying the Himar-1 transposon packaged in the genome of a temperature-sensitive derivative of mycobacteriophage TM4 [28].
Growth media as described in Subheading 2.1, items 2–6.
MBTA : Add 4.7 g 7H9 base and 7 g Bacto agar to 900 mL of deionized water. Heat to dissolve the agar. Dispense 100 mL in bottles, autoclave and store at room temperature.
2.3 Mapping Transposon Insertion Sites in Biofilm-Defective Mutants
Taq DNA polymerase.
10× buffer for Taq DNA polymerase (with MgCl 2).
10 mM dNTP mix.
Dimethyl sulfoxide (DMSO).
Primer 1: GGCCAGCGAGCTAACGAGACNNNNGTTGC.
Primer 2: CGCTTCCTCGTGCTTTACGGTATCG.
Primer 3: GGCCAGCGAGCTAACGAGAC.
Primer 4: GGCCAGCGAGCTAACGAGAC.
3 Methods
3.1 Biofilm Assay in a 96-Well Format
Inoculate a frozen stock of desired mycobacterial strain in 10 mL of 7H9ADCTw (see Notes 2–4).
Incubate the cultures at 37 °C till the OD600 reaches ∼1.0 for M. smegmatis (see Note 5).
Inoculate 20 mL of biofilm media with 20 μL of M. smegmatis culture obtained from step 2 (see Note 6).
Dispense 200 μL of the bacterial suspension from step 3 in each well of a 96-well plate, and incubate the plates in humidified conditions at 30 °C for M. smegmatis (see Note 7).
A film of bacteria is visible for M. smegmatis cultures after 3 days of incubation that matures to robust and textured pellicles in 4–5 days (see Note 8).
3.2 Construction and Screening of Transposon Library
Inoculate a frozen stock of M. smegmatis, mc 2 155, in 100 mL 7H9ADCTw.
Incubate the cells at 37 °C on a shaker incubator till the OD600 is about 0.8–1.0.
Mix 0.3 mL of cells, 100–1,000 plaque forming units (PFU) of phAE781 and 3 mL of MBTA pre-warmed to 42 °C. Vortex the mixture briefly and pour it evenly on an 85 mm petri dish containing 7H10ADC agar. Repeat this for at least 15–20 plates (see Notes 9 and 10).
Incubate the plates at 30 °C for 2–3 days, until a confluence of plaques is seen (see Note 11).
Overlay each of the plates with 4 mL of phage buffer and store the plates at 4 °C for a minimum of 4 h, or a maximum of overnight.
Pool the liquid with phages (phAE781) from the plates. Remove bacterial contaminants by filtering the lysate through 0.22 μm membrane filter. The phage stock can be kept at 4 °C up to 1 month without significant loss of viability.
Titer the phage stock by spotting tenfold serial dilutions on a lawn of M. smegmatis on 7H10ADC agar at 30 °C. A good stock will have greater than 1010 PFU/mL.
Ensure the thermal sensitivity of the phages by spotting tenfold serial dilutions on a lawn of M. smegmatis and incubating the plate at 37 °C for 1–2 days. The number of plaques should be reduced by several orders of magnitude.
Inoculate a frozen stock of M. smegmatis into 100 mL of 7H9ADCTw.
Incubate at 37 °C till the OD600 reaches ∼1.0 (∼6 × 108 cfu/mL).
Centrifuge the cells (3,000 × g at room temperature for 10 min), and resuspend the pellet in 100 mL 7H9 base (no Tween 80).
Incubate at 37 °C for 3 h to wash off residual Tween 80 from the bacterial surface (see Note 12).
Centrifuge the cells (3,000 × g at room temperature for 10 min) and resuspend the pellet in 0.1 volume (10 mL) of 7H9 base (no Tween 80) pre-warmed at 37 °C.
Add phAE781 at a multiplicity of infection (MOI) of 10. For example, 1 mL of ∼1011 PFU for ∼1010 cfu in a suspension is optimum.
Incubate the mixture at 37 °C for 30 min (see Note 13).
Transfer the phage-bacteria mixture into a 250 mL sterile bottle containing 90 mL of pre-warmed (37 °C) 7H9ADCTw. Incubate the content at 37 °C for 3 h (see Note 14).
Centrifuge the cells (3,000 × g at room temperature) for 10 min, resuspend the pellet in 1 mL of 7H9Tw and plate the transductants on 7H10ADCTw with 40 μg/mL of kanamycin.
Incubate the plates till colonies appear (see Note 15).
Determine the quality of the library by identifying transposon insertion sites in 10–12 random colonies by method described in Subheading 3.3. About 30–50 % of the colonies with unique junction sequences are desirable in a well-represented library. If library is satisfactory, proceed with the screening steps described below (see Note 16).
Stack two sets of thirty 96-well plates marked numerically, such as stack A1-30 and stack B1-30. Using a multichannel pipette, dispense 200 μL of 7H9ADCTw with kanamycin in each well of the plates in stack A. Keep stack B plates aside for later use.
Inoculate single isolated colonies individually from M. smegmatis library (step 15) in each well of the plates in stack A.
Wrap the plates with Parafilm thoroughly and incubate them without shaking in humidified conditions at 37 °C for 2 days to obtain primary cultures of each clone. Dispersed growth should be observed after incubation. As controls, inoculate a wild-type strain and a known biofilm mutant in separate 96-well plates to obtain their primary cultures (see Note 17).
In plates B1-30, dispense 200 μL of biofilm medium using a multichannel pipette (see Note 6).
Using multichannel pipette, inoculate about 5 μL of bacteria from the stack A plates into the exact same positions in stack B plates. Keep the first stack secured at either 4 °C. As controls, inoculate 5 μL of wild-type or known biofilm-negative strains from primary cultures.
Wrap the stack B plates and place them at 37 °C incubator under humidified conditions (see Note 18).
Observe the plates after 2 days. A thin film of bacteria should be visible for wild-type M. smegmatis cultures (Figs. 1a and 2a). The negative controls (biofilm-defective mutants) should show little to no growth at the air–medium surface (Figs. 1b and 2b). The biofilms of transposon mutants can possibly range from normal to severe deficiencies. However, each mutant with altered phenotype must be tested in a secondary screen (steps 25–27) to rule out false positives (see Notes 8, 19 and 20).
Identify mutant clones with altered biofilms from the primary screen in stack A plates, and streak out the clones on 7H10ADC plates with 40 μg/mL of kanamycin. Incubate at 37 °C till colonies appear.
Inoculate a single colony of a mutant from the plates in a 50 mL polystyrene conical tube containing 5 mL 7H9ADCTw medium with 40 μg/mL of kanamycin, and culture the cells at 37 °C till saturation. As controls, inoculate wild-type strain and a known biofilm-deficient strain for the respective species.
For secondary screen of M. smegmatis mutants, inoculate 10 μL of cells in 10 mL of biofilm media in a 65 mm petri dish. Incubate at 37 °C and compare the biofilm development of the mutants with controls after 2 days (see Notes 18 and 21).
Fig. 1.
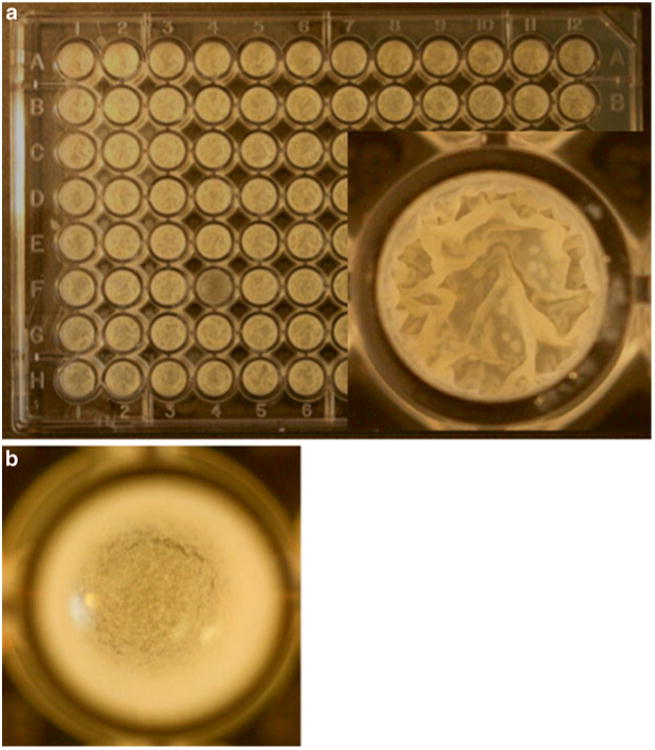
(a) Growth of M. smegmatis mc2 155 biofilms in a 96-well format after 5-days of Incubation at 30 °C. A top-down view of one of the typical wells is magnified in the inset, which shows the robust pellicles on the air–media interface. (b) A top-down view of mc2155: Δlsr-2 biofilms in a well of a 96-well plate
Fig. 2.
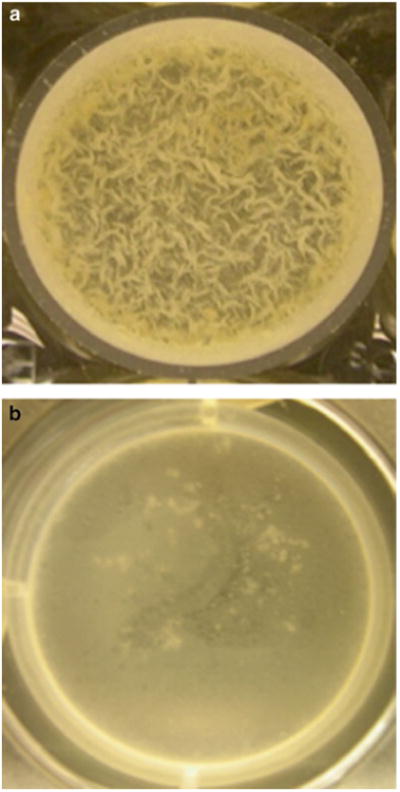
A top-down view of biofilms of M. tuberculosis mc27000 (a), and mc27000: ΔhelY (b) on air–media interface in a well of a 96-well plate after 3-week incubation at 37 °C incubation
3.3 Mapping Transposon Insertion Sites in the Biofilm-Defective Mutants
For mutants with confirmed biofilm deficiency (Subheading 3.2), resuspend a loopful of cells from the streak (Subheading 3.2) in a microfuge tube containing 200 μL of sterile water.
Vortex the suspensions briefly (∼30 s) and place in 95 °C heating block for 15 min (with occasional flicking every 5 min). Place them on ice.
- Use 5 μL of the content from each tube to set up a two-step degenerate nested PCR reactions as following:
- First PCR
- Reaction contents:
- 5 μL of 10× PCR buffer (with MgCl2)
- 5 μL of DNA
- 1 μL of 10 mM dNTPs
- 2.5 μL of DMSO
- 1 unit of Taq DNA polymerase
- 0.5 μL of 100 μM primer 1
- 0.5 μL of 100 μM primer 2
-
34.5 μL H2 O2Reaction conditions: 95 °C for 5 min, 5 Cycles of (95 °C for 30 s, 30 °C for 30 s, 72 °C for 30 s), 25 cycles of (95 °C for 30 s, 38 °C for 30 s, 72 °C for 30 s), 72 °C for 7 min.
- Second PCR
- Reaction contents:
- 5 μL of 10× PCR buffer (with MgCl2)
- 2 μL of the content from the first PCR 1 μL of 10 mM dNTPs
- 2.5 μL of DMSO
- 1 unit of Taq DNA polymerase
- 0.5 μL of 100 μM primer 3
- 0.5 μL of 100 μM primer 4
- 39.5 μL of H2 O2
- Reaction conditions: 95 °C for 5 min, 30 Cycles of (95 °C for 30 s, 56 °C for 30 s, 72 °C for 30 s), 72 °C for 7 min.
Purify the PCR products from the second reaction and sequence using Primer 4.
A typical sequencing read will represent the transposon insertion site with a sequence reading, (transposon DNA) CAGCCAACC TGTT/A(mycobacterial DNA)
4 Notes
Experiments with M. tuberculosis H37Rv require a Biosafety Level 3 laboratory (BSL-3). Because BSL-3 facilities are highly restrictive and may not be readily accessible to investigators, an attenuated derivative of H37Rv, mc2 7000, was constructed. mc2 7000 contains deletions in the region of difference 1 (RD1) locus and panCD operons, which render the strain incapable of growth in many mammalian hosts [7]. As a result, the strain can be safely used in a Biosafety Level 3 (BSL-2) laboratory.
ADC enrichment containing oleic acid (OADC) is used for M. tuberculosis strains. For culturing mc2 7000, add 1 mL of 10 % d -pantothenate stock in 1 L of medium (final concentration of 100 μg/mL).
The protocol described here was developed for a high efficiency transformable strain of M. smegmatis, mc2 155 [29]. Some of the strains in the ATCC collection may be naturally deficient in biofilm formation and must be verified before initiating the screen.
The protocol for M. tuberculosis biofilms was developed using an attenuated strain, mc2 7000 [7]. Because panCD mutation confers pantothenate auxotrophy in mc2 7000, the strain requires pantothenate supplementation (100 μg/mL) in the medium. However, the protocol described in this chapter can be adopted for any strain of M. tuberculosis. All virulent strains of M. tuberculosis must be cultured under BSL-3 containment.
For M. tuberculosis use culture with OD600 ∼ 0.2–0.4. It is important to use an early log phase culture of M. tuberculosis to achieve reproducibility in these biofilm assays. A late log phase or early stationary phase culture often has inconsistent growth patterns of biofilms.
For M. tuberculosis, inoculate 20 mL of Sauton's media with 0.2 mL of primary culture. While biofilms of M. smegmatis are cultured in a modified M63 media (called biofilm media), M. tuberculosis biofilms are best obtained in Sauton's media.
Incubate M. tuberculosis biofilms at 37 °C. Moreover, incubation under humidified conditions is particularly critical for growing M. tuberculosis biofilms in 96-well plates to avoid risk of liquid evaporation during the extended incubation at 37 °C. Keeping the plates undisturbed during incubation is important to avoid well-to-well contamination and to allow attachment of bacteria to the substrata.
For M. tuberculosis, it takes about 3 weeks to form biofilms, and an additional 2 weeks for full maturation (Figs. 1a and 2a). A longer incubation period (4–5 weeks) is necessary for complete maturation of M. tuberculosis biofilms. However, in the 96-well format excessive bacterial outgrowth associated with mature biofilms increases the risk of well-to-well contamination. Therefore, an early (2–3 weeks) stage when a thin film is formed at the interface is best for mutant screening.
phAE781 is a recombinant TM4ts carrying shuttle phasmid carrying Himar-1 transposon, packaged into a TM4ts phage (phAE159). At permissive temperature (30 °C), the recombinant phage carrying transposable elements can replicate in M. smegmatis, while at restrictive temperature (37 °C) it delivers the transposon in mycobacteria, but does not replicate.
MBTA should be kept at around 42 °C to avoid loss of bacterial or phage viability, while keeping the top agar in liquid form.
For a high titer stock, a confluence of plaques with clearly distinct boundaries on a bacterial lawn is optimum. A totally clear plate due to excessive phage input produces low titer stocks.
Incubation time to wash off Tween 80 from M. tuberculosis is 16 h.
Incubation time for phage infection in M. tuberculosis is 3 h.
Outgrowth incubation time for M. tuberculosis is 16–18 h.
Often it is necessary to calibrate the plating volume for the transductants to achieve well-separated 200–300 colonies on an 85 mm petri dish. In a typical scenario, 10, 50, 100, and 200 μL of transductants are diluted with 7H9Tw to a final volume of 500 μL, and plated on separate plates. It takes about 2–3 days for M. smegmatis, and 3–4 weeks for M. tuberculosis transductants form colonies.
We have described methods for screening ∼3,000 independent mutants, although the method can be scaled up.
Incubate M. tuberculosis at 37 °C for about 3 weeks.
M. smegmatis produces greater biomass when forming biofilms at 30 °C, and this temperature is usually preferred for studying M. smegmatis biofilms. However, 37 °C is preferred for screening transposon mutants of M. smegmatis, because residual transducing phages (phAE781) in the primary colonies can inhibit bacterial growth at 30 °C.
It is important to be mindful of the distinction between a growth-deficient mutant and a biofilm-deficient mutant. While the former grows slowly under all conditions, the latter will show deficiency only when grown under biofilm-specific conditions, without apparent growth defects in planktonic form. The slow-growing mutants are likely to appear as false positives in the primary screen and therefore must be resolved in the secondary screen.
Occurrence of false positives in the 96-well assay is usually more frequent in M. tuberculosis than M. smegmatis. M. tuberculosis is quite fastidious and even slight variations in growth conditions across the wells, particularly with respect to head-space air, can significantly impact in the timing of biofilm formation for otherwise normal cells.
For secondary screening of M. tuberculosis mutants, inoculate 250 μL of cells in a 250 mL bottle containing 25 mL of Sauton's medium. Tighten the lid firmly and incubate undisturbed at 37 °C for 3 weeks, after which loosen the lids and follow the growth of the mutant biofilms for the next 2 weeks. Compare the biofilms with a wild-type control in a secondary screen.
Acknowledgments
This work was supported by NIH grants AI079288 to A.O., AI064494 to G.F.H., and AI026170 and AI097548 to W.R.J.
References
- 1.Vlamakis H, Chai Y, Beauregard P, Losick R, Kolter R. Sticking together: building a biofilm the Bacillus subtilis way. Nat Rev Microbiol. 2013;11(3):157–168. doi: 10.1038/nrmicro2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mulcahy LR, Isabella VM, Lewis K. Pseudomonas aeruginosa biofilms in disease. Microb Ecol. 2013 doi: 10.1007/s00248-013-0297-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hall-Stoodley L, Costerton JW, Stoodley P. Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol. 2004;2(2):95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- 4.Islam MS, Richards JP, Ojha AK. Targeting drug tolerance in mycobacteria: a perspective from mycobacterial biofilms. Expert Rev Anti Infect Ther. 2012;10(9):1055–1066. doi: 10.1586/eri.12.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ojha A, Hatfull GF. The role of iron in Mycobacterium smegmatis biofilm formation: the exochelin siderophore is essential in limiting iron conditions for biofilm formation but not for planktonic growth. Mol Microbiol. 2007;66(2):468–483. doi: 10.1111/j.1365-2958.2007.05935.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ojha A, Anand M, Bhatt A, Kremer L, Jacobs WR, Jr, Hatfull GF. GroEL1: a dedicated chaperone involved in mycolic acid biosynthesis during biofilm formation in mycobacteria. Cell. 2005;123(5):861–873. doi: 10.1016/j.cell.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 7.Ojha AK, Baughn AD, Sambandan D, Hsu T, Trivelli X, Guerardel Y, Alahari A, Kremer L, Jacobs WR, Jr, Hatfull GF. Growth of Mycobacterium tuberculosis biofilms containing free mycolic acids and harbouring drug- tolerant bacteria. Mol Microbiol. 2008;69(1):164–174. doi: 10.1111/j.1365-2958.2008.06274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O'Toole GA, Pratt LA, Watnick PI, Newman DK, Weaver VB, Kolter R. Genetic approaches to study of biofilms. Methods Enzymol. 1999;310:91–109. doi: 10.1016/s0076-6879(99)10008-9. [DOI] [PubMed] [Google Scholar]
- 9.Kolter R, Greenberg EP. Microbial sciences: the superficial life of microbes. Nature. 2006;441(7091):300–302. doi: 10.1038/441300a. [DOI] [PubMed] [Google Scholar]
- 10.Xu KD, Stewart PS, Xia F, Huang CT, McFeters GA. Spatial physiological heterogeneity in Pseudomonas aeruginosa biofilm is determined by oxygen availability. Appl Environ Microbiol. 1998;64(10):4035–4039. doi: 10.1128/aem.64.10.4035-4039.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Werner E, Roe F, Bugnicourt A, Franklin MJ, Heydorn A, Molin S, Pitts B, Stewart PS. Stratified growth in Pseudomonas aeruginosa biofilms. Appl Environ Microbiol. 2004;70(10):6188–6196. doi: 10.1128/AEM.70.10.6188-6196.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vlamakis H, Aguilar C, Losick R, Kolter R. Control of cell fate by the formation of an architecturally complex bacterial community. Genes Dev. 2008;22(7):945–953. doi: 10.1101/gad.1645008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kolter R, Losick R. One for all and all for one. Science. 1998;280(5361):226–227. doi: 10.1126/science.280.5361.226. [DOI] [PubMed] [Google Scholar]
- 14.Serra DO, Hengge R. Stress responses go 3D—the spatial order of physiological differentiation in bacterial macrocolony biofilms. Environ Microbiol. 2014 doi: 10.1111/1462-2920.12483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stoodley P, Sauer K, Davies DG, Coster ton JW. biofilms as complex differentiated communities. Annu Rev Microbiol. 2002;56:187–209. doi: 10.1146/annurev.micro.56.012302.160705. [DOI] [PubMed] [Google Scholar]
- 16.Pang JM, Layre E, Sweet L, Sherrid A, Moody DB, Ojha A, Sherman DR. The polyketide Pks1 contributes to biofilm formation in Mycobacterium tuberculosis. J Bacteriol. 2012;194(3):715–721. doi: 10.1128/JB.06304-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fux CA, Costerton JW, Stewart PS, Stoodley P. Survival strategies of infectious biofilms. Trends Microbiol. 2005;13(1):34–40. doi: 10.1016/j.tim.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 18.Nickel JC, Ruseska I, Wright JB, Coster ton JW. Tobramycin resistance of Pseudomonas aeruginosa cells growing as a biofilm on urinary catheter material. Antimicrob Agents Chemother. 1985;27(4):619–624. doi: 10.1128/aac.27.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bjarnsholt T, Ciofu O, Molin S, Givskov M, Hoiby N. Applying insights from biofilm biology to drug development—can a new approach be developed? Nat Rev Drug Discov. 2013;12(10):791–808. doi: 10.1038/nrd4000. [DOI] [PubMed] [Google Scholar]
- 20.WHO. The burden of diseases caused by TB. Global Tuberculosis Report. 2012:8–28. [Google Scholar]
- 21.O'Garra A, Redford PS, McNab FW, Bloom CI, Wilkinson RJ, Berry MP. The immune response in tuberculosis. Annu Rev Immunol. 2013;31:475–527. doi: 10.1146/annurev-immunol-032712-095939. [DOI] [PubMed] [Google Scholar]
- 22.Tufariello JM, Chan J, Flynn JL. Latent tuberculosis: mechanisms of host and bacillus that contribute to persistent infection. Lancet Infect Dis. 2003;3(9):578–590. doi: 10.1016/s1473-3099(03)00741-2. [DOI] [PubMed] [Google Scholar]
- 23.Zumla A, Nahid P, Cole ST. Advances in the development of new tuberculosis drugs and treatment regimens. Nat Rev Drug Discov. 2013;12(5):388–404. doi: 10.1038/nrd4001. [DOI] [PubMed] [Google Scholar]
- 24.Sambandan D, Dao DN, Weinrick BC, Vilcheze C, Gurcha SS, Ojha A, Kremer L, Besra GS, Hatfull GF, Jacobs WR., Jr Keto-mycolic acid-dependent pellicle formation confers tolerance to drug-sensitive Mycobacterium tuberculosis. MBio. 2013;4(3):e00222–13. doi: 10.1128/mBio.00222-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Recht J, Kolter R. Glycopeptidolipid acetylation affects sliding motility and biofilm formation in Mycobacterium smegmatis. J Bacteriol. 2001;183(19):5718–5724. doi: 10.1128/JB.183.19.5718-5724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bardarov S, Bardarov S, Jr, Pavelka MS, Jr, Sambandamurthy V, Larsen M, Tufariello J, Chan J, Hatfull G, Jacobs WR., Jr Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology. 2002;148(Pt 10):3007–3017. doi: 10.1099/00221287-148-10-3007. [DOI] [PubMed] [Google Scholar]
- 27.Bardarov SS, Bardarov SS, Jr, Jacobs WR., Jr Transposon mutagenesis in mycobacteria using conditionally replicating mycobacteriophages. Methods Mol Med. 2001;54:43–57. doi: 10.1385/1-59259-147-7:043. [DOI] [PubMed] [Google Scholar]
- 28.Bardarov S, Kriakov J, Carriere C, Yu S, Vaamonde C, McAdam RA, Bloom BR, Hatfull GF, Jacobs WR., Jr Conditionally replicating mycobacteriophages: a system for transposon delivery to Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 1997;94(20):10961–10966. doi: 10.1073/pnas.94.20.10961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Snapper SB, Melton RE, Mustafa S, Kieser T, Jacobs WR., Jr Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol Microbiol. 1990;4(11):1911–1919. doi: 10.1111/j.1365-2958.1990.tb02040.x. [DOI] [PubMed] [Google Scholar]