

Abstract
Recent studies have suggested that nitric oxide (NO) binding to hemoglobin (Hb) may lead to the inhibition of sickle cell fiber formation and the dissolution of sickle cell fibers. NO can react with Hb in at least 3 ways: 1) formation of Hb(II)NO, 2) formation of methemoglobin, and 3) formation of S-nitrosohemoglobin, through nitrosylation of the β93 Cys residue. In this study, the role of β93 Cys in the mechanism of sickle cell fiber inhibition is investigated through chemical modification with N-ethylmaleimide. UV resonance Raman, FT-IR and electrospray ionization mass spectroscopic methods in conjunction with equilibrium solubility and kinetic studies are used to characterize the effect of β93 Cys modification on Hb S fiber formation. Both FT-IR spectroscopy and electrospray mass spectrometry results demonstrate that modification can occur at both the β93 and α104 Cys residues under relatively mild reaction conditions. Equilibrium solubility measurements reveal that singly-modified Hb at the β93 position leads to increased amounts of fiber formation relative to unmodified or doubly-modified Hb S. Kinetic studies confirm that modification of only the β93 residue leads to a faster onset of polymerization. UV resonance Raman results indicate that modification of the α104 residue in addition to the β93 residue significantly perturbs the α1β2 interface, while modification of only β93 does not. These results in conjunction with the equilibrium solubility and kinetic measurements are suggestive that modification of the α104 Cys residue and not the β93 Cys residue leads to T-state destabilization and inhibition of fiber formation. These findings have implications for understanding the mechanism of NO binding to Hb and NO inhibition of Hb S fiber formation.
Keywords: Sickle Cell Disease, Hemoglobin, NO, Fiber Formation, S-nitrosylhemoglobin, Thiol Modification, UV resonance Raman
Introduction
Sickle cell disease is a genetic disorder caused by a single point mutation at the β6 position of hemoglobin (Hb) (β6 Glu→Val), which creates a hydrophobic patch on the surface of the molecule that, under deoxygenating conditions, leads to the formation of long fibers [1-3]. Nitric oxide has been implicated as a potential treatment for sickle cell disease (SCD), either by increasing the oxygen affinity of sickle cell erythrocytes or by vasodilation [4-7]. In addition, a common treatment for SCD, hydroxyurea (HU) stimulates production of Hb F and potentially leads to formation of NO. Long term effects of HU therapy are attributed to Hb F production, while shorter term effects were attributed to NO production [8-10]. At present, the mechanism of NO action with hemoglobin particularly with respect to inhibition of sickle cell fiber formation is unknown.
The reactions of NO with Hb are complex and can proceed by at least three competing pathways: formation of S-nitrosohemoglobin (SNO-Hb), oxidation of the heme iron (Fe 2+ → Fe3+), and formation of nitrosylhemoglobin (HbNO). Partially liganded NO samples can result in the formation of α-nitrosylhemoglobin, in which the molecule retains a T- or deoxy quaternary state structure. Partially liganded HbS-NO does not significantly increase fiber solubility, as it does not stabilize or promote the formation of the R quaternary state [11]. These results call into question mechanisms, which require increased O2 affinity as a consequence of HbNO formation.
Other investigators have determined that limited exposure of deoxy Hb to NO leads to nitrosylation of β-subunit hemes and subsequent oxygenation leads to the formation of SNO-Hb [12]. Formation of SNO-Hb primarily occurs when NO binds to the β93 Cys residues [13-15] and the S-nitroso form of hemoglobin has been observed to exhibit increased O2 affinity [16].
A recent model for NO action, proposed by Stamler and co-workers [17, 18] suggests that Hb quaternary state, and thus O2 affinity, is mediated by the allosteric transfer of NO from the heme irons to the β93 Cys residues. This model stems from the observation that SNO-Hb formation is favored when Hb is in the R- or fully liganded state, while HbNO formation is favored when Hb is in the T-state [17]. In this model, NO transport is related to oxygen tension, which implies that a detectable concentration of NO exists in the blood, and that NO activity is preserved by binding to Hb [19]. The inhibitory effect in SCD is attributed to the formation of SNO-Hb, which stabilizes the R-state and increases HbS fiber solubility. Delivery of NO to the vasculature is achieved by changes in allosteric state and is dependent on the degree of O2 saturation.
The work of Gladwin and co-workers did not find evidence for the allosteric cycling of NO from the heme iron to the β93 Cys residues and they determined that generation of Cys-modified Hb did not influence the reaction kinetics [20-22]. These experiments were not supportive of the allosteric cycling model, and suggested that β93 Cys plays a minor role in the regulation of NO activity by Hb. These and other findings have led to an alternate model, in which Hb acts as a nitrite reductase and nitrite is a storage pool for NO. In this model deoxy R-state hemes exhibit greater reductase activity than deoxy T-state hemes, leading to maximal rates of NO production when Hb is partially saturated with O2 to a level of 40-60% [22-25]. Release of NO under these conditions leads to vasodilation; thus, NO release and vasodilation are intrinsically coupled to the allosteric state of Hb. Exposure to nitrite also leads to the formation of measurable amounts of SNO-Hb and an arterial to venous gradient of nitrite [24]. The organ chamber bioassay experiments of Stamler and co-workers, however, are suggestive that the reaction of nitrite and deoxy Hb does not relax blood vessels unless SNO-Hb is formed [26] and that SNO-Hb formation involves a Fe(III)NO precursor [27]. One significant difference between the two models lies in the role of SNO-Hb as an intermediate in the process of NO delivery. At present, the function of the β93 Cys residue in the regulation of NO activity, blood flow and dissolution of Hb S fibers remains a point of debate [28].
In this study, formation of the nitrosothiol as an inhibitor of sickle cell fiber formation is addressed by chemical modification with N-ethylmaleimide (NEM). The choice of NEM as the modifying agent stems from its specific reactivity with Cys sulfhydryl groups under conditions of neutral pH [29]. In addition, previous work had indicated that only the β93 Cys residues were reactive out of the six Cys residues present in an Hb tetramer [30-32]. Chemical modification of β93 Cys simulates SNO formation at β93 without the possibility of other reactions, such as formation of HbNO or methemoglobin. This study specifically examines how NEM modification at β93 Cys affects Hb structure and sickle cell fiber formation.
The structure of the NEM-modified Hb is analyzed using UV resonance Raman spectroscopy, which has been used extensively to characterize the structure of Hb quaternary states and Hb S fibers [33, 34]. The choice of UVRR spectroscopy stems from the fact that it has proven to be an extremely versatile method for studying Hb structure and dynamics. Spiro and co-workers have previously identified spectral features diagnostic of the critical α1β2 interface [33], which are monitored as a function of quaternary state and fiber formation. In addition, Phe signals, reflective of local environment, increase in intensity with fiber formation, and are used as monitors of the fiber state [34, 35]. Thus, one advantage of UVRR spectroscopy over other spectroscopic techniques is the ability to selectively probe both the α1β2 subunit interface, and the intermolecular 1β2–2β1 interaction that promotes Hb S fiber formation.
The current work supports a model in which NEM modification occurs at the α104 Cys residue in addition to β93, as demonstrated by FT-IR, electrospray ionization mass spectroscopy and reversed-phased liquid chromatography/tandem mass spectrometry (LC-ESI-MS/MS). Equilibrium solubility and kinetic data indicate that modification of only β93 enhances Hb S fiber formation while α104 modification counteracts that effect. UVRR measurements of the structure of NEM-modified samples are indicative of weaker H-bonding at the α1β2 interface, in the doubly-modified samples, which destabilizes the T-state, when compared to native Hb S. These effects are discussed in light of Hb S fiber formation and inhibition.
Materials and Methods
Hemoglobin Preparation
Hb S was isolated from the blood of SS and AS patients, following the procedure as described [36].
Thiol Modification
All chemical modifications of the –SH residues of Hb A and Hb S were performed using NEM (Sigma), in 0.1 M potassium phosphate buffer, pH 7.1, at room temperature [29, 37]. Thiol modification of hemoglobin was accomplished by 2 different procedures, hereafter referred to as NEM low (NEM-L) and NEM high (NEM-H). Hemoglobin was reacted with NEM at a 2:1 NEM:Hb ratio for 3 hours in the NEM-L modification method [33, 34], and a NEM:Hb ratio of 5:1 for 4 hours in the NEM-H method [37]. The concentration of protein ranged from 1.5 mM Hb to 3.5 mM Hb. Protein concentrations are reported with respect to tetrameric Hb for all experiments. Following reaction, excess NEM was removed via gel filtration on a G-25 column (Pharmacia) and the samples were dialyzed overnight at 4°C against a 0.1 M potassium phosphate buffer, pH 7.1.
Reactive Thiol Quantitation
The extent of thiol modification was determined using PMB [38], and by a Thiol and Sulfide Quantitation Kit (Molecular Probes, OR) [39]. The concentration of reactive Cys residues was determined following the procedure of Park et al. [40]. Quantitation measurements were performed in a 0.1 M potassium phosphate buffer, pH 7.1, on HbCO, NEM-L Hb and NEM-H Hb at room temperature. The absorbance at 250 nm (Boyer method), and 412 nm (Thiol Quantitation Kit), was measured using a Beckman DU 650 spectrophotometer (Beckman). The concentration of active Cys residues was determined by comparison with a standard curve constructed with known L-Cys concentrations. Quantitation of reactive thiols in unmodified and modified Hb A and Hb S was done on at least 3 independent samples. Within the accuracy of the experiment, Hb A and Hb S behaved exactly the same with respect to Cys reactivity.
Fourier Transform Infrared Spectroscopy
FT-IR analysis was performed on Hb A, NEM-L Hb A, and NEM-H Hb A. Samples were prepared in 0.1 M potassium phosphate buffer, pH 7.1, and concentrated to approximately 6 mM Hb, using 30,000 MWCO Centriprep concentrators (Amicon). Samples were maintained in the CO form by purging samples thoroughly with CO during concentration. FT-IR spectra were obtained using a Bruker FT-IR spectrophotometer equipped with a MCT liquid nitrogen-cooled detector and Opus NT programming spectra software (spectrophotometer was made available to us courtesy of Prof. Sean Decatur, Mt. Holyoke College). The optical bandpass for the experiments was 4000 to 200 cm-1. Sample data were collected using a demountable cell holder with 0.5 mm pathlength CaF2 windows, at a constant temperature of 10 °C. A total of 512 interferograms from each sample were averaged. Spectra were initially obtained as percent transmittance and converted to absorption. A buffer spectrum was subtracted from each sample spectrum. All spectral analyses were performed with GramsAI spectral analysis software (ThermoGalactic, Salem, NH).
Mass Spectrometry
One-dimension electrospray ionization mass spectrometry (ESI-MS) was performed on a TOF analyzer mass spectrometer (Keck Biotechnology Resource Laboratory, Yale University, New Haven, CT). Samples of NEM-L Hb, NEM-H Hb and Hb A were prepared in a 0.1 M potassium phosphate, pH 7.1 buffer, at a concentration of 10 μM Hb.
For reversed-phase electrospray ionization tandem mass spectrometry (LC-ESI-MS/MS) NEM-L and NEM-H samples at a concentration of 2 mM Hb were denatured with 8 M urea in the presence of 1 mM L-Cys overnight. Following denaturation, the samples were diluted to 100 μM in 20 mM Tris, 5 mM EDTA buffer, pH 8.3. Samples were simultaneously digested with 20 μM sequencing grade trypsin (Promega, Madison, WI), and 20 μM sequencing grade chymotrypsin (Roche Diagnostics, Indianapolis, IN) for 24 hours at 37°C. Following digestion, the samples were purified using C18 Ultramicrospin columns (Nest Group, Southborough, MA). Reversed-phase microcapillary liquid chromatography followed by tandem mass spectrometry was performed as described [41] with the following modifications. All samples were loaded onto manually pulled fused silica microcapillary columns (0.75 μm ID) using a pressure cell (Brechbuhler, Huston, TX). A precolumn was not used, and the column resin was exclusively 200A C18 resin. Three CID (collision-induced dissociation) scans were alternated with each survey scan over a 90 minute gradient of acetonitrile from 5% to 35%, followed by a 10 minute gradient from 35% to 80% acetonitrile. Data analysis and identification of peptide fragments were performed using the Sequest and Bioworks software (Thermo Electron Corporation, Waltham, MA), and Microsoft Excel (Microsoft Corporation, Seattle, WA).
Equilibrium Solubility
Equilibrium solubility measurements were performed on Hb S, NEM-L Hb S, and NEM-H Hb S. Deoxy Hb S samples at a concentration of approximately 1 mM Hb were prepared from HbCO by photolysis under an N2 atmosphere at 4 °C, in a 1.0 M potassium phosphate buffer, pH 7.1, with 10 mM sodium dithionite. Samples were photolyzed for at least 2 hours to ensure complete deoxygenation. Formation of deoxy Hb was confirmed by visible absorbance at 555 nm (ε555= 12.5 mM-1cm-1). Deoxygenated samples were transferred into N2-purged vials through an N2-purged cannula needle. Concentration determinations were done after photolysis by transferring a 10 μL aliquot of each sample into 1000 μl of CO-purged buffer, in an N2 atmosphere, and fitting the resulting absorbance spectrum with HbCO, deoxyHb, and metHb basis spectra [42]. Deoxy samples (95% or greater) were incubated for 24 hours at room temperature. After 24 hours, the samples were centrifuged at 10,000 ×g for 3 hours, followed by filtration through a 0.2 um filter (Pall Corporation), to remove any unpelleted aggregates [43]. Following centrifugation and filtration, the total Hb concentration of the supernatant was determined following the procedure described for the initial concentration determination. Supersaturation ratio was determined as follows, S = Cinitial/Csoluble. Reported ratios are the average of at least 4 independent experiments.
Kinetics of Hb S fiber formation
Deoxy samples, prepared as above, were transferred via an N2-purged cannula needle into N2- purged 1.0 mm pathlength glass cuvettes. Fiber formation was initiated by a temperature jump from 10 to 35 °C, and absorbance at 700 nm was monitored at 30 second intervals for a total of 6000 seconds. The initial 15% of the progress curves were fit to the following equation, which results from the integration of the linearized rate equations of the double nucleation mechanism [44, 45]:
| (1) |
where A0 and Amax are the initial and final absorbances, and Af and B are the shape and rate parameters used to define the initial portion of the polymerization curve. Reported values result from an average of at least 3 independent experiments. Absorbance curves were also fit to a sigmoidal equation:
| (2) |
Where A1 and A2 are the initial and final absorbance and x0 is the point at which 50% of the total absorbance change has occurred; a time which reflects both the delay time and rapid gelation stages [46].
UV resonance Raman Spectroscopy
A Q-switched, Nd:YLF pumped Ti:sapphire laser system (Quantronix, New York) was used to generate the excitation wavelengths, by frequency quadrupling the IR output of the Ti:sapphire laser using two BBO crystals, as previously described [47]. All spectra were collected with an acquisition time of 15 minutes, and the spectra shown represent at least 3 hours of averaged data. Samples were held in a continuously spinning, gastight, aluminum disk with a polished sapphire window (Esco Products Inc., NJ). Deoxy samples were prepared as for solubility experiments. Hb S fiber samples were prepared by temperature jump from 10 °C to 35 °C, and fiber formation was monitored by increasing sample turbidity at 700 nm. Hb S tetramer samples at a concentration of 0.25 mM Hb were continuously cooled with N2 gas passed through a dry ice/2-propanol bath, while Hb S fibers at a concentration of either 3.0 mM Hb in 0.1 M phosphate or 1.0 mM Hb with 1.0 M phosphate were examined at room temperature. Sample position was adjusted every 15 minutes, and the entire sample volume was changed after 1 hour of laser exposure. The Phe band occurring at 2020 cm-1, was used to perform spectral subtractions, as previously described [34]. Data manipulation and analysis were performed using GramsAI spectral analysis software (ThermoGalactic, Salem, NH).
Results
NEM Modification of Hb: Quantitation of extent and location
To investigate the mechanism of β93 modification in the dissolution of Hb S fibers, NEM modification of the Cys residues in Hb was performed. Since nitrosylation can occur at either the heme iron or at the Cys residues, and can lead to metHb formation, NEM modification was used as a model for NO binding to the β93 Cys residue. A distinct advantage of chemical modification is that the direct effect of Cys modification can be examined without formation of HbNO or metHb.
Two modification procedures were utilized to compare the effects of Cys modification with high and low levels of NEM; these procedures are referred to as NEM-H and NEM-L, respectively. The NEM-L modification protocol was developed by monitoring the decrease in NEM absorbance at 305 nm [29] (data not shown), while the NEM-H protocol was taken from Ho and co-workers [37]. Cys reactivity under these conditions was examined by both the Boyer method [38] and through a papain-based assay, in which a disulfide-inhibited derivative of papain is stoichiometrically reduced in the presence of thiols and enzyme activity is measured spectrophotometrically with L-BAPNA [39]. Both methods indicated that Cys reactivity for Hb A and Hb S was the same within error and slightly greater than the expected value of 2 reactive Cys/Hb tetramer (Table 1). Overall, these results demonstrated that Cys reactivity was reduced upon reaction with NEM, and the number of reactive thiols decreased with longer reaction times and increased reagent as expected; nevertheless, for both the NEM-L and the NEM-H modification procedures these quantitation results were suggestive of incomplete modification of β93 Cys residues (Table 1).
Table 1. Reactive Cys residues per Hb tetramer.
| Boyera | L-BAPNAb | |
|---|---|---|
|
|
||
| Hb A | 2.2 ± 0.4 | 2.5 ± 0.5 |
| Hb S | 1.71 ± 0.3 | 2.4 ± 0.3 |
| NEM-L Hb A | 1.74 ± 0.5 | 1.5 ± 0.7 |
| NEM-L Hb S | n/a | 1.8 ± 0.4 |
| NEM-H Hb A | 0.82 ± 0.1 | 0.9 ± 0.4 |
| NEM-H Hb S | n/a | 1.2 ± 0.1 |
Average from 3 independent measurements,
Average from 4 independent measurements
FT-IR Spectroscopy
Given the relatively high error associated with the determination of Cys reactivity by both the Boyer and L-BAPNA methods (Table 1) and to ensure complete modification of the β93 Cys residue, FT-IR spectroscopy was employed to measure both the extent and location of NEM modification. The IR spectra obtained are consistent with those previously reported [48, 49] (Figure 1) in which three S-H absorption bands are observed at 2566, 2553, and 2588 cm-1, and are assigned to the β112, α104, and β93 Cys residues, respectively. The S-H band at 2588 cm-1 assigned to β93 was not observed in either the NEM-H Hb or NEM-L Hb spectrum (Figure 1), indicating that complete modification of the β93 Cys was accomplished by both modification protocols, in contrast to the concentration of reactive -SH determined by the Boyer and L-BAPNA methods.
Figure 1.

FT-IR spectra of the S-H stretching region. FT-IR spectra of Hb A (top), NEM-L Hb A (center) and NEM-H Hb A (bottom) in the 2500-2700 cm-1 S-H stretching region are shown. Sample concentration was 6-7 mM Hb, in a 0.1 M potassium phosphate buffer, pH 7.1. Curve fits were performed using a mixed Gaussian/Lorentzian peak fitting function and are shown with dotted lines. Spectra have been arbitrarily displaced vertically.
Analyses of modified and unmodified Hb FT-IR spectra with respect to peak area, height, and width were performed to determine whether Cys residues other than β93 were also modified, and if the extent of Cys modification was dependent on the concentration of NEM and the reaction time. Previous work had suggested that the other Cys residues should not be modified by the relatively mild reaction conditions used in this study [15, 29, 37, 50-52]. The peak areas of the α140 and β112 S-H absorption bands, as determined from the fitting of the data, were compared and the ratio was found to be similar for Hb A and NEM-L Hb A (Figure 2). However, analysis of the NEM-H Hb spectra consistently indicated a decrease of 25% in the α104/β112 S-H band peak area ratio, when compared with either Hb A or NEM-L Hb, respectively (Figure 2). The source of the decreased peak area ratio could either result from a decrease in the absorbance of the α104 Cys residues, an increase in the β112 Cys absorbance, or both. The ESI-MS results, as described below, suggest that the α104 Cys residues are the source of the peak ratio decrease. We chose to examine a peak area ratio rather than an absolute intensity because of the baseline corrections and buffer subtractions applied to the spectra. Peak height and peak width ratios of the α104 and β112 S-H bands yield the same qualitative results (data not shown).
Figure 2.

Modification at other Cys residues from FT-IR peak area absorbance. Ratio of the α104 to β112 FT-IR absorption bands in terms of peak area are shown. The intensity and width of the fitted curves were used to calculate peak area. Original data is shown in figure 1.
Electrospray Ionization Mass Spectrometry
To further examine the number of sites of NEM modification per Hb chain, ESI-MS was used, since it can sensitively differentiate mass changes as a result of NEM modification [14]. In both NEM-H Hb and NEM-L Hb samples, a peak was observed at 15,993 Da, corresponding to the mass of a β chain with one NEM molecule. In some preparations, a low intensity peak at 15,867 Da, corresponding to the mass of an unmodified β chain, was observed for NEM-L (Figure 3). The percent intensity of this peak is within the error level for the measurement (12 ± 16). A peak corresponding to the mass of unmodified β chain was not observed for NEM-H Hb samples (Table 2). Thus, both the ESI-MS and FT-IR data consistently indicate that the β93 Cys residue is completely modified by the NEM-H protocol, and almost 90% modified by the NEM-L protocol, in contrast to the Boyer and L-BAPNA methods of quantitation.
Figure 3.

Histogram of the percentage of chain labeling as determined by ESMS. Percentages were determined relative to unmodified Hb A. Samples of NEM-L Hb, NEM-H Hb and Hb A were prepared in a 0.1 M potassium phosphate, pH 7.1 buffer, at a concentration of 10 μM Hb. Data is also shown in Table 2.
Table 2. ESMS determination of α and β chain molecular weight.
| α chaina 15,125 D | β chaina 15,867 D | α chain modifieda 15,252 Db | β chain modifieda 15,992 Dc | |
|---|---|---|---|---|
|
|
||||
| Hb A | 100 | 100 | — | — |
| NEM-L | 88 ± 7 | 12 ± 16 | 12 ± 7 | 88 ± 17 |
| NEM-H | 67 ± 8 | 0 | 33 ± 11 | 100 |
Percentage of chain present relative to total.
MW = 1 α chain + 1 NEM.
MW = 1 β chain + 1 NEM
In the ESI-MS spectra, the unmodified α chain is the dominant species present in all samples, however, a peak at 15,253 Da, corresponding to an α chain with one NEM group, was present in the NEM-H Hb samples. The peak intensities of individual species were compared, and these analyses revealed that 33 ± 11% of the total α chain intensity of NEM-H Hb corresponds to modified α chain, while only 12 ± 7% of the total α chain intensity of NEM-L Hb can be assigned to modified α chain (Table 2). Modification of the α-chain was considered to occur at the α104 Cys residue only because of the selective reactivity of the NEM group for Cys residues under the reaction conditions used [29-32].
Reversed-Phase Liquid Chromatography Electrospray Ionization Tandem Mass Spectrometry (LC-ESI-MS/MS)
To verify the site of NEM modification, LC-ESI-MS/MS was performed. The three most intense peptide ions in each MS1 survey scan of eluting peptides were selected for collision-induced dissociation to generate MS2 spectra. Sequest software was then used to match the resulting MS2 mass spectra with all possible theoretical peptides from the human hemoglobin sequence (PDB accession: 2hbs), using a dynamic search that identifies peptides with unmodified cysteine residues and those containing NEM-modified cysteine residues (Cys + 125.13 Da). Analysis of Hb β chains confirms that the β93 Cys residue is the primary site of NEM modification. By this method, a small amount of modification is observed at the β112 Cys residue. The ESI-MS results are suggestive that modification at this site is less than 5% (see above). Analysis of Hb α chains confirms that modification is occurring at the α104 Cys residues. For both Hb α and β chains, NEM modification at sites other than Cys residues was not observed. Thus, FT-IR, ESI-MS, and tandem MS results consistently indicate complete modification of β93 Cys by the NEM-L and NEM-H modification methods. In addition, these results point to partial modification of the α104 Cys residue with little to no modification of Cys β112 by the NEM-H method.
How does NEM modification affect Hb S fiber formation?
Equilibrium Solubility
To determine the affect of NEM modification on Hb S fiber formation, equilibrium solubility measurements were performed. Solubility is described as the concentration of deoxy Hb S present in the supernatant after fibers are allowed to form and sedimented by centrifugation. The ratio of initial concentration (C0) to the soluble fraction (Cs) is known as the supersaturation ratio and is expressed as: S ≡ C0/Cs [53]. The solubility and the supersaturation ratio are a measure of the relative propensity of Hb molecules to form fibers. These measurements were performed in a 1.0 M phosphate buffer to minimize the amount of sample used. Fiber formation in high phosphate buffers has been characterized extensively [54] and thus, provides a good comparison point for our measurements. Measurements were performed on Hb S, NEM-L Hb S and NEM-H Hb S samples at a concentration of approximately 1 mM Hb (6.4 g/dl). Under these conditions, we determined a supersaturation ratio for Hb S of 1.29 ± 0.12, which is consistent with previously reported values obtained under similar conditions [54] (Table 3). The overall solubility of NEM-L Hb S is considerably lower than Hb S (0.72 mM vs. 1.11 mM) and this is also reflected in the supersaturation ratio for NEM-L Hb S (1.83 ± 0.14). These findings are indicative of a greater degree of fiber formation in the NEM-L samples with respect to Hb S. In contrast, the solubility of NEM-H is equivalent to that of deoxy Hb S; although the supersaturation ratio is slightly lower than that observed for Hb S (1.11 vs. 1.29 – Table 3). The larger supersaturation ratio determined for NEM-L Hb relative to NEM-H Hb is suggestive that modification at β93 enhances fiber formation, whereas the additional NEM modification at α104 renders the protein more soluble.
Table 3.
Nucleation and polymerization parameters determined from equilibrium solubility experiments and kinetic progress curves.
| Co (mM Hb) | Cs (mM Hb) | S1 | i*2 | j*3 | C (mM Hb) | Td 15% (s) | Td 50% (s) | Af (mM) | B (s-1) | |
|---|---|---|---|---|---|---|---|---|---|---|
|
|
||||||||||
| Hb S | 1.44 ± 0.14 | 1.11 ± 0.13 | 1.29 ± 0.12 | 6.18 | 7.72 | 1.2 ± 0.11 | 886 ± 79 | 1286 ± 105 | 1.8 × 10-6 | 0.20 |
| NEM-L | 1.32 ± 0.12 | 0.72 ± 0.11 | 1.83 ± 0.14 | 4.19 | 5.23 | 1.1 ± 0.10 | 294 ± 67 | 473 ± 113 | 6.1 × 10-6 | 0.25 |
| NEM-H | 1.24 ± 0.10 | 1.12 ± 0.15 | 1.11 ± 0.13 | 8.51 | 10.64 | 1.2 ± 0.12 | 1589 ± 93 | 1781 ± 128 | 1.4 × 10-6 | 0.14 |
S is the supersaturation ratio; S = Co/Cs [53].
i* is the size of the homogeneous nucleus; i* = -(4RT + δ1μPC)/RT ln S where δ1 describes the fraction of intermolecular bonds in the nucleus relative to the infinite polymer, μPC is the chemical potential contribution to the infinite polymer from the bonding free energy between molecules. Both of these parameters are taken from Ferrone et al. [45].
j* is the size of the heterogeneous nucleus; j* = i* + (1/ln S) [45]
Calculated values for homogeneous and heterogeneous nuclei, i* and j*, respectively, using S [44, 45, 55] are also consistent with the determined delay times (Table 3), in which the smallest nucleus sizes are observed for NEM-L. Specifically, if the solubility is decreased, the tendency to aggregate increases and this is reflected in smaller nucleus sizes [45].
Kinetic Studies
The difference in behavior of NEM-L Hb and NEM-H Hb was further explored by monitoring the progress curves of fiber formation using sample turbidity. For all three Hbs, the curves are characterized by an initial period, in which little to no fiber formation is observed, followed by a period of rapid fiber formation (Figure 4). From these curves it is apparent that NEM-L Hb S exhibits the fastest onset to fiber formation, followed by Hb S and NEM-H Hb S. The data were analyzed through consideration of the double nucleation model [44, 45], in which a hyperbolic function, the linear solution to the rate equations, is used to describe the initial portions of the progress curves. The initial 15% of the progress curves yielded excellent fits to this model (not shown), demonstrating that this model adequately describes the data. Delay times were also determined considering the sigmoidicity of the curves, in which a T50 time corresponds to the time at which 50% of the fibers are formed [46]. Either form of analysis qualitatively yields the same results, in which the shortest delay times are observed for NEM-L and the longest delay times are observed for NEM-H (Table 3).
Figure 4.

Kinetic progress curves of Hb S, NEM-L and NEM-H polymerization. Protein concentration was 1.0 mM Hb in 1.0 M potassium phosphate, pH 7.1. Polymerization was initiated by temperature jump from 10-35 °C, and absorbance was measured every 30 seconds.
Differences in Hb and phosphate concentrations and the amount of the temperature jump prohibit exact comparisons of delay times, nevertheless we find that our delay times are generally consistent with those determined previously for Hb S [54]. These progress curves (Figure 4) and associated delay times (Table 3) correlate well with the equilibrium solubility measurements and are suggestive that NEM-L Hb accelerates fiber formation, while NEM-H inhibits it.
Structure of NEM modified Hb S probed by UVRR spectroscopy
Given these differences in physical properties of the NEM-H Hb and NEM-L Hb modified samples, we elected to elucidate the structural differences using UVRR spectroscopy. These measurements were performed under conditions of low and high phosphate concentration and we find that the same structural effects are observed under both conditions. We employ an excitation wavelength of 215 nm to selectively enhance contributions from Phe and Tyr residues, and an excitation wavelength of 230 nm to enhance the signal from Tyr and Trp residues. When an excitation wavelength of 215 nm is used, the spectroscopic signal consists of an average of all the Phe residues; therefore, to resolve the signal from those residues that experience a change in environment or structure upon quaternary state change or fiber formation, we generate difference spectra between the states.
The 1β1-2β2 interaction: Local environment of the β85 Phe in the T-state
The 215 nm-excited spectrum (Supporting information: Figure 1) is dominated by Phe and Tyr ring-stretching modes in the 1585-1614 cm-1 spectral region [34, 56]. In the 215 nm-excited T-R difference spectra, negative peaks are observed at 1605 and 1585 cm-1, which correspond to the Y8a, F8b and F8a vibrational modes of Phe and Tyr residues (Supporting information: Figure 2). An additional negative band is observed at 1550 cm-1, which arises from the W3 mode of Trp residues. The decreased intensity of Tyr and Trp modes is consistent with T-state H-bond formation, as has been previously reported [57]. The intensity decrease of Phe modes in the T-state probably results from an increase in the solvent exposure of one or more Phe residues, since the intensities of some Phe vibrational modes are diagnostic of local environments, where a higher intensity is indicative of a more hydrophobic environment [34, 35]. X-ray crystal structure determinations have suggested that the β85 Phe is more solvent exposed in the T state, and the change in intensity is mainly attributed to that residue [3, 58, 59].
In general, Hb S, NEM-H Hb and NEM-L Hb yield similar T-R difference spectra, and the similarity is suggestive that the overall structures of the T- and R-states, as monitored by Phe vibrational modes, are similar for NEM-modified and unmodified Hb S. Subtle differences in local environment are determined from a comparison of the T-states, which reveal that the Phe environment in NEM-L Hb is more hydrophobic than NEM-H Hb or Hb S, as shown by the positive peaks at 1587 (F8b) and 1607 cm-1 (F8a) in T-T difference spectra (Figure 5). In the NHT - ST spectrum negative peaks are observed at 1587 cm-1 and 1607 cm-1, which is indicative that the Phe environment in NEM-H Hb S is more solvent exposed relative to Hb S. Thus, based on the UVRR results, the ordering of the relative hydrophobicity of Phe residues in the T-state for the different Hb molecules is NEM-L Hb S > Hb S>NEM-H Hb S. This ordering is consistent with the relative propensity to form fibers as determined from the equilibrium solubility and kinetic measurements (see above).
Figure 5.

Comparison of 215 nm excited T-T difference spectra. NEM-LT Hb- Hb ST (top), NEM-HT Hb -Hb ST (center) and NEM-LT Hb-NEM-HT Hb (bottom) difference spectra are shown. T-state samples were 1.0 mM Hb, in a 1.0 M potassium phosphate buffer, pH 7.1, and were prepared as described in Materials and Methods.
The fiber state
Changes in the fiber state are probed by generating fiber-T state (F-T) difference spectra for Hb S, NEM-H Hb, and NEM-L Hb (Figure 6). When an excitation wavelength of 215 nm is used, the F-T spectra are dominated by large positive peaks at 1603 and 1585 cm-1, suggestive of an increase in Phe signal intensity upon fiber formation [34]. A comparison of the relative intensities of the F8b band, with respect to the 1000 cm-1 Phe F12 band reveals that the relative intensity of the F8b band of NEM-L Hb is 64 ± 3% higher than Hb S, and 59 ± 3 % higher than NEM-H Hb. The intensity increase is indicative of a more hydrophobic local environment for NEM-L Hb Phe residues, relative to either Hb S or NEM-H Hb.
Figure 6.

Comparison of 215 nm-excited UVRR F-T difference spectra. F-T difference spectra of Hb S (top), NEM-H Hb (center) and NEM-L Hb (bottom), were obtained with 215 nm excitation. T-state samples were 1.0 mM Hb, in a 1.0 M potassium phosphate buffer, pH 7.1. Fiber samples were 1.0 mM Hb, in a 0.1 M potassium phosphate buffer, pH 7.1. Fiber formation was initiated by a temperature jump from 10 to 35 °C.
The α1β2 interface: Perturbation of H-bonds in the NEM-H T-state
The vibrational modes enhanced at 230 nm correspond primarily to the Tyr and Trp residues in hemoglobin (Supporting information: Figure 3) and the difference spectra monitor the hydrogen bond interactions at the α1β2 interface. In the T-R spectrum of Hb S (Figure 7), the double sigmoidal feature seen at high frequency (>1500 cm-1) is attributed to Tyr Y8a and Y8b modes. These modes are known to shift to higher frequency upon T state formation, as a result of the formation of the α42Tyr-β99Asp and α140Tyr-α93Val hydrogen bonds in the T state [33, 60, 61]. In the NEM-H Hb T-R spectrum (Figure 7), the Tyr high frequency double derivative shape is significantly diminished. The negative peak detected at 1608 cm-1 in the Hb S T-R difference spectra is not observed in NEM-H spectra, because of the smaller frequency shift, which is indicative of weaker Tyr H-bonds. The Y8a frequency shift is largest in Hb S (1.2 cm-1) and smallest in NEM-H Hb (0.4 cm-1) (Figure 8). The Y9a band also does not exhibit a frequency shift upon formation of the T-state in NEM-H Hb, as shown by the absence of the derivative shape in the difference spectrum, and the detection of the peak frequency at 1177 cm-1 as opposed to 1182 cm-1 where it is observed for NEM-L Hb and Hb S (Figure 7). These observations consistently point to a disruption of Tyr H-bonds in NEM-H Hb.
Figure 7.

Comparison of 230 nm-excited UVRR T-R difference spectra. T-R difference spectra of Hb S (top), NEM-H Hb (center) and NEM-L Hb (bottom), were obtained with 230 nm excitation. T- and R-state samples were 1.0 mM Hb, in a 1.0 M potassium phosphate buffer, pH 7.1.
Figure 8.

Comparison of frequency shifts of Tyr vibrational modes. Frequency shifts of the Tyr Y8a vibrational mode of Hb S, NEM-L Hb and NEM-H Hb upon formation of the T- and fiber states are shown. Frequencies were determined from parent spectra obtained with 230 nm excitation (Supporting information: Figure 3), and measured relative to R-state spectra. Sample conditions are as described in Figure 7.
In addition to the smaller frequency shifts, the NEM-H Hb T-R difference spectra also exhibit a decrease in intensity at 1550 cm-1 (Figure 7). This peak arises from the W3 Trp vibrational mode and in previous work the intensity increase was attributed to formation of the intersubunit β237 Trp-α194 Asp H bond [33]. This peak is clearly resolved in both the Hb S and NEM-L Hb T-R difference spectra; however, it appears as a shoulder in the NEM-H Hb T-R difference spectra. The reduced intensity is indicative of a weaker intersubunit H-bond. Thus, the UVRR difference spectra are suggestive that the NEM-H Hb T-state, particularly the α1β2 interface, is perturbed relative to Hb S and NEM-L Hb, potentially because of the α104 Cys modification.
The fiber state
The Hb S, NEM-H Hb and NEM-L Hb F-R difference spectra obtained with 230 nm excitation (Figure 9) are consistent with those previously reported [34]. Increased frequency shifts and higher intensities of the Tyr and Trp modes respectively, are reflective of stronger α1β2 intersubunit H-bonds in the fiber state, relative to either the R- or the T-state (Supporting information: Figure 3). Smaller Tyr frequency shifts are observed in the fiber state for the NEM-H Hb sample (1.0 cm-1) relative to Hb S (2.1 cm-1) and NEM-L (2.0 cm-1). In addition, the W3 peak at 1550 cm-1 is not observed in the NEM-H Hb F-R difference spectra, which is suggestive of considerable weakening of the critical β37-α94 T-state H-bond. These results imply that the α1β2 interface in the T- and fiber states of NEM-H Hb is significantly perturbed relative to Hb S and NEM-L Hb. Since NEM-H Hb is modified at the α104 Cys residue in addition to the β93, we suggest that modification at α104 is more perturbing to the overall T-quaternary state structure than modification at the β93 Cys only.
Figure 9.

Comparison of 230 nm-excited UVRR F-R difference spectra. F-R difference spectra of Hb S (top), NEM-H Hb (center) and NEM-L Hb (bottom), obtained with 230 nm excitation. Samples were 1.0 mM Hb, in 1.0 M potassium phosphate buffer, pH 7.1. Fibers were formed by initiating a temperature jump from 10-35 °C.
Discussion
The α104 Cys residue is modified by NEM
In this investigation, as a model for the effect of S-nitrosothiol formation, Cys residues are chemically modified and the effect on Hb S polymerization is monitored. A variety of different methods, including FT-IR spectroscopy, ESI-MS and tandem MS, indicate NEM modification occurs at both β93 and α104 Cys residues and that the α104 residue is moderately reactive under relatively standard conditions [37]. Previously, modifications at Hb Cys residues other than β93 were only observed at high concentrations of modifying reagent [14, 15, 31]. Benesch and co-workers [31] observed that titration with an 100-fold molar excess of PMB results in the observation of 7.5 reactive –SH groups per mole of Hb, while lower PMB concentrations yield a value of 2.4 reactive –SH groups per mole of Hb. More recently, Ferranti and co-workers [14] demonstrated by ESI-MS that a 100-fold excess of NO relative to Hb leads to S-nitrosothiol formation at the α104 and β112 Cys residues. An important observation from the current work is that under conditions previously thought to only lead to β93 modification [37], modification of the α104 Cys residue also occurs (Figure 10). Furthermore, the physical and structural properties of the doubly-modified Hb are considerably different from Hb S and β93-modified Hb.
Figure 10.
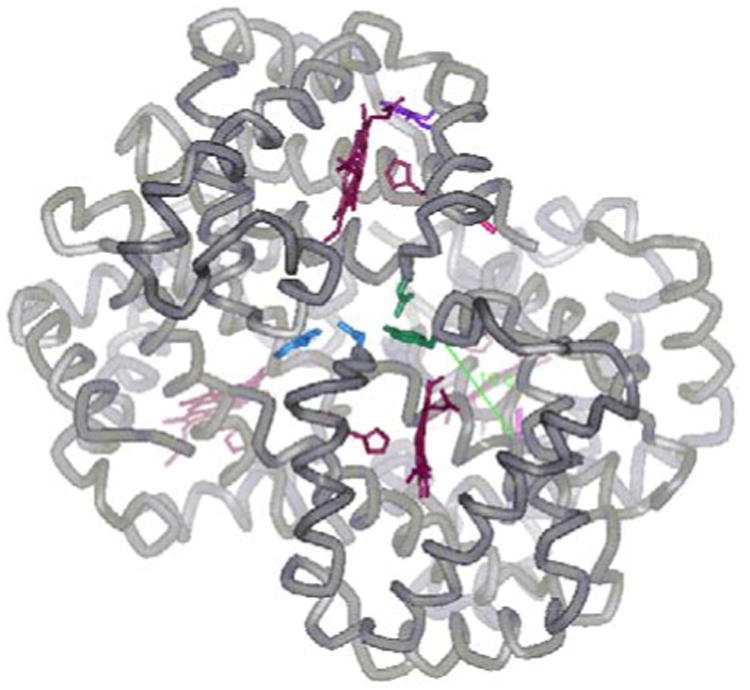
The α1β2 interface. The α1β2 interface is depicted, with the α104 Cys highlighted in light pink, and the β93 Cys highlighted in dark pink. The residues forming intersubunit H-bonds (α42 Tyr and β99 Asp) and (β37 Trp and α94 Asp) are highlighted in green and blue respectively. The β85 Phe and β88 Leu residues of the 1β1-2β2 donor-acceptor interaction are shown in purple. Heme groups and coordinating His residues are shown in dark red. The Cα-Cα distance from the α104 Cys to the α42 Tyr (16.6 Å) is shown in green. Diagram was made using WebLab ViewerPro and the coordinates from the deoxy Hb S X-ray crystal structure (2HBS.PDB).
NEM modification of β93 Cys promotes Hb S fiber formation
Equilibrium measurements reveal that the solubility of NEM-L Hb S is reduced relative to deoxy Hb S and NEM-H Hb S. Kinetic measurements confirm that the onset of fiber formation of NEM-L Hb S is faster than either Hb S or NEM-H Hb S. In contrast to the current work, previous studies [51] observed that the delay times for polymerization of deoxy Hb S modified by NEM were approximately 1.5 times longer than that of native deoxy Hb S. Additionally, Garel et al. [50] observed inhibition of erythrocyte sickling in NEM-modified Hb, as measured by an increase in P50 and by equilibrium solubility. In both instances, the inhibition of the sickling effect was attributed to a destabilization of the T-state and consequent increase in oxygen affinity [50]. These solubilities and the increased delay times obtained for NEM-modified Hb S [50, 51, 62] are consistent with the values obtained by us for NEM-H Hb S.
NMR investigations of glutathione-modified Hb detected a disruption of the α42-β99 H-bond at the α1β2 interface and a localized perturbation of this interface [63]. These findings, which correlate well with current UVRR results on NEM-H Hb S, in conjunction with the solubility and kinetic studies, are strongly suggestive that the doubly-modified Cys species was present in the earlier work, in which inhibition of polymerization was observed.
We propose that the differences between our observations and those reported previously, stem from differences in the level and location of Cys modification. The modifying conditions used by Ho and co-workers [37] correspond to those used for the NEM-H Hb sample, in which approximately 33% of the α-chains are modified. In contrast, less than 10% of the α104 Cys residues are modified in the NEM-L Hb samples. Thus, we suggest that in many of the earlier studies in which only modification of the β93 residues was considered, in fact some modification of the α104 Cys residues was also occurring, and leading to the observed T-state destabilization and increased O2 affinity.
Comparisons of NEM-L Hb and Hb S UVRR spectra are not indicative of large structural perturbations because of the β93 modification and particularly, the α1β2 interface is not perturbed. Because UVRR spectra do not specifically monitor His residues, the presence of the β146 His - β94 Asp interaction cannot be determined; although NMR and X-ray crystallography measurements have suggested that this critical salt bridge is disrupted in thiol-modified Hb [13, 37, 63].
The UVRR spectra obtained with 215 nm excitation are indicative of an increase in hydrophobicity of the local environment of the β85 Phe residue in NEM-L Hb S. We attribute the increase in relative hydrophobicity to the NEM modification, which is in close proximity to this residue. This increase in local hydrophobicity potentially leads to the increased amounts of fiber formation observed in our solubility determinations. In previous studies other thiolmodifying agents have been shown to enhance polymerization and this enhancement results in part from the effect the reagent has on the intermolecular interactions in the Hb S polymer [64]. Thus, the enhanced fiber formation observed for the β93 only modified Hb S is attributed to stronger intermolecular interactions of individual Hb tetramers because of the modifying group.
Earlier studies had suggested that in the formation of fibers, the location of the modified β93 residue with respect to the β6 Val involved in the donor-acceptor interaction determined whether it would have an inhibitory effect [62, 65, 66]. Interestingly, the inhibition of fiber formation was attributed to modified β93 residues located cis to the β6 Val residue which disrupts α-chain intermolecular contacts in the double strands involving the α40, α45 and α47 residues [65, 66]. The UVRR measurements specifically probe the β85 Phe residue in the acceptor pocket and thus, these results are suggestive that modified β93 residues located trans to the β6 Val residue are responsible for the fiber enhancement effect observed in this work.
In previous work where only NEM-modification of β93 residues was observed, the level of modification was usually 50% or less [15, 16]. In the current work, ESI-MS data are indicative of greater than 85% modification of the β93 residues. In the earlier studies with low levels of β93 Cys modification [15, 16], we speculate that the inhibitory effect detected for modification at the cis position dominates over the enhancement effect at the trans position.
NEM modification of the α104 Cys destabilizes the T-state and inhibits fiber formation
Equilibrium solubility and kinetic measurements of NEM-L Hb S demonstrate that fiber formation is increased relative to Hb S, as shown by an increased supersaturation ratio, lower solubility and shorter delay time (Table 3). However, the additional modification at the α104 Cys residue appears to counteract the effect of the modification at the β93 Cys residue alone, as shown by the similar solubilities and supersaturation ratios of Hb S and NEM-H Hb S. UVRR measurements potentially provide a molecular basis for understanding the source of this inhibition. Specifically, these studies reveal that critical α1β2 intersubunit H-bonds, formed by Trp β37 and Tyr α42, are significantly weaker or disrupted in NEM-H Hb (Figure 7) and these changes are propagated in the fiber state (Figure 9). Thus, NEM modification of α104 Cys appears to mediate long-range interactions that destabilize the T-state and inhibit fiber formation, since fiber formation requires the T-state (Figure 10). In earlier studies [37, 50, 51], which examined the modification effects of NEM and other thiol reagents on Hb stability and Hb S fiber formation, the increase in O2 affinity and inhibition of fiber formation was attributed to T-state destabilization. We suggest that these effects mainly result from the additional modification at α104. Modification at β93 could contribute to the effects observed in NEM-H Hb; however, our measurements on samples with primarily β93 modified and not α104 do not show any evidence for perturbation of the α1β2 interface.
A previous UVRR study, which examined the effect of NEM modification on Hb structure, had concluded that the modification perturbs the “switch” region Asp β99 – Tyr α42 H-bond [67]. These findings are consistent with the current UVRR observations for NEM-H Hb, where the frequency shift associated with this H-bond is considerably reduced. We suggest that the modification procedures used in the previous study also led to some modification of the α104 Cys residues. Interestingly, in the previous study Cys modifications were also performed with polyethylene glycol-maleimide based reagents, which did not show the same perturbation of the α1β2 interface. This finding is similar to what is observed in the current work for the NEM-L Hb in which only the β93 Cys residues are modified, and suggest that the bulkier mixed disulfide reagents containing polyethyleneglycol were only able to modify the β93 Cys residues and not the more buried α104 Cys residues. Thus, the current findings are completely consistent with earlier work and further, provide a molecular explanation for the structural differences observed for the different modifying groups.
Implications for NO binding and reactivity
The role of the β93 Cys residue in the regulation of NO delivery and Hb S fiber formation has been the focus of considerable study. Bonaventura and co-workers have observed that S-nitrosylated Hb A and unpolymerized Hb S exhibit an increase in oxygen affinity, and that the presence of 30% SNO-Hb S decreases Hb S polymerization, even at high Hb S concentrations [15]. Formation of SNO-Hb has been linked to vasodilation [5], as well as increased oxygen affinity [16] both of which would indicate S-nitrosylated Hb S has potential to be an effective treatment for sickle cell disease. The current results suggest that a complex interplay of competing effects is potentially associated with thiol modification in Hb, in which the level and location of Cys modification need to be explicitly considered. Nevertheless, the current study is suggestive that modification at the α104 residue may have a potent inhibitory effect and therefore, may be a good target for anti-sickling agents.
Undoubtedly, the differences in physical and chemical properties between NEM and NO need to be considered in any assessment of the effects of Cys modification. The larger size and hydrophobicity of the NEM group may lead to more pronounced effects as compared to NO, particularly with respect to the increased amounts of fiber formation observed for the β93 only modified samples. S-nitrosylation of Cys residues is primarily observed under conditions where the concentration of NO is significantly smaller than Hb [18] and thus, the current results are suggestive that S-nitrosylation would primarily occur at β93. However, the smaller size of NO relative to NEM suggests that it may be more reactive than NEM at the α104 position; although NEM may be more perturbing as a modifying group. The current findings, which demonstrate that α104 modification disrupts the α1β2 interface and inhibits Hb S polymerization, in conjunction with previous reports that SNO-Hb increases O2 affinity, are suggestive that α104 may be a site of NO action in addition to β93. Future studies will examine the effect of S-nitrosylation at α104 with respect to Hb allostery and ligand binding.
Supplementary Material
Supporting Information Available. Figures depicting the 215 nm exited UVRR parent spectra of Hb S, NEM-L Hb and NEM-H Hb (Figure 1), the T-R difference spectra of Hb S, NEM-L Hb, and NEM-H Hb in high and low phosphate buffer (Figure 2), the T-T difference spectra of Hb S, NEM-L Hb, and NEM-H Hb in low phosphate buffer (Figure 3), and the F-T difference spectra of Hb S, NEM-L Hb, and NEM-H Hb in low phosphate buffer (Figure 4). Figures depicting the 230 nm excited UVRR parent spectra of Hb S, NEM-L Hb, and NEM-H Hb (Figure 5), the T-R difference spectra of Hb S, NEM-L Hb, and NEM-H Hb in low phosphate buffer (Figure 6), and the F-R difference spectra of Hb S, NEM-L Hb, and NEM-H Hb in low phosphate buffer (Figure 7). A table containing a list of all the labeled peptides identified by LC-ESI-MS/MS is also given.
Acknowledgments
This work was supported by a grant from the Patrick and Catherine Weldon Donaghue Medical Research Foundation. K. M. K. gratefully acknowledges support from an NIH training grant in Molecular Biophysics (GM08271). C. K. R. acknowledges support from the Howard Hughes Medical Institute.
We are grateful to Drs. Kenneth Bridges (Brigham and Womens Hospital), C. Alvin Head, and Pedro Montero-Huerta (Medical College of Georgia) for bringing this research problem to our attention and for providing us with sickle cell blood. We thank Prof. Sean Decatur (Mt. Holyoke College) and his laboratory for the use of the FT-IR spectrometer, and for assisting us with the acquisition and analysis of the FT-IR data. We thank Julian Villar for assistance with the tandem MS experiments and data analysis.
Abbreviations used are
- SCD
Sickle Cell Disease
- Hb
Hemoglobin
- NO
Nitric Oxide
- SNO-Hb
S-nitrosohemoglobin
- HbNO
nitrosylhemoglobin
- HbCO
carbonmononxyhemoglobin
- HU
hydroxyurea
- NEM
N-ethylmaleimide
- UVRR
UV resonance Raman
- FT-IR
Fourier transform infrared
- ESI-MS
electrospray ionization mass spectrometry
- LC-ESI-MS/MS
reversed phase liquid chromatography electrospray ionization tandem mass spectrometry
- CID
collision-induced dissociation
- PMB
p-mercuribenzoate
- L-BAPNA
N-benzoyl-L-arginine p-nitroanilide hydrochloride
- BBO
barium borate
- MWCO
molecular weight cut-off
- NEM-L
NEM low modified Hb
- NEM-H
NEM high modified Hb
References
- 1.Eaton WA, Hofrichter J. Hemoglobin S gelation and sickle cell disease. Blood. 1987;70:1245–1266. [PubMed] [Google Scholar]
- 2.Eaton WA, Hofrichter J. Sickle cell hemoglobin polymerization. Adv Protein Chem. 1990;40:63–279. doi: 10.1016/s0065-3233(08)60287-9. [DOI] [PubMed] [Google Scholar]
- 3.Padlan EA, Love WE. Refined Crystal Structure of Deoxyhemoglobin S I. Restrained Least-Squares Refinement at 3.0-Å Resolution. J Biol Chem. 1985;260:8272–8279. doi: 10.2210/pdb1hbs/pdb. [DOI] [PubMed] [Google Scholar]
- 4.Head CA, Brugnara C, Martinez-Ruiz R, Kacmarek RM, Bridges KR, Kuter D, Bloch KD, Zapol WM. Low concentrations of nitric oxide increase oxygen affinity of sickle erythrocytes in vitro and in vivo. J Clin Invest. 1997;100:1193–1198. doi: 10.1172/JCI119631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gladwin MT, Schechter AN, Shelhamer JH, Pannell LK, Conway DA, Hrinczenko BW, Nichols JS, Pease-Fye ME, Noguchi CT, Rodgers GP, Ognibene FP. Inhaled nitric oxide augments nitric oxide transport on sickle cell hemoglobin without affecting oxygen affinity. J Clin Invest. 1999;104:937–945. doi: 10.1172/JCI7637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vichinsky E. New therapies in sickle cell disease. Lancet. 2002;360:629–631. doi: 10.1016/S0140-6736(02)09776-3. [DOI] [PubMed] [Google Scholar]
- 7.Gladwin MT, Ognibene FP, Shelhamer JH, Pease-Fye ME, Noguchi CT, Rodgers GP, Schechter AN. Nitric Oxide Transport on Sickle Cell Hemoglobin: Where Does it Bind? Free Rad Res. 2001;35:175–180. doi: 10.1080/10715760100300721. [DOI] [PubMed] [Google Scholar]
- 8.Huang J, Hadimani SB, Rupon JW, Ballas SK, Kim-Shapiro DB, King SB. Iron nitrosyl hemoglobin formation from the reactions of hemoglobin and hydroxyurea. Biochemistry. 2002;41:2466–2474. doi: 10.1021/bi011470o. [DOI] [PubMed] [Google Scholar]
- 9.Kim-Shapiro DB, King SB, Bonifant CL, Kolibash CP, Ballas SK. Time-resolved absorption study of the reaction of hydroxyurea with sickle cell hemoglobin. Biochim Biophys Acta. 1998;1380:64–74. doi: 10.1016/s0304-4165(97)00132-3. [DOI] [PubMed] [Google Scholar]
- 10.Rupon JW, Domingo SR, Smith SV, Gummadi BK, Shields H, Ballas SK, King SB, Kim-Shapiro DB. The reactions of myoglobin, normal adult hemoglobin, sickle cell hemoglobin and hemin with hydroxyurea. Biophys Chem. 2000;84:1–11. doi: 10.1016/s0301-4622(99)00132-5. [DOI] [PubMed] [Google Scholar]
- 11.Xu X, Lockamy VL, Chen K, Huang Z, Shields H, King SB, Ballas SK, Nichols JS, Gladwin MT, Noguchi CT, Schechter AN, Kim-Shapiro DB. Effects of Iron Nitrosylation on Sickle Cell Hemoglobin Solubility. J Biol Chem. 2002;277:36787–36792. doi: 10.1074/jbc.M205350200. [DOI] [PubMed] [Google Scholar]
- 12.Luchsinger BP, Rich EN, Gow AJ, Williams EM, Stamler JS, Singel DJ. Routes to S-nitroso-hemoglobin formation with heme redox and preferential reactivity in the beta subunits. Proc Natl Acad Sci U S A. 2003;100:461–466. doi: 10.1073/pnas.0233287100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan NL, Rogers PH, Arnone A. Crystal structure of the S-nitroso form of liganded human hemoglobin. Biochemistry. 1998;37:16459–16464. doi: 10.1021/bi9816711. [DOI] [PubMed] [Google Scholar]
- 14.Ferranti P, Malorni A, Mamone G, Sannolo N, Marino G. Characterisation of S-nitrosohaemoglobin by mass spectrometry. FEBS Lett. 1997;400:19–24. doi: 10.1016/s0014-5793(96)01258-6. [DOI] [PubMed] [Google Scholar]
- 15.Bonaventura C, Godette G, Ferruzzi G, Tesh S, Stevens RD, Henkens R. Responses of normal and sickle cell hemoglobin to S-nitrosocysteine: implications for therapeutic applications of NO in treatment of sickle cell disease. Biophys Chem. 2002;98:165–181. doi: 10.1016/s0301-4622(02)00092-3. [DOI] [PubMed] [Google Scholar]
- 16.Bonaventura C, Ferruzzi G, Tesh S, Stevens RD. Effects of S-nitrosation on oxygen binding by normal and sickle cell hemoglobin. J Biol Chem. 1999;274:24742–24748. doi: 10.1074/jbc.274.35.24742. [DOI] [PubMed] [Google Scholar]
- 17.Jia L, Bonaventura C, Bonaventura J, Stamler JS. S-nitrosohaemoglobin: a dynamic activity of blood involved in vascular control. Nature. 1996;380:221–226. doi: 10.1038/380221a0. [DOI] [PubMed] [Google Scholar]
- 18.Singel DJ, Stamler JS. Chemical physiology of blood flow regulation by red blood cells: the role of nitric oxide and S-nitrosohemoglobin. Annu Rev Physiol. 2005;67:99–145. doi: 10.1146/annurev.physiol.67.060603.090918. [DOI] [PubMed] [Google Scholar]
- 19.Gow AJ, Luchsinger BP, Pawloski JR, Singel DJ, Stamler JS. The oxyhemoglobin reaction of nitric oxide. Proc Natl Acad Sci USA. 1999;96:9027–9032. doi: 10.1073/pnas.96.16.9027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gladwin MT, Jack J, Lancaster R, Freeman BA, Schechter AN. Nitric oxide's reactions with hemoglobin: a view through the SNO-storm. Nature Med. 2003;9:496–500. doi: 10.1038/nm0503-496. [DOI] [PubMed] [Google Scholar]
- 21.Xu X, Cho M, Spencer NY, Patel N, Huang Z, Shields H, King SB, Gladwin MT, Hogg N, Kim-Shapiro DB. Measurements of nitric oxide on the heme iron and beta-93 thiol of human hemoglobin during cycles of oxygenation and deoxygenation. Proc Natl Acad Sci U S A. 2003;100:11303–11308. doi: 10.1073/pnas.2033883100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S, Yang BK, Waclawiw MA, Zalos G, Xu X, Huang KT, Shields H, Kim-Shapiro DB, Schechter AN, Richard I, Cannon O, Gladwin MT. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nature Med. 2003;9:1498–1505. doi: 10.1038/nm954. [DOI] [PubMed] [Google Scholar]
- 23.Huang KT, Keszler A, Patel N, Patel RP, Gladwin MT, Kim-Shapiro DB, Hogg N. The Reaction between Nitrite and Deoxyhemoglobin. J Biol Chem. 2005;280:31126–31131. doi: 10.1074/jbc.M501496200. [DOI] [PubMed] [Google Scholar]
- 24.Kim-Shapiro DB, Gladwin MT, Patel RP, Hogg N. The reaction between nitrite and hemoglobin: the role of nitrite in hemoglobin-mediated hypoxic vasodilation. J Inorg Biochem. 2005;99:237–246. doi: 10.1016/j.jinorgbio.2004.10.034. [DOI] [PubMed] [Google Scholar]
- 25.Huang Z, Shiva S, Kim-Shapiro DB, Patel RP, Ringwood LA, Irby CE, Huang KT, Ho C, Hogg N, Schechter AN, Gladwin MT. Enzymatic Function of Hemoglobin as a Nitrite Reductase that Produces NO under Allosteric Control. J Clin Invest. 2005;115:2099–2107. doi: 10.1172/JCI24650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luchsinger BP, Rich EN, Yan Y, Williams EM, Stamler JS, Singel DJ. Assessments of the chemistry and vasodilatory activity of nitrite with hemoglobin under physiologically relevant conditions. J Inorg Biochem. 2005;99:912–921. doi: 10.1016/j.jinorgbio.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 27.Angelo M, Singel DJ, Stamler JS. An S-nitrosothiol (SNO) synthase function of hemoglobin that utilizes nitrite as a substrate. Proc Natl Acad Sci U S A. 2006;103:8366–8371. doi: 10.1073/pnas.0600942103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allen BW, Piantadosi CA. How do red blood cells cause hypoxic vasodilation? The SNO-hemoglobin paradigm. Am J Physiol Heart Circ Physiol. 2006;291:H1507–1512. doi: 10.1152/ajpheart.00310.2006. [DOI] [PubMed] [Google Scholar]
- 29.Riordan J, Vallee BL. Reactions with N-Ethylmaleimide and p-Mercuricbenzoate. Meth Enzymol. 1972:446–449. doi: 10.1016/S0076-6879(72)25040-6. [DOI] [PubMed] [Google Scholar]
- 30.Riggs A. The binding of N-ethylmaleimide by human hemoglobin and its effect upon the oxygen equilibrium. J Biol Chem. 1961;236:1948–1954. [PubMed] [Google Scholar]
- 31.Benesch R, Benesch RE. Determination of--SH groups in proteins. Methods Biochem Anal. 1962;10:43–70. doi: 10.1002/9780470110270.ch2. [DOI] [PubMed] [Google Scholar]
- 32.Guidotti G. Studies on the chemistry of hemoglobin. I. The reactive sulfhydryl groups. J Biol Chem. 1967;242:3673–3684. [PubMed] [Google Scholar]
- 33.Rodgers KR, Su C, Subramaniam S, Spiro TG. Hemoglobin R to T structural dynamics from simultaneous monitoring of tyrosine and tryptophan time-resolved UV resonance Raman signals. J Am Chem Soc. 1992;114:3697–3709. [Google Scholar]
- 34.Sokolov L, Mukerji I. Structure of Sickle Cell Hemoglobin Fibers Probed with UV Resonance Raman Spectroscopy. J Phys Chem B. 2000;104:10835–10843. [Google Scholar]
- 35.Hildebrandt PG, Copeland RA, Spiro TG, Otlewski J, Laskowski M, Jr, Prendergast FG. Tyrosine hydrogen-bonding and environmental effects in proteins probed by ultraviolet resonance Raman spectroscopy. Biochemistry. 1988;27:5426–5433. doi: 10.1021/bi00415a007. [DOI] [PubMed] [Google Scholar]
- 36.Yohe ME, Sheffield KM, Mukerji I. Solubility of fluoromethemoglobin S: effect of phosphate and temperature on polymerization. Biophys J. 2000;78:3218–3226. doi: 10.1016/S0006-3495(00)76858-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng Y, Shen TJ, Simplaceanu V, Ho C. Ligand binding properties and structural studies of recombinant and chemically modified hemoglobins altered at beta 93 cysteine. Biochemistry. 2002;41:11901–11913. doi: 10.1021/bi0202880. [DOI] [PubMed] [Google Scholar]
- 38.Boyer PD. Spectrophotometric Study of the Reaction of Protein Sulfhydryl Groups with Organic Mercurials. J Am Chem Soc. 1954;76:4331–4337. [Google Scholar]
- 39.Haughland RP. Molecular Probes Handbook of Fluorescent Probes and Research Products. Invitrogen Detection Technologies; Eugene: 2002. [Google Scholar]
- 40.Park S, Hayes BL, Marankan F, Mulhearn DC, Wanna L, Mesecar AD, Santarsiero BD, Johnson ME, Venton DL. Regioselective covalent modification of hemoglobin in search of antisickling agents. J Med Chem. 2003;46:936–953. doi: 10.1021/jm020361k. [DOI] [PubMed] [Google Scholar]
- 41.Lee H, Yi EC, Wen B, Reily TP, Pohl L, Nelson S, Aebersold R, Goodlett DR. Optimization of reversed-phase microcapillary liquid chromatography for quantitative proteomics. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;803:101–110. doi: 10.1016/j.jchromb.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 42.Huang Z, Louderback JG, Goyal M, Azizi F, King SB, Kim-Shapiro DB. Nitric oxide binding to oxygenated hemoglobin under physiological conditions. Biochim Biophys Acta. 2001;1568:252–260. doi: 10.1016/s0304-4165(01)00227-6. [DOI] [PubMed] [Google Scholar]
- 43.Adachi K, Asakura T. Effect of Liganded Hemoglobin S and Hemoglobin A on the Aggregation of Deoxy-hemoglobin S. J Biol Chem. 1982;257:5738–5744. [PubMed] [Google Scholar]
- 44.Ferrone FA, Hofrichter J, Eaton WA. Kinetics of sickle hemoglobin polymerization. Studies using temperature-jump and laser photolysis techniques. J Mol Biol. 1985;183:591–610. doi: 10.1016/0022-2836(85)90174-3. [DOI] [PubMed] [Google Scholar]
- 45.Ferrone FA, Hofrichter J, Eaton WA. Kinetics of sickle hemoglobin polymerization. A double nucleation mechanism. J Mol Biol. 1985;183:611–631. doi: 10.1016/0022-2836(85)90175-5. [DOI] [PubMed] [Google Scholar]
- 46.Fasanmade AA. Gelation kinetics of dilute hemoglobin from sickle cell anemia patients. Hemoglobin. 1996;20:415–428. doi: 10.3109/03630269609005845. [DOI] [PubMed] [Google Scholar]
- 47.Sokolov L, Mukerji I. Conformational Changes in FmetHbS Probed with UV Resonance Raman and Fluorescence Spectroscopic Methods. J Phys Chem B. 1998;102:8314–8319. [Google Scholar]
- 48.Moh PP, Fiamingo FG, Alben JO. Conformational sensitivity of beta-93 cysteine SH to ligation of hemoglobin observed by FT-IR spectroscopy. Biochemistry. 1987;26:6243–6249. doi: 10.1021/bi00393a044. [DOI] [PubMed] [Google Scholar]
- 49.Bare GH, Alben JO, Bromberg PA. Sulfhydryl groups in hemoglobin. A new molecular probe at the alpha1 beta 1 interface studied by Fourier transform infrared spectroscopy. Biochemistry. 1975;14:1578–1583. doi: 10.1021/bi00679a005. [DOI] [PubMed] [Google Scholar]
- 50.Garel MC, Domenget C, Galacteros F, Martin-Caburi J, Beuzard Y. Inhibition of erythrocyte sickling by thiol reagents. Mol Pharmacol. 1984;26:559–565. [PubMed] [Google Scholar]
- 51.Domenget C, Garel MC, Rhoda MD, Caburi-Martin J, Galacteros F, Beuzard Y. Kinetics of polymerization of hemoglobin S modified by thiol reagents and by oxidation. Biochim Biophys Acta. 1985;830:71–79. doi: 10.1016/0167-4838(85)90133-5. [DOI] [PubMed] [Google Scholar]
- 52.Perutz MF, Muirhead H, Mazzarella L, Crowther RA, Greer J, Kilmartin JV. Identification of residues responsible for the alkaline Bohr effect in haemoglobin. Nature. 1969;222:1240–1243. doi: 10.1038/2221240a0. [DOI] [PubMed] [Google Scholar]
- 53.Hofrichter J, Ross PD, Eaton WA. Supersaturation in Sickle Cell Hemoglobin Solutions. Proc Natl Acad Sci U S A. 1976;73:3035–3039. doi: 10.1073/pnas.73.9.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adachi K, Asakura T. Nucleation-controlled aggregation of deoxyhemoglobin S. Possible difference in the size of nuclei in different phosphate concentrations. J Biol Chem. 1979;254:7765–7771. [PubMed] [Google Scholar]
- 55.Ferrone FA. Nucleation: the connections between equilibrium and kinetic behavior. Methods Enzymol. 2006;412:285–299. doi: 10.1016/S0076-6879(06)12017-0. [DOI] [PubMed] [Google Scholar]
- 56.Austin JC, Jordan T, Spiro TG. Ultraviolet Resonance Raman Studies of Proteins and Related Model Compounds. In: Clark RJH, Hester RE, editors. Biomolecular Spectroscopy. John Wiley and Sons Ltd.; New York: 1993. pp. 55–127. [Google Scholar]
- 57.Jayaraman V, Rodgers KR, Mukerji I, Spiro TG. R and T states of fluoromethemoglobin probed by ultraviolet resonance Raman spectroscopy. Biochemistry. 1993;32:4547–4551. doi: 10.1021/bi00068a009. [DOI] [PubMed] [Google Scholar]
- 58.Harrington DJ, Adachi K, Royer WE., Jr Crystal structure of deoxy-human hemoglobin beta6 Glu → Trp. Implications for the structure and formation of the sickle cell fiber. J Biol Chem. 1998;273:32690–32696. doi: 10.1074/jbc.273.49.32690. [DOI] [PubMed] [Google Scholar]
- 59.Perutz MF, Fermi G, Luisi B, Shaanan B, Liddington RC. Stereochemistry of cooperative mechanisms in hemoglobin. Cold Spring Harb Symp Quant Biol. 1987;52:555–565. doi: 10.1101/sqb.1987.052.01.063. [DOI] [PubMed] [Google Scholar]
- 60.Huang S, Peterson ES, Ho C, Friedman JM. Quaternary structure sensitive tyrosine interactions in hemoglobin: a UV resonance Raman study of the double mutant rHb (beta99Asp→Asn, alpha42Tyr→Asp) Biochemistry. 1997;36:6197–6206. doi: 10.1021/bi970018v. [DOI] [PubMed] [Google Scholar]
- 61.Nagai M, Wajcman H, Lahary A, Nakatsukasa T, Nagatomo S, Kitagawa T. Quaternary Structure Sensitive Tyrosine Residues in Human Hemoglobin: UV Resonance Raman Studies of Mutants at α140, β35 and β145 Tyrosine. Biochemistry. 1999;38:1243–1251. doi: 10.1021/bi982269p. [DOI] [PubMed] [Google Scholar]
- 62.Garel MC, Domenget C, Caburi-Martin J, Prehu C, Galacteros F, Beuzard Y. Covalent binding of glutathione to hemoglobin. I. Inhibition of hemoglobin S polymerization. J Biol Chem. 1986;261:14704–14709. [PubMed] [Google Scholar]
- 63.Craescu CT, Poyart C, Schaeffer C, Garel MC, Kister J, Beuzard Y. Covalent binding of glutathione to hemoglobin. II. Functional consequences and structural changes reflected in NMR spectra. J Biol Chem. 1986;261:14710–14716. [PubMed] [Google Scholar]
- 64.Garel MC, Caburi-Martin J, Domenget C, Kister J, Craescu CT, Poyart C, Beuzard Y. Changes of polymerization and conformation of hemoglobin S induced by thiol reagents. Biochim Biophys Acta. 1990;1041:133–140. doi: 10.1016/0167-4838(90)90056-l. [DOI] [PubMed] [Google Scholar]
- 65.Caburi-Martin J, Garel MC, Domenget C, Prehu C, Beuzard Y. Contact inhibition within hemoglobin S polymer by thiol reagents. Biochim Biophys Acta. 1986;874:82–89. doi: 10.1016/0167-4838(86)90105-6. [DOI] [PubMed] [Google Scholar]
- 66.Wodak SJ, De Coen JL, Edelstein SJ, Demarne H, Beuzard Y. Modification of human hemoglobin by glutathione. III. Perturbations of hemoglobin conformation analyzed by computer modeling. J Biol Chem. 1986;261:14717–14724. [PubMed] [Google Scholar]
- 67.Juszczak LJ, Manjula B, Bonaventura C, Acharya SA, Friedman JM. UV resonance Raman study of beta93-modified hemoglobin A: chemical modifier-specific effects and added influences of attached poly(ethylene glycol) chains. Biochemistry. 2002;41:376–385. doi: 10.1021/bi011212r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available. Figures depicting the 215 nm exited UVRR parent spectra of Hb S, NEM-L Hb and NEM-H Hb (Figure 1), the T-R difference spectra of Hb S, NEM-L Hb, and NEM-H Hb in high and low phosphate buffer (Figure 2), the T-T difference spectra of Hb S, NEM-L Hb, and NEM-H Hb in low phosphate buffer (Figure 3), and the F-T difference spectra of Hb S, NEM-L Hb, and NEM-H Hb in low phosphate buffer (Figure 4). Figures depicting the 230 nm excited UVRR parent spectra of Hb S, NEM-L Hb, and NEM-H Hb (Figure 5), the T-R difference spectra of Hb S, NEM-L Hb, and NEM-H Hb in low phosphate buffer (Figure 6), and the F-R difference spectra of Hb S, NEM-L Hb, and NEM-H Hb in low phosphate buffer (Figure 7). A table containing a list of all the labeled peptides identified by LC-ESI-MS/MS is also given.