

Abstract
Diabetes not only increases the risk but also worsens the motor and cognitive recovery after stroke, which is the leading cause of disability worldwide. Repair after stroke requires coordinated communication among various cell types in the central nervous system as well as circulating cells. Vascular restoration is critical for the enhancement of neurogenesis and neuroplasticity. Given that vascular disease is a major component of all complications associated with diabetes including stroke, this review will focus on cellular communications that are important for vascular restoration in the context of diabetes.
Introduction
The increasing incidence of diabetes is only the tip of the global health and social problems the disease poses. Elevated and uncontrolled blood glucose has an avalanche effect leading to debilitating complications, all which are underlined by small or large vascular disease (Association, 2014). Central nervous system (CNS) complications of diabetes including stroke and cognitive impairment are by far the least studied complications of diabetes. In addition to large vessels affected by accelerated atherosclerosis, it is increasingly recognized that small artery disease also contributes to ischemic injury of the brain (Ergul et al., 2012b). The combination of small artery disease, small infarcts and diabetes is a triple threat for the occurrence as well as the motor and cognitive recovery after stroke, which is the leading cause of disability worldwide. Therefore, better understanding of the regulation of endogenous repair mechanisms of the brain under physiological and pathophysiological conditions is fundamental to stroke research.
Vascular protection and restoration is critical for stroke recovery not only for the improvement of cerebral blood flow and blood brain barrier (BBB) integrity but also for the enhancement of neurogenesis and neuroplasticity in the angiogenic microenvironment (Ergul et al., 2012a). Our understanding of the brain repair process after stroke in diabetes is very limited. Recent studies showed that type 2 diabetes stimulates dysfunctional cerebral angiogenesis (Prakash et al., 2012; Prakash et al., 2013a). After stroke, while control animals are able to launch reparative neovascularization in both ischemic and nonischemic hemispheres, diabetic animals develop vasoregression, which is associated with poor functional recovery (Ergul et al., 2014; Prakash et al., 2013b). Impaired angiogenesis after stroke has been reported in both type 1 and type 2 diabetes (Cui et al., 2011; Ye et al., 2011). These studies showed that while vessel density is increased in the ipsilateral hemisphere 14 days after stroke, vessels fail to mature. A recent study further confirmed these results (Poittevin et al., 2014). Neovascularization, which encompasses remodeling of existing vessels, angiogenesis and barriergenesis, is a very complex process that requires coordination of cell-to-cell interactions (Ergul et al., 2014). This cellular communication is not limited to signals among vascular cells such as endothelial cell-vascular smooth muscle cell or tip cells-stalk cells but indeed includes a network of vascular cells, surrounding resident cells of the brain including pericytes, neurons, glia, and oligodendrocytes as well as circulating blood and bone marrow cells (Figure 1). In diabetes, impaired reparative angiogenesis may be due to impaired activation of growth factors, failure in vessel maturation (lack of pericyte coverage) and reduced circulating progenitor cells (Gallagher et al., 2007). Thus, in this review, we will first define vascular repair and restoration and then systematically discuss the interactions between endothelial cells and other cell types that contribute to this process in the context of diabetes.
Figure 1. A schematic representation of the bidirectional interactions between various CNS and peripheral cell types that contribute to neurovascular repair after stroke.

Diabetes impedes vascular restoration and functional recovery.
A. Vascular Repair and Restoration
Ischemic damage to the cerebrovasculature ignites a series of events, both local and systemic, leading to a repair process with both positive and negative consequences in the long term. Stimulation of angiogenesis and vasculogenesis can improve perfusion but chronic expression of growth factors and cytokines may lead to neointimal proliferation and atherosclerosis (Zhang and Xu, 2014). Vascular repair starts with a combination of events including arteriogenesis, angiogenesis and vasculogenesis and leads to vascular restoration with stabilized and functional vessels necessary for recovery from ischemic stroke. Immediately upon vessel occlusion, there is an increase in shear stress in the surrounding vessels, acting as a trigger for arteriogenesis. The formation of new collateral vessels from existing structures serves to maintain blood flow to the penumbral tissue and limit the damage. On a molecular level, nitric oxide (NO) has been shown to be critically important in mediating the vasodilation and formation of functional collaterals (Hoefer et al., 2006). A key feature in the persistence of the new collaterals is the recruitment of circulating monocytes/macrophages and these cells have been shown to be necessary for the maturation and growth of the collateral vessels. Endothelial and smooth muscle cell proliferation and migration follows, leading to the formation of new or expanded vessels. In the case of effective collateral formation, the larger diameter vessels relieve the shear stress and provide feedback to stop the process. With defective collateralization, as is seen in diabetes, the stimulus for proliferation remains, making the likelihood of pathologic angiogenesis more likely (Abaci et al., 1999).
Angiogenesis genes are upregulated within minutes of the onset of cerebral ischemia, involving all cell types in the neurovascular unit (endothelial, neurons, astrocytes, etc). These cells together release growth factors including vascular endothelial growth factor (VEGF), brain-derived neurotrophic factor (BNDF), basic fibroblast growth factor (bFGF), and platelet derived growth factor (PDGF) which stimulate endothelial proliferation, migration and tube formation and locally produced chemokines attract circulating and resident progenitor cells to support the process (Hermann et al., 2013). Enhanced production of proteases (MMPs) from the injured cells contributes to destroy the basement membrane and allow new vessel sprouting. Angiogenesis is followed by barriergenesis, a process in which endothelial cells mature and establish selective permeability properties (Lee et al., 2003; Rieckmann and Engelhardt, 2003). Although most investigation of angiogenesis has been focused on the ischemic border zone after cerebral ischemia, there is emerging evidence that areas remote from the lesion also undergo angiogenic changes and this has been linked to other plasticity markers involved in recovery (Ergul et al., 2012a; Guan et al., 2011).
Vascular restoration is not only important for maintenance of perfusion of “at risk” tissue. In the aftermath of an acute ischemic stroke, neurogenesis in the subventricular zones of both hemispheres leads to the production of neuronal precursor cells (NPCs) that home to the area of injury and enhance recovery (Jin et al., 2001). Migration of NPCs occurs in close association with blood vessels, however, and it has been shown that co-transplantation of NPCs with vascular progenitor cells (VPCs) is dramatically more effective that either cell type alone in improving recovery after experimental stroke (Li et al., 2014a).
B. CNS Cell-to-Cell Interactions in Vascular Restoration
In this section, we will discuss the interaction between endothelial cells and resident cells of CNS with respect to vascular restoration starting with pericytes which are the most widely studied cells in this regard. Each subsection is organized to summarize the role(s) of a particular cell type in vascular repair/restoration, how this role is altered after stroke and finally what is known about this interaction after stroke in diabetes.
1. Pericytes
Pericytes are multifunctional cells found on pre-capillary arterioles, capillaries, and post-capillary venules (Bonkowski et al., 2011; Dalkara et al., 2011; Dore-Duffy, 2014; Hill et al., 2014; Winkler et al., 2011). They are embedded within the endothelial basement membrane (Attwell et al., 2010; Dalkara et al., 2011). While pericytes are distributed throughout the body, they are more abundant in the CNS and retina, in both of which blood barrier function is very important (Dalkara et al., 2011; Sa-Pereira et al., 2012). Their unique localization along the vasculature, while keeping in contact with astrocytes and possibly directly with neurons, strongly suggests their participation in cell-to-cell communication (Bonkowski et al., 2011; Jojart et al., 1984). Indeed, pericytes contribute to the induction and maintenance of the BBB, microvascular stability, endothelial cell survival and angiogenesis, neurovascular coupling and blood flow regulation, and clearance of extracellular molecules by phagocytosis (Armulik et al., 2010; Bell et al., 2010; Diaz-Flores et al., 2009; Gerhardt and Betsholtz, 2003; Li et al., 2011; Ozerdem and Stallcup, 2003). Furthermore, pericytes may function as pluripotent stem cells (Bonkowski et al., 2011). Clearly, all of these actions are critical for the neurovascular repair process after ischemic brain injury and changes in the microenvironment mediated by stroke and/or diabetes can impact pericyte interactions with other cells in the CNS that ultimately affect these protective functions.
For the purpose of this review, we will focus on the dynamic role of pericytes in angiogenesis. At initiation of angiogenesis, pericytes migrate away from endothelial cells and secrete proangiogenic growth factors like VEGF-A (Bonkowski et al., 2011; Dore-Duffy and LaManna, 2007). In addition, they secrete MMPs such as MMP2 and 9 that degrade basement membrane proteins enabling the endothelial cells to sprout (Virgintino et al., 2007). Later in the process, pericytes contribute to the termination of angiogenesis and stabilization of newly formed vessels through barriergenesis (Lee et al., 2003; Rieckmann and Engelhardt, 2003). Pericytes are recruited in response to PDGF-β secreted by endothelial cells and through the activation of PDGF-R β, they proliferate and enhance the interaction with endothelial cells (Figure 2). Seminal studies with PDGF-R β and PDGF-β knock-out mice that are basically devoid of pericytes provided key information in this field (Dalkara et al., 2011; Enge et al., 2002; Winkler et al., 2011). Together with activation of transforming growth factor-β (TGF-β) and angiopoietin-1 (Ang-1), expression of N-cadherin, integrins and gap junctions, especially connexin 43 (CX43) is stimulated. As vessel stabilization occurs, pericytes, along with astrocytes, secrete matrix proteins (Bonkowski et al., 2011; Diaz-Flores et al., 2009; Stratman et al., 2009).
Figure 2. Possible mechanisms contributing to weakened pericyte-endothelial cell interactions in diabetes.

A. At the initiation of angiogenesis, pericytes migrate away from endothelial cells and secrete proangiogenic growth factors like VEGF-A. Pericytes are later recruited in response to PDGF-β secreted by endothelial cells and through the activation of PDGF-R β, they proliferate and enhance the interaction with endothelial cells. Pericytes contribute to the termination of angiogenesis and stabilization of newly formed vessels through barriergenesis. Activation of TGF-β and Ang-1, stimulates expression of N-cadherin, integrins and gap junctions, especially connexin 43 (CX43). As vessel stabilization occurs, pericytes, along with astrocytes, secrete matrix proteins. B. In diabetes, pericyte recruitment, attachment and maturation are impaired. Increased phosphorylated pPDGFR-β, decreased PDGFR-β expression and or altereations in Ang-1 and Ang-2 signaling may contribute to loss of communication between these two cell types.
The role(s) pericytes play in stroke injury and recovery is an understudied area of research. It has long been speculated that pericytes are involved in regulation of neurovascular coupling and cerebral blood flow. While it is still debatable, recent studies suggest that pericytes exhibit a prolonged contraction in response to ischemia, contributing to perfusion deficits after stroke (Yemisci et al., 2009). Hall et al also reported that ischemia mediates capillary constriction by pericytes which leads to pericyte death exacerbating the ischemic injury (Hall et al., 2014). As discussed above, pericytes may provide the initial trigger for an angiogenic response. Severity of the loss of pericytes after an ischemic event may have important implications for stroke recovery but direct evidence is lacking.
The impact of diabetes on pericyte functions in angiogenesis and microvascular integrity has been mostly studied in diabetic retinopathy as pericyte loss is a hallmark of early changes in the disease (Dalkara et al., 2011; Ejaz et al., 2008, Winkler, 2011 #21). Evidence suggests that there is a critical threshold for pericyte coverage that determines the fate of endothelial cells (Ejaz et al., 2008; Winkler et al., 2011). When pericyte number is decreased, there is a proliferative response leading to pathological angiogenesis. When pericyte loss exceeds 50%, endothelial cells start undergoing apoptosis and vasoregression. A recent study suggested that a potential link between diabetes and pericyte deficiency may be due to phosphorylation of PDGF-Rβ under hyperglycemic conditions which renders the receptor less responsive to PDGF-β stimulation (Geraldes et al., 2009). It has also been suggested that angiopoietin-2 (Ang-2) may contribute to pericyte loss in diabetes. Ang-2 is increased in retinal endothelial cells as well as in the vitreous of patients with diabetic retinopathy (Park et al., 2014). A recent study showed that Ang-2 induces pericyte apoptosis in the retina by upregulation of α3β1 integrin signaling. Studies also suggest that CX-43 expression, another important protein for endothelial-pericyte communication, is decreased in diabetes (Li et al., 2003).
The effect(s) of diabetes on CNS pericytes are not fully understood. We recently showed that diabetes promotes pathological angiogenesis in the brain and this is associated with decreased pericyte coverage (Figure 3) (Prakash et al., 2012). Our studies indicate that Ang-2 expression is increased in brain microvascular endothelial cells isolated from these animals which may contribute to decreased pericytes in this model (unpublished data). It is highly possible that diabetes can mediate PDGF-Rβ phosphorylation or decreased CX-43 expression in CNS pericytes as reported in the retina (Figure 2).
Figure 3. Diabetes reduces pericytes in the brain.
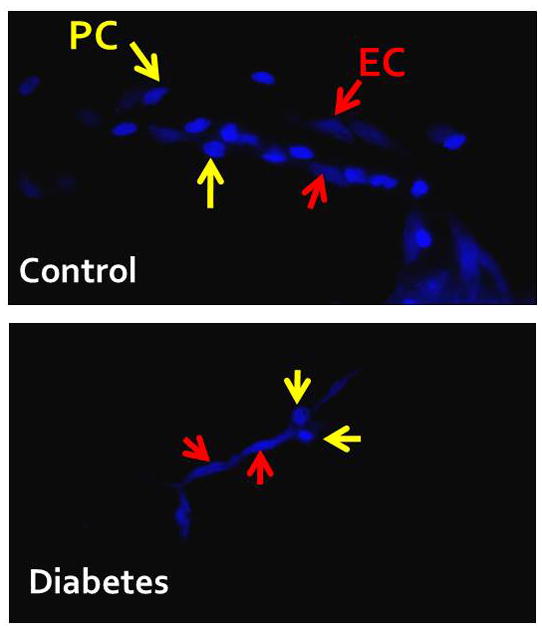
Brain capillaries were isolated from control and diabetic Goto-Kakizaki rats 6–7 weeks after the onset of diabetes. Samples were allowed to adhere to cover slips and stained with nuclear stain DAPI. To determine the number of pericytes (PC), round nuclei were counted versus endothelial cells (EC) that have elongated nuclei.
As discussed above, vascular repair is impaired and there is substantial vasoregression after stroke in diabetes. Our results suggest that this vascular loss occurs in the presence of VEGF and it involves peroxynitrite-mediated apoptosis of endothelial cells (Abdelsaid et al., 2014). The role of pericytes in this response is practically unknown. In light of the reports that pericyte density is an important determinant of the vascular reparative response, one can speculate that extensive loss of pericytes contributes to post-stroke cerebral vasoregression in diabetes.
2. Glial Cells
The most prominent cell found in the CNS is the astrocyte. A strong body of literature indicates that astrocytes not only provide structural support for neurons and endothelial cells but also are involved in the regulation of neuronal metabolism and cerebral blood flow (CBF), and enable synapse formation and BBB integrity via direct contact with endothelial cells with their end-feet (Barres, 2008; Liebner et al., 2011; Ransom and Ransom, 2012; Ullian et al., 2004). One reason they may be able to accomplish these tasks is they coordinate neurovascular communication with their numerous processes. We will briefly discuss the role(s) of astrocytes in angiogenesis and barriergenesis. Many angiogenic growth factors such as VEGF-A, BDNF and insulin like growth factor-1 (IGF-1) are secreted by astrocytes (Fulmer et al., 2014; Lee et al., 2009; Li et al., 2014d; Lopez-Lopez et al., 2004). An earlier study reported that meteorin, a protein highly abundant in astrocytes, stimulates expression and secretion of antiangiogenic proteins thrombospondin-1 (TSP-1) and TSP-2 in an autocrine manner which then inhibit angiogenesis by the surrounding endothelial cells (Park et al., 2008). It appears that astrocytes and pericytes are both involved in the formation and maintenance of capillary like structures by endothelial cells (Itoh et al., 2011). Studies have also shown that astrocytes are necessary for barriergenesis and directly regulate factors modulating BBB formation, such as tight junction proteins, SSeCKS, Ang-1 and TGFβ (Bonkowski et al., 2011; Dore-Duffy and LaManna, 2007; Dore-Duffy, 2014; Garcia et al., 2004; Lee et al., 2003; Rieckmann and Engelhardt, 2003; Rubin et al., 1991).
Astrocytes exhibit a multimodal neurovascular response when confronted with insults such as diabetes and stroke. The role of astrocytes in the neurovascular repair process after stroke is complex and controversial. While reactive astrocytes can contribute to glial scar formation and secrete factors that inhibit axonal regeneration, it is also possible that reactive astrocytes protect neurons by taking up excess glutamate, secreting neurotrophic factors and providing a barrier that limits expansion of damage (Faulkner et al., 2004; Hansson and Ronnback, 2003; Li et al., 2014c; Liu et al., 2014; Mazzanti et al., 2001). A recent study provided evidence that attenuated astrocytic reactivity can impair axonal remodeling and functional recovery (Liu et al., 2014). A central role for reactive astrocytes in mediating CNS plasticity and recovery after stroke was recently reviewed (Li et al., 2014c) but how astrocytes contribute to vascular plasticity is not fully understood. Interestingly, angiostatic proteins TSP-1 and TSP-2, which also have synaptogenic properties, are increased mainly in astrocytes after stroke (Liauw et al., 2008; Lin et al., 2003). It has been postulated that upregulation of these proteins are responsible for the resolution of postischemic angiogenesis (Liauw et al., 2008; Lin et al., 2003) but a clear role in vascular repair is not known.
Supporting the concept that reactive astrocytes contribute to the recovery process, Jing et al. reported that while ischemia caused a proliferation of astrocytes in the penumbra region in control animals, there was a significantly lower amount of reactive astrocytes in diabetic animals which display worse outcome (Jing et al., 2013). This decrease in astrocytes was associated with greater BBB damage and the brain repair process was stunted. They also identified regions where astrocyte infiltration was greater, and there was a significant improvement in vascular and neural reconstruction. Similarly, Kumari and colleagues reported that robust increases in astrocytic and microglial activation observed in normal mice are blunted in diabetic mice after ischemic injury and this is accompanied by decreased and delayed expression of IGF-1 by these cells (Kumari et al., 2007). As mentioned above, in addition to its neuroprotective properties, IGF-1 also stimulates angiogenesis. It is known that post stroke angiogenesis is impaired in aged animals and is associated with concurrent decreases in IGF-1 (Ingraham et al., 2008; Sonntag et al., 1999). Modulation of IGF-1 may be another mechanism by which glial cells contribute to repair after stroke, especially in the diabetic setting. In contrast to these studies, we found amplified astrocyte reactivity in their type 2 diabetic model with an abundance of thin processes and a reduced soma size after stroke (Prakash et al., 2013b). These changes in astrocyte morphology were associated with vasoregression and poorer outcomes in diabetes. These differences in the activity levels of astrocytes may stem from the use of different models of diabetes with varying disease severity as well as the severity of the ischemic injury. Nevertheless, these studies suggest that modulation of astrocytic reactivity may contribute to the impairment of vascular repair in diabetes. Interestingly, as pointed out by Liu and colleagues, there are different forms of astrocytes in the CNS (Liu et al., 2014). Differential role(s) of these astrocytes in vascular repair deserves further investigation.
Microglia are bone marrow-derived macrophages that reside in the CNS. In addition to their immune functions, they closely interact with the vasculature. Indeed, studies with different in vivo models in which microglia were lacking demonstrated a critical role for microglia in angiogenesis in the brain (Arnold and Betsholtz, 2013; Fantin et al., 2010). A recent report provided evidence that conditioned medium from BV2 microglial cells promotes angiogenesis via upregulation of angiogenic factors ephrin-A3 and ephrin-A4 in endothelial cells. Moreover, they showed that tumor necrosis factor (TNF)-α secreted from microglia mediates this effect (Li et al., 2014b). While studying the effect of epidermal growth factor treatment of neurogenesis in the supraventricular zone (SVZ), Lindberg et al showed critical role of microglia in the angiogenic response in this neurogenic niche (Lindberg et al., 2014).
Microglia are activated after stroke. The pattern of activation depends on the localization with respect to infarct and involves polarization of microglia either into a protective M2 alternatively activated phenotype or proinflammatory M1 classically activated phenotype (Lee et al., 2014). Collectively, the balance can determine the impact of microglia on neurovascular repair. In this regard, information on the role of microglia in angiogenic response after stroke is limited. It has been reported galectin-3 (Gal-3), an angiogenic protein that is primarily localized to microglia, is increased after ischemic injury and augments endothelial proliferation and microvessel density (Lalancette-Hebert et al., 2012; Yan et al., 2009). Yan and colleagues recently reported that Gal-3 secreted by BV2 microglial cells enhances angiogenic potential of neighboring endothelial cells via activation of integrin-linked kinase signaling (Wesley et al., 2013). Microglia and endothelial cell interactions after stroke in diabetic setting are even less understood. As discussed above, Kumari and colleagues reported blunted astrocyte/microglial activation after ischemic injury in diabetes and this was associated with decreased and delayed secretion of TNF-α and IGF-1 (Kumari et al., 2007). Since microglial TNF-α has been shown to enhance angiogenesis (Li et al., 2014b), it is possible that this may be another mechanism contributing to impaired repair and vasoregression after stroke in diabetes.
3. Neurons
Nerves and blood vessels are arguably the backbone of communication throughout living systems. As such, it is not surprising that neurons and endothelial cells are in a bidirectional communication to orchestrate the repair process after ischemic injury. It is increasingly recognized that neurogenesis and synaptogenesis occur in concert with angiogenesis in experimental models of recovery after ischemic brain injury (Ergul et al., 2012a; Navaratna et al., 2009; Xiong et al., 2010). A recent study demonstrated that endothelial cells transplanted into the brain promote vasculogenesis and enhance neurogenesis further providing support for this concept (Ishikawa et al., 2013).
One of the most studied growth factor involved with both neuronal and endothelial cell survival is VEGF-A. VEGF-A and its receptor VEGFR2 has been proven to aid in the migration and differentiation of endothelial cells as well as in vasculogenesis, and angiogenesis (Dumont et al., 1995; Gerhardt et al., 2003; Kearney et al., 2004; Ruhrberg, 2003; Shalaby et al., 1995). Originally isolated from the vasculature, it is also known that neurons express VEGF and its receptors. There are many in vitro and in vivo studies indicating how VEGF-A regulates neuronal proliferation, size, survival and guidance (Bocker-Meffert et al., 2002; Erskine et al., 2011; Jin et al., 2002; Khaibullina et al., 2004; Rosenstein et al., 2003; Ruiz de Almodovar et al., 2010; Ruiz de Almodovar et al., 2011; Silverman et al., 1999). A classic and powerful example of the interaction between angiogenic and neurotrophic effects of growth factors is the song bird studies in which increased VEGF-induced angiogenesis has been shown to promote proper development of young neurons in the higher vocal center via the release of BDNF (Hartog et al., 2009; Louissaint et al., 2002; Rasika et al., 1994).
Several other growth factors have been implemented in this bidirectional communication. BDNF is synthesized by both neurons and endothelial cells and has neurotrophic and angiogenic effects (Alhusban et al., 2013; Kermani et al., 2005). Endothelial cell conditioned media can significantly increase the viability of neurons after various insults including hypoxia, amyloid-β challenge, oxidative stress, or endoplasmic reticulum stress and this is mediated by BDNF (Guo et al., 2008). An interesting recent study provided evidence that neuronal TGFβ is also involved in brain angiogenesis. Neuronal knock-out of TGFβ receptor 2 altered the profile of proangiogenic factors secreted by neurons such as VEGF, IGF-1 and FGF. These changes were associated with atypical blood vessel formation (Hellbach et al., 2014). Nerve growth factor (NGF) is another neurotropin with opposing endothelial actions. While tropomyosin-related kinase A (TrkA) receptor activation by NGF promotes endothelial integrity and averts pericyte damage, NGF precursor protein, proNGF, mediates endothelial cell death by p75NTR (Hammes et al., 1995; Kim et al., 2004).
After stroke, numerous studies has shown that VEGF and its receptors are upregulated not only in endothelial cells but in neurons and astrocytes as well (Beck et al., 2002; Brockington et al., 2006; Forsythe et al., 1996; Hai et al., 2003; Jin et al., 2000; Lennmyr et al., 1998; Lennmyr et al., 2005; Shin et al., 2008; Tolosa et al., 2008; Wang et al., 2004) and associated with improved neurogenesis, angiogenesis and functional outcome (Jin et al., 2002; Li et al., 2009; Wang et al., 2007).
Axonal guidance proteins have been shown to contribute to both endothelial and neuronal repair. One such protein is netrin-1 (Lu et al., 2011; Lu et al., 2012; Sun et al., 2011; Wu et al., 2008). Studies using netrin-1 deficient mice showed vast blood vessel impairment (Park et al., 2004). Following stroke, netrin-1 reduces infarct size, attenuates neuronal death, strengthens functional outcome, prompts neuronal migration, and fosters angiogenesis. Another neuronal guidance molecule that affects angiogenesis is semaphorin 3A (Sema3A). In ischemic retinopathies, Sema3A binds to neuropilin-1 (Nrp-1) and inhibits endothelial cell motility and angiogenesis (Joyal et al., 2011; Klagsbrun and Eichmann, 2005; Mamluk et al., 2002; Miao et al., 1999). However, the roles of these axonal guidance proteins in diabetes and especially in recovery after stroke are not known.
Our understanding of neuron-endothelial cell-astrocyte interactions that can affect neuronal and vascular repair after stroke in diabetes is very limited. While we know that diabetes worsens functional recovery and this is associated with cerebral vasoregression and infarct expansion (Ergul et al., 2014; Li et al., 2013; Prakash et al., 2013b), underlying mechanisms remain unknown. We have evidence that this vasoregression occurs in the presence of VEGF (Abdelsaid et al., 2014) suggesting that microenvironment is critical for protective and reparative functions of growth factors involved in recovery. Evidence suggests that oxidative and nitrative stress diverts survival effects of VEGF to a proapoptotic pathway in endothelial cells in diabetes. Whether VEGF signaling is affected in a similar way in neurons remains to be determined. Microenvironment can also affect the generation of restorative growth factors. For example in the eye, oxidative stress inactivates MMP-7, which cleaves the precursor protein, proNGF, to its mature form, NGF, resulting in excess proNGF which causes neuronal and vascular injury in the retina via p75NTR signaling (Al-Gayyar et al., 2011; Bruno and Cuello, 2006). A similar paradigm may exist for BDNF in diabetes. Using an ELISA which detects both pro and mature forms, we reported greater BDNF levels in the brains of diabetic GK rats. Yet, the outcome of stroke was poor in this model (Li et al., 2010; Li et al., 2013). We now have data that proBDNF and p75NTR levels are greater at baseline and increased even further after stroke in diabetic animals. A recent study reported that proBDNF negatively regulates neuronal remodeling (Yang et al., 2014). Another change with regard to BDNF effects may involve TrkB receptors, which are activated by BDNF and neuroprotective. A recent study showed that advanced glycation end-products degrade neuronal TrkB receptor via upregulation of MMP-9 (Navaratna et al., 2013). It is of great interest to determine the localization of these signals and whether these mechanisms contribute to vasoregression and/or impaired neuronal repair after stroke in diabetes.
4. Oligodendrocytes
Oligodendrocytes are known for myelinating axons and therefore play an important purpose in signal transmission among the neurovascular network. However, their contribution to neurovascular repair and interaction with other cells in this process is far less understood. Once stroke occurs, there is a rapid loss of oligodendrocytes due to ischemia-related oxidative stress, glutamate excitotoxicity and depolarization (Arai and Lo, 2009; Dewar et al., 2003; Micu et al., 2006). Oligodendrocyte progenitor cells work rapidly to restore demyelinated axons (Gensert and Goldman, 1997). Endothelial cells have even been shown in vitro to promote the production and sustenance of oligodendrocyte progenitor cells by Akt and Src signaling pathways by releasing the trophic factors such as bFGF and BDNF (Arai and Lo, 2009). Recent studies by Jiang et al. also show that oligogenic activity and oligodendrocyte progenitor maturation typically occur in the peri-infarct basal ganglia moderately concurrent with increased vessel density. Another interesting finding by Arai K et al. is that oligodendrocyte progenitor cell may not always play the role of the protagonist during injury recovery since they release MMP-9 (Arai and Lo, 2009). MMP9 is linked to BBB breakdown during white matter injury thus potentially aggravates disease states, once again proving that glial activation can be either advantageous, by protecting healthy cells and aiding in the restoration of injured ones, or deleterious, by exacerbating the disease state and impeding regenerative growth (Giaume et al., 2007; Heneka et al., 2010).
Astrocytes are also involved in the myelination process as increased astrocyte infiltration directly correlated with increased remyelination (Jing et al., 2013). This is not surprising considering oligodendrocyte myelination is promoted by astrocyte-secreted cytokine leukemia inhibitory factor (Ishibashi et al., 2006). The interesting discovery from Li PA’s group is that, while stroke conditions induce demyelination and inhibit astrocyte proliferation and mobility, diabetes worsens this demyelination and prolongs recovery (Jing et al., 2013).
C. Vascular Restoration and Interactions with Circulating Cells
In this section, we will discuss the role of circulating cells in pathogenesis and recovery after diabetic stroke. We will also highlight their possible role in neovascularization and how it could be altered in the diabetic milieu. Circulating cells comprise all cell types in the systemic circulation that can infiltrate the brain after ischemic stroke and/or modify stroke outcome. This includes immune cells and stem/progenitor cells. In addition, we will also discuss microparticles that are secreted by circulating cells and have been shown to impact stroke outcome.
1. Immune cells
Several studies have shown that diabetes affects the immune response. In one of the earlier studies, Panes et al. investigated the systemic immune cell recruitment after peripheral ischemia reperfusion injury in diabetes (Panes et al., 1996). They compared leukocyte endothelial cell adhesive interaction in the mesenteric venules of control, hyperglycemic and streptozotocin (STZ) induced type 1 diabetic rats. Diabetic rats showed increased number of rolling but not adherent or emigrating leukocytes at baseline. Ischemia-reperfusion elicited larger increases in leukocyte adhesion and emigration in diabetic but not acutely hyperglycemic rats. These effects were mediated through CD11/CD18–ICAM-1 interactions (adhesion) and P-selectin (rolling). Other studies showed decreased membrane fluidity (Masuda et al., 1990), and enhanced superoxide production (Freedman and Hatchell, 1992) by granulocytes isolated from diabetic animals. Clinically, neutrophils isolated from diabetic patients produced significantly greater amounts of superoxide anions, which correlated negatively with patients’ glycemic control (Karima et al., 2005; Wierusz-Wysocka et al., 1987). Moreover, neutrophils isolated from healthy adults are activated under elevated glucose levels (Kummer et al., 2007). Similarly, monocytes isolated from type 2 diabetic patients showed a pro-inflammatory profile while monocytes from healthy volunteers showed increased secretion of the inflammatory cytokine, TNF-α when cultured under high glucose conditions (Gonzalez et al., 2012). These findings demonstrated that immune cells are activated in the diabetic milieu and with high glucose levels. Thus, diabetic subjects might be more prone to immune mediated tissue injury after stroke.
Based on the previously mentioned studies, it is conceivable that diabetes affect the immune response to stroke as well. While the effect of stroke on immune cells has been examined in different studies, reports studying the immune cell function in diabetic stroke are only beginning to emerge. These studies are listed in Table 1. As obvious from the table, the studies are few and mostly recent (only came out in the last 4 years). Interventional studies using circulating cells in diabetic stroke are listed in Table 2. The effect of diabetes on systemic cells’ functions after stroke is highlighted in Figure 4.
Table 1.
Articles studying circulating cells response in diabetic stroke models
| Author | Animal model | Stroke model | Circulating cells response | Stroke outcome | Follow up time |
|---|---|---|---|---|---|
| (Ritter et al., 2011) | Zucker Diabetic Fatty (ZDF) rats – type 2 diabetic | Intraluminal filament model - 2 h MCAO | Increased neutrophil activation, adhesion and decreased shear rate. Increased expression of CD11b and sICAM | Worse infarct size and neurologic function. | 4 h after reperfusion |
| (Chen et al., 2011a) | Type 2 diabetic db/db mice | Intraluminal filament model - Permanent MCAO | In diabetic animals, BM-derived EPCs were reduced and dysfunctional as measured by reduced tube formation ability in vitro. The levels of circulating EPC-MPs and EMPs were increased in db/db mice. | Larger infarct volume | 48 h after MCAO |
| (Kim et al., 2014) | Diet induced type 2 diabetes in mice | Intraluminal filament model - 30 min MCAO | Increased MCP-1 expression in peritoneal immune cells. Blunted inflammatory response in diabetic brain manifested by decreased MCP-1 RNA. Attenuated LPS-stimulated inflammatory response in macrophages. | Exacerbate d ischemic brain injury and infarct. | 3 days post ischemia |
MCAO: middle cerebral artery occlusion; CD11b: cluster of differentiation molecule b; sICAM; soluble intercellular adhesion molecule; BM: bone marrow; EPCs: endothelial progenitor cells; EPC-MPs; endothelial progenitor cell-derived microparticles; MCP-1: monocyte chemoattractant protein 1; LPS: lipopolysaccharide.
Table 2.
Interventional studies in diabetic stroke using circulating cells
| Author | Animal model | Stroke model | Treatment | Stroke outcome | Follow up time |
|---|---|---|---|---|---|
| (Chen et al., 2011a) | Type 2 diabetic db/db mice | Intraluminal filament model - Permanent MCAO | EPCs preincubated with db/+ MPs or db/db MPs were infused 2 h after MCAO | infusion of EPCs preincubated with db/+ MPs increased the level of cEPCs and reduced ischemic damage, and these beneficial effects were reduced or lost in EPCs preincubated with db/db MPs. | 48 h |
| (Chen et al., 2011b) | STZ induced type 1 diabetic Wistar rats | Intraluminal filament model - 2 h MCAO | IV injection of BMSC at 24 h after MCAO | Increased mortality, BBB leakage and brain hemorrhage | 14 days after stroke |
| (Chen et al., 2012) | Type 2 diabetic db/db mice | Intraluminal filament model - Permanent MCAO | Infusion of Ad-CXCR4 primed EPCs | Increased level of circulating EPCs and CXCR4 expression in the brain. Decreased infarct volume and neurological deficit with treatment |
7 days after stroke |
| (Yan et al., 2013) | STZ induced type 1 diabetic Wistar rats | Intraluminal filament model - 2 h MCAO | IV injection of BMSC at 24 h after MCAO and Niaspan 40 mg/kg orally daily for 14 days | Combination treatment decreased BBB leakage but did not improve infarct volume or functional outcome. | 14 days after stroke |
| (Ye et al., 2013) | STZ induced type 1 diabetic Wistar rats | Intraluminal filament model - 2 h MCAO | IV injection of BMSC at 24 h after MCAO and Niaspan 40 mg/kg orally daily for 14 days | Combination treatment increased white matter remodeling and synaptic plasticity. | 14 days after stroke |
| (Yan et al., 2014) | STZ induced type 1 diabetic Wistar rats | Intraluminal filament model - 2 h MCAO | IV injection of HUCBCs at 24 h post-MCAO | Treatment improved functional outcome and enhanced vascular and white matter remodeling without affecting the infarct volume. | 14 days after stroke |
| (Bai et al., 2014) | Type 2 diabetic db/db mice | Photothrom botic stroke - Permanent MCAO | Infusion of BM derived EPCs 24 h after stroke. | Less pronounced angiogenic response and delayed behavioral recovery compared to wild-type controls. | 21 days after stroke |
MCAO: middle cerebral artery occlusion; STZ: streptozotocin; EPCs: endothelial progenitor cells; MPs: microparticles: BMSC: bone marrow stromal cells; Ad: adenovirus; CXCR4: chemokine receptor type 4; HUCBCs: human umbilical cord blood cells.
Figure 4. Effect of diabetes on systemic cells’ functions after stroke.

Bone marrow endothelial progenitor cells (EPC) produce angiogenic mediators and are incorporated in the newly formed vessels at the ischemic border zone after stroke. Diabetes reduces EPC count and angiogenic response. Diabetes increase neutrophil recruitment, activation and adhesion in the diabetic brain after stroke leading to increased inflammation. Diabetics have elevated levels of platelet-derived microparticles (PMP) putting them at risk of increased coagulation after ischemic stroke.
Neutrophils, the most abundant type of granulocyte, are among the first cells to be recruited to the brain after stroke (Jin et al., 2010) and have been associated with worsened outcome clinically (Kim et al., 2012; Price et al., 2004) and experimentally (Matsuo et al., 1994; Shiga et al., 1991). Ritter et al. examined neutrophil adhesion in type 2 diabetes and stroke using Zucker Diabetic Fatty (ZDF) rats subjected to 2 h middle cerebral artery occlusion (MCAO) (Ritter et al., 2011). Diabetic rats showed increased neutrophil adhesion and decreased shear rate. This was associated with increased expression of CD11b (a measure of neutrophil activation) and soluble intercellular adhesion molecule (sICAM – a ligand for neutrophil adhesion) and correlated with increased inflammatory genes expression, thus leading to poor stroke outcome.
Among the chemotactic factors involved in attracting immune cells to the site of injury after stroke is monocyte chemotactic protein-1 (MCP-1). MCP-1 gene polymorphism was strongly associated with susceptibility to ischemic stroke. This polymorphism tended to be higher in diabetic patients (Buraczynska et al., 2010). Kim et al. have recently examined how diabetes alters the inflammatory response and stroke outcome (Kim et al., 2014). Using diet induced type 2 diabetes in mice, the authors showed an increase in MCP-1 in peritoneal immune cells collected from diabetic mice before and after stroke. Interestingly, they found a blunted inflammatory response in the diabetic brain before stroke and early at 6 h post-stroke manifested by decreased MCP-1 RNA compared to controls. This correlated with attenuated lipopolysaccharide (LPS)-stimulated inflammatory response in diabetic macrophages suggesting a deregulation of the inflammatory cascade in diabetic animals. Despite this, diabetic animals showed exacerbated ischemic brain injury.
While immune cell recruitment has been shown to worsen stroke outcome, it can also play a beneficial role through promoting neovascularization. As mentioned earlier, monocyte/macrophage recruitment is necessary for arteriogenesis and collateral vessel maturation. MCP-1 and Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) have been shown to increase arteriogenesis and collateralization after femoral artery occlusion through increased monocyte/macrophage survival (Buschmann et al., 2001; Ito et al., 1997). Similarly, GM-CSF enhances arteriogenesis and leptomeningeal collateral growth in the hypo-perfused rodent brain through monocyte/macrophage recruitment (Buschmann et al., 2003; Todo et al., 2008). Clinically, different monocyte subtypes were associated with either tissue injury or repair and angiogenesis after stroke (Urra et al., 2009). In diabetes, peripheral neovascularization and collateral vessel development is impaired secondary to reduced monocyte recruitment. This is believed to be due to defective VEGF receptor signaling in these cells (Carr et al., 2011; Tchaikovski et al., 2009; Waltenberger, 2001). However, how diabetes affects the immune cell mediated neovascularization in the brain is yet to be discovered.
2. Stem/progenitor cells
Different stem/progenitor cells have been shown to play a beneficial role in ischemic stroke recovery. These cells are not terminally differentiated and include endothelial progenitor cells, and bone marrow stromal cells.
a. Endothelial progenitor cells
Circulating endothelial progenitor cells (EPCs) represent a specific type of bone marrow derived immature cells that have the ability to differentiate to endothelial cells (Yoder, 2012). EPCs can be isolated from bone marrow, cord or peripheral blood. EPCs express surface markers similar to hematopoietic and endothelial cells and are recruited to the endothelium following hypoxia or ischemia. EPCs characterization is difficult and they are generally identified by staining for CD34, CD133, or VEGFR2 using flow cytometry. These cells play an important role in angiogenesis and vascular repair (Zhao et al., 2013). These functions can be impaired under the oxidative environment of diabetes.
The clinical importance of endothelial progenitor cells in endogenous vascular repair has been highlighted early on in coronary artery disease (Schmidt-Lucke et al., 2005; Werner et al., 2005). Similarly, ischemic stroke patients with increased circulating EPCs levels show better functional outcome at 3 months (Marti-Fabregas et al., 2013; Sobrino et al., 2007), while fewer number of EPCs on admission is an independent risk factor for poor outcome at 6-month (Tsai et al., 2014). Acute statin therapy or pretreatment, which are known to improve stroke outcome, have been shown to increase circulating EPCs levels in the first week after stroke (Marti-Fabregas et al., 2013; Sobrino et al., 2012).
Experimentally, EPCs have been shown to participate in neovascularization, endothelial repair and vascular restoration after ischemic stroke. A study by Zhang et al. investigated the contribution of EPCs to neovascularization after stroke (Zhang et al., 2002). Using embolic stroke mouse model and bone marrow transplantation, they could show the incorporation of EPCs in the newly formed vessels in the ischemic border zone. This highlights the contribution of endogenous EPCs to ischemic recovery through vasculogenesis. Interestingly, administration of EPC conditioned media achieved similar proangiogenic response and functional recovery suggesting that EPCs mediate some of their actions via growth factors secretion (Rosell et al., 2013).
EPCs are reduced and dysfunctional under diabetic milieu. Diabetic patients with stroke and atherosclerotic intracranial artery stenosis have reduced circulating EPCs compared to respective non-diabetics (Liu et al., 2013). Experimentally, type 2 diabetic db/db mice show reduced angiogenic response to bone marrow-derived EPC infusion after stroke (Bai et al., 2014). Taken together, EPC-mediated angiogenic and vascular restorative actions seem to be impaired in diabetic stroke.
b. Bone marrow stromal cells
Exogenously administered bone marrow stromal cells (BMSCs) have been shown to enhance regeneration and functional recovery after stroke through promoting angiogenesis and vascular stabilization (Chen et al., 2001; Zacharek et al., 2007). When administered to type 1 diabetic rats after stroke and followed up for 14 days, BMSCs therapy could not elicit functional recovery and was associated with higher mortality, BBB leakage, and brain hemorrhage. BMSCs treatment also increased cerebral arteriosclerosis after stroke in these rats as manifested by internal carotid artery neointimal formation and cerebral arteriole narrowing/occlusion (Chen et al., 2011b). In a subsequent study, the authors adopted a combination treatment of BMSC with 40 mg/kg of Niaspan administered daily for 14 days after stroke (Yan et al., 2013). Niaspan is a prolonged release formulation of niacin (vitamin B3), which has anti-atherosclerotic properties by increasing high-density lipoprotein (HDL) levels and decreasing low-density lipoprotein (LDL) levels. The combination treatment reduced the atherosclerotic effects of BMSCs in diabetic rats and enhanced white matter remodeling and synaptic protein expression (Ye et al., 2013). However, it did not improve functional outcome. The same group later tested human umbilical cord blood cells (HUCBCs) under the same experimental conditions (Yan et al., 2014). They found that HUCBCs could improve functional outcome and enhanced white matter and vascular remodeling.
3. Microparticles
Microparticles (MPs) are membrane bound vesicles (100 to 1000 nm in diameter) that are released by budding from different cell types (Mause and Weber, 2010). They are present physiologically, with 70 to 90 % of circulating MPs being derived from platelets. MPs are considered a form of communication between cells since they enclose different signaling molecules, and thus, can influence the function and phenotype of recipient cells. Moreover, they can be tracked to their parent cells through analysis of surface antigens. MPs can be increased under different pathological conditions such as autoimmune disorders, atherosclerosis, cancers, and infection (Mause and Weber, 2010).
Platelet derived MPs (PMPs) are elevated acutely after cerebral infarction and correlate with the presence of intracranial atherothrombotic lesions (Kuriyama et al., 2010). Apart from the procoagulative properties of PMPs, these particles are thought to contain growth factors that may promote brain cells survival and neurovascular restoration. Experimentally, PMPs were shown to be involved in the neuroprotection conferred by remote ischemic conditioning after stroke (Shan et al., 2013). Similarly, exogenous administration of platelet derived MPs was shown to induce angiogenesis and neurogenesis after permanent MCAO in rats and was associated with behavioral recovery at 90 days in rats (Hayon et al., 2012a). In vitro, treatment of neural stem cells with platelet derived MPs increased their survival and led to larger neurospheres. Moreover, the treatment also increased the differentiation potential of neural stem cells (NSC) to glia and neurons. These effects were attributed to various growth factors contained within the PMP (Hayon et al., 2012b). Interestingly, in these studies, ischemia was induced using suture occlusion and it would be very interesting to examine the role of microparticles in the more clinically relevant embolic model to assess their procoagulative potential.
Type 2 diabetic patients have elevated levels of MP which correlate with diabetic complications (Tsimerman et al., 2011) such as vascular dysfunction (Feng et al., 2010), diabetic nephropathy (Omoto et al., 1999), diabetic retinopathy (Ogata et al., 2005; Ogata et al., 2006), myocardial infarction (Morel et al., 2004), atherosclerosis (Nomura et al., 1995), coronary artery disease (Nomura, 2009), and possibly stroke. There is one preclinical study in which Chen et al. (Chen et al., 2011a) investigated the function of circulating endothelial progenitor cells (cEPCs) and cellular membrane microparticles (MPs) in diabetic stroke using db/db mice, a genetic model of type 2 diabetes with a point mutation in the leptin receptor. Bone marrow-derived EPCs were reduced in db/db mice and dysfunctional as measured by reduced tube formation ability in vitro. In contrast, the levels of EPC-derived MPs were increased in these mice after stroke and correlated with fasting blood glucose and cerebral ischemic damage. The same group investigated the effect of EPCs priming with chemokine receptor type 4 (CXCR4) using adenovirus. The CXCR4-primed EPCs therapy was able to reduce the ischemic brain damage and promote repair in db/db mice (Chen et al., 2012).
Conclusions
The reasons why years of stroke research has failed to identify new therapeutic approaches are multifactorial. However, there are 2 major points that all researchers agree upon. First, stroke research should not only focus on neurons and look at repair and recovery mechanisms from a broader perspective. With the development of neurovascular unit concept, there is more intense investigation of cell-to-cell communications within the ischemic and peri-infarct area to meet this need. Recent studies suggest that nonischemic hemisphere also contributes to the repair and recovery following stroke (Ergul et al., 2012a; Guan et al., 2011; Prakash et al., 2013b). As summarized above, circulating cells are also involved in this response. Collectively, we propose that cellular communications leading to recovery are very complex and include whole body responses. Second, most therapeutic targets have been identified in otherwise healthy and young animals without comorbidities that are common to stroke patients. Most if not all studies discussed in this review used only male animals. Diabetes is the most rapidly increasing comorbidity in acute ischemic stroke. As we move forward to better understand the role(s) of cell-to-cell communications in neuronal repair and vascular restoration, diabetes and sex should be included in experimental models.
Acknowledgments
Adviye Ergul is a Research Career Scientist at the Charlie Norwood Veterans Affairs Medical Center in Augusta, Georgia. This work was supported in part by VA Merit Award (BX000347), VA Research Career Scientists Award, and NIH awards (NS083559, NS083559S1 and S2) to Adviye Ergul; VA Merit Award (BX000891) and NIH award (NS063965) to Susan C. Fagan. The contents do not represent the views of the Department of Veterans Affairs or the United States Government.
References
- Abaci A, et al. Effect of diabetes mellitus on formation of coronary collateral vessels. Circulation. 1999;99:2239–42. doi: 10.1161/01.cir.99.17.2239. [DOI] [PubMed] [Google Scholar]
- Abdelsaid M, et al. Metformin Treatment in Post-stroke Period Prevents Nitrative Stress and Restores Angiogenic Signaling in the Brain in Diabetes. Diabetes. 2014 doi: 10.2337/db14-1423. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Gayyar MM, et al. Thioredoxin interacting protein is a novel mediator of retinal inflammation and neurotoxicity. Br J Pharmacol. 2011;164:170–80. doi: 10.1111/j.1476-5381.2011.01336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhusban A, et al. AT1 receptor antagonism is proangiogenic in the brain: BDNF a novel mediator. J Pharmacol Exp Ther. 2013;344:348–59. doi: 10.1124/jpet.112.197483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai K, Lo EH. An oligovascular niche: cerebral endothelial cells promote the survival and proliferation of oligodendrocyte precursor cells. J Neurosci. 2009;29:4351–5. doi: 10.1523/JNEUROSCI.0035-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armulik A, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–61. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- Arnold T, Betsholtz C. Correction: The importance of microglia in the development of the vasculature in the central nervous system. Vasc Cell. 2013;5:12. doi: 10.1186/2045-824X-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Association, A.D. National diabetes fact sheet. 2014 http://www.diabetes.org/diabetes-statistics.jsp.
- Attwell D, et al. Glial and neuronal control of brain blood flow. Nature. 2010;468:232–43. doi: 10.1038/nature09613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai YY, et al. Image-guided pro-angiogenic therapy in diabetic stroke mouse models using a multi-modal nanoprobe. Theranostics. 2014;4:787–97. doi: 10.7150/thno.9525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron. 2008;60:430–40. doi: 10.1016/j.neuron.2008.10.013. [DOI] [PubMed] [Google Scholar]
- Beck H, et al. Cell type-specific expression of neuropilins in an MCA-occlusion model in mice suggests a potential role in post-ischemic brain remodeling. J Neuropathol Exp Neurol. 2002;61:339–50. doi: 10.1093/jnen/61.4.339. [DOI] [PubMed] [Google Scholar]
- Bell RD, et al. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68:409–27. doi: 10.1016/j.neuron.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocker-Meffert S, et al. Erythropoietin and VEGF promote neural outgrowth from retinal explants in postnatal rats. Invest Ophthalmol Vis Sci. 2002;43:2021–6. [PubMed] [Google Scholar]
- Bonkowski D, et al. The CNS microvascular pericyte: pericyte-astrocyte crosstalk in the regulation of tissue survival. Fluids Barriers CNS. 2011;8:8. doi: 10.1186/2045-8118-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockington A, et al. Expression of vascular endothelial growth factor and its receptors in the central nervous system in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2006;65:26–36. doi: 10.1097/01.jnen.0000196134.51217.74. [DOI] [PubMed] [Google Scholar]
- Bruno MA, Cuello AC. Activity-dependent release of precursor nerve growth factor, conversion to mature nerve growth factor, and its degradation by a protease cascade. Proc Natl Acad Sci U S A. 2006;103:6735–40. doi: 10.1073/pnas.0510645103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buraczynska K, et al. Monocyte chemoattractant protein (MCP-1) A-2518G gene polymorphism in stroke patients with different comorbidities. Clin Biochem. 2010;43:1421–6. doi: 10.1016/j.clinbiochem.2010.09.011. [DOI] [PubMed] [Google Scholar]
- Buschmann IR, et al. GM-CSF: a strong arteriogenic factor acting by amplification of monocyte function. Atherosclerosis. 2001;159:343–56. doi: 10.1016/s0021-9150(01)00637-2. [DOI] [PubMed] [Google Scholar]
- Buschmann IR, et al. Therapeutic induction of arteriogenesis in hypoperfused rat brain via granulocyte-macrophage colony-stimulating factor. Circulation. 2003;108:610–5. doi: 10.1161/01.CIR.0000074209.17561.99. [DOI] [PubMed] [Google Scholar]
- Carr CL, et al. Dysregulated selectin expression and monocyte recruitment during ischemia-related vascular remodeling in diabetes mellitus. Arterioscler Thromb Vasc Biol. 2011;31:2526–33. doi: 10.1161/ATVBAHA.111.230177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, et al. Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats. Stroke. 2001;32:1005–11. doi: 10.1161/01.str.32.4.1005. [DOI] [PubMed] [Google Scholar]
- Chen J, et al. Circulating endothelial progenitor cells and cellular membrane microparticles in db/db diabetic mouse: possible implications in cerebral ischemic damage. Am J Physiol Endocrinol Metab. 2011a;301:E62–71. doi: 10.1152/ajpendo.00026.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, et al. Adverse effects of bone marrow stromal cell treatment of stroke in diabetic rats. Stroke. 2011b;42:3551–8. doi: 10.1161/STROKEAHA.111.627174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, et al. Transfusion of CXCR4-primed endothelial progenitor cells reduces cerebral ischemic damage and promotes repair in db/db diabetic mice. PLoS One. 2012;7:e50105. doi: 10.1371/journal.pone.0050105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X, et al. Angiopoietin/Tie2 pathway mediates type 2 diabetes induced vascular damage after cerebral stroke. Neurobiol Dis. 2011;43:285–92. doi: 10.1016/j.nbd.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalkara T, Gursoy-Ozdemir Y, Yemisci M. Brain microvascular pericytes in health and disease. Acta Neuropathol. 2011;122:1–9. doi: 10.1007/s00401-011-0847-6. [DOI] [PubMed] [Google Scholar]
- Dewar D, Underhill SM, Goldberg MP. Oligodendrocytes and ischemic brain injury. J Cereb Blood Flow Metab. 2003;23:263–74. doi: 10.1097/01.WCB.0000053472.41007.F9. [DOI] [PubMed] [Google Scholar]
- Diaz-Flores L, et al. Pericytes. Morphofunction, interactions and pathology in a quiescent and activated mesenchymal cell niche. Histol Histopathol. 2009;24:909–69. doi: 10.14670/HH-24.909. [DOI] [PubMed] [Google Scholar]
- Dore-Duffy P, LaManna JC. Physiologic angiodynamics in the brain. Antioxid Redox Signal. 2007;9:1363–71. doi: 10.1089/ars.2007.1713. [DOI] [PubMed] [Google Scholar]
- Dore-Duffy P. Pericytes and adaptive angioplasticity: the role of tumor necrosis factor-like weak inducer of apoptosis (TWEAK) Methods Mol Biol. 2014;1135:35–52. doi: 10.1007/978-1-4939-0320-7_4. [DOI] [PubMed] [Google Scholar]
- Dumont DJ, et al. Vascularization of the mouse embryo: a study of flk-1, tek, tie, and vascular endothelial growth factor expression during development. Dev Dyn. 1995;203:80–92. doi: 10.1002/aja.1002030109. [DOI] [PubMed] [Google Scholar]
- Ejaz S, et al. Importance of pericytes and mechanisms of pericyte loss during diabetes retinopathy. Diabetes Obes Metab. 2008;10:53–63. doi: 10.1111/j.1463-1326.2007.00795.x. [DOI] [PubMed] [Google Scholar]
- Enge M, et al. Endothelium-specific platelet-derived growth factor-B ablation mimics diabetic retinopathy. EMBO J. 2002;21:4307–16. doi: 10.1093/emboj/cdf418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ergul A, Alhusban A, Fagan SC. Angiogenesis: a harmonized target for recovery after stroke. Stroke. 2012a;43:2270–4. doi: 10.1161/STROKEAHA.111.642710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ergul A, et al. Cerebrovascular complications of diabetes: focus on stroke. Endocr Metab Immune Disord Drug Targets. 2012b;12:148–58. doi: 10.2174/187153012800493477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ergul A, et al. Cerebral neovascularization in diabetes: implications for stroke recovery and beyond. J Cereb Blood Flow Metab. 2014 doi: 10.1038/jcbfm.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erskine L, et al. VEGF signaling through neuropilin 1 guides commissural axon crossing at the optic chiasm. Neuron. 2011;70:951–65. doi: 10.1016/j.neuron.2011.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantin A, et al. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood. 2010;116:829–40. doi: 10.1182/blood-2009-12-257832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner JR, et al. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24:2143–55. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng B, et al. Circulating level of microparticles and their correlation with arterial elasticity and endothelium-dependent dilation in patients with type 2 diabetes mellitus. Atherosclerosis. 2010;208:264–9. doi: 10.1016/j.atherosclerosis.2009.06.037. [DOI] [PubMed] [Google Scholar]
- Forsythe JA, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–13. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman SF, Hatchell DL. Enhanced superoxide radical production by stimulated polymorphonuclear leukocytes in a cat model of diabetes. Exp Eye Res. 1992;55:767–73. doi: 10.1016/0014-4835(92)90181-q. [DOI] [PubMed] [Google Scholar]
- Fulmer CG, et al. Astrocyte-derived BDNF supports myelin protein synthesis after cuprizone-induced demyelination. J Neurosci. 2014;34:8186–96. doi: 10.1523/JNEUROSCI.4267-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher KA, et al. Diabetic impairments in NO-mediated endothelial progenitor cell mobilization and homing are reversed by hyperoxia and SDF-1 alpha. J Clin Invest. 2007;117:1249–59. doi: 10.1172/JCI29710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia CM, et al. Endothelial cell-astrocyte interactions and TGF beta are required for induction of blood-neural barrier properties. Brain Res Dev Brain Res. 2004;152:25–38. doi: 10.1016/j.devbrainres.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Gensert JM, Goldman JE. Endogenous progenitors remyelinate demyelinated axons in the adult CNS. Neuron. 1997;19:197–203. doi: 10.1016/s0896-6273(00)80359-1. [DOI] [PubMed] [Google Scholar]
- Geraldes P, et al. Activation of PKC-delta and SHP-1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy. Nat Med. 2009;15:1298–306. doi: 10.1038/nm.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhardt H, Betsholtz C. Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res. 2003;314:15–23. doi: 10.1007/s00441-003-0745-x. [DOI] [PubMed] [Google Scholar]
- Gerhardt H, et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol. 2003;161:1163–77. doi: 10.1083/jcb.200302047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaume C, et al. Glia: the fulcrum of brain diseases. Cell Death Differ. 2007;14:1324–35. doi: 10.1038/sj.cdd.4402144. [DOI] [PubMed] [Google Scholar]
- Gonzalez Y, et al. High glucose concentrations induce TNF-alpha production through the down-regulation of CD33 in primary human monocytes. BMC Immunology. 2012;13:19. doi: 10.1186/1471-2172-13-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan W, et al. Vascular protection by angiotensin receptor antagonism involves differential VEGF expression in both hemispheres after experimental stroke. PLoS One. 2011;6:e24551. doi: 10.1371/journal.pone.0024551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, et al. Neuroprotection via matrix-trophic coupling between cerebral endothelial cells and neurons. Proc Natl Acad Sci U S A. 2008;105:7582–7. doi: 10.1073/pnas.0801105105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hai J, et al. Vascular endothelial growth factor expression and angiogenesis induced by chronic cerebral hypoperfusion in rat brain. Neurosurgery. 2003;53:963–70. doi: 10.1227/01.neu.0000083594.10117.7a. discussion 970–2. [DOI] [PubMed] [Google Scholar]
- Hall CN, et al. Capillary pericytes regulate cerebral blood flow in health and disease. Nature. 2014;508:55–60. doi: 10.1038/nature13165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes HP, Federoff HJ, Brownlee M. Nerve growth factor prevents both neuroretinal programmed cell death and capillary pathology in experimental diabetes. Mol Med. 1995;1:527–34. [PMC free article] [PubMed] [Google Scholar]
- Hansson E, Ronnback L. Glial neuronal signaling in the central nervous system. FASEB J. 2003;17:341–8. doi: 10.1096/fj.02-0429rev. [DOI] [PubMed] [Google Scholar]
- Hartog TE, et al. Brain-derived neurotrophic factor signaling in the HVC is required for testosterone-induced song of female canaries. J Neurosci. 2009;29:15511–9. doi: 10.1523/JNEUROSCI.2564-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayon Y, et al. Platelet microparticles induce angiogenesis and neurogenesis after cerebral ischemia. Curr Neurovasc Res. 2012a;9:185–92. doi: 10.2174/156720212801619018. [DOI] [PubMed] [Google Scholar]
- Hayon Y, et al. Platelet microparticles promote neural stem cell proliferation, survival and differentiation. J Mol Neurosci. 2012b;47:659–65. doi: 10.1007/s12031-012-9711-y. [DOI] [PubMed] [Google Scholar]
- Hellbach N, et al. Neural deletion of Tgfbr2 impairs angiogenesis through an altered secretome. Hum Mol Genet. 2014;23:6177–90. doi: 10.1093/hmg/ddu338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, et al. Neuroinflammatory processes in Alzheimer’s disease. J Neural Transm. 2010;117:919–47. doi: 10.1007/s00702-010-0438-z. [DOI] [PubMed] [Google Scholar]
- Hermann DM, Buga AM, Popa-Wagner A. Neurovascular remodeling in the aged ischemic brain. J Neural Transm. 2013 doi: 10.1007/s00702-013-1148-0. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Hill J, et al. Emerging roles of pericytes in the regulation of the neurovascular unit in health and disease. J Neuroimmune Pharmacol. 2014;9:591–605. doi: 10.1007/s11481-014-9557-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoefer IE, Piek JJ, Pasterkamp G. Pharmaceutical interventions to influence arteriogenesis: new concepts to treat ischemic heart disease. Curr Med Chem. 2006;13:979–87. doi: 10.2174/092986706776360996. [DOI] [PubMed] [Google Scholar]
- Ingraham JP, et al. Aging reduces hypoxia-induced microvascular growth in the rodent hippocampus. J Gerontol A Biol Sci Med Sci. 2008;63:12–20. doi: 10.1093/gerona/63.1.12. [DOI] [PubMed] [Google Scholar]
- Ishibashi T, et al. Astrocytes promote myelination in response to electrical impulses. Neuron. 2006;49:823–32. doi: 10.1016/j.neuron.2006.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, et al. Vasculogenesis in experimental stroke after human cerebral endothelial cell transplantation. Stroke. 2013;44:3473–81. doi: 10.1161/STROKEAHA.113.001943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito WD, et al. Monocyte chemotactic protein-1 increases collateral and peripheral conductance after femoral artery occlusion. Circ Res. 1997;80:829–37. doi: 10.1161/01.res.80.6.829. [DOI] [PubMed] [Google Scholar]
- Itoh Y, et al. Astrocytes and pericytes cooperatively maintain a capillary-like structure composed of endothelial cells on gel matrix. Brain Res. 2011;1406:74–83. doi: 10.1016/j.brainres.2011.06.039. [DOI] [PubMed] [Google Scholar]
- Jin K, et al. Neurogenesis in dentate subgranular zone and rostral subventricular zone after focal cerebral ischemia in the rat. Proc Natl Acad Sci U S A. 2001;98:4710–5. doi: 10.1073/pnas.081011098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, et al. Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivo. Proc Natl Acad Sci U S A. 2002;99:11946–50. doi: 10.1073/pnas.182296499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin KL, et al. Induction of vascular endothelial growth factor and hypoxia-inducible factor-1alpha by global ischemia in rat brain. Neuroscience. 2000;99:577–85. doi: 10.1016/s0306-4522(00)00207-4. [DOI] [PubMed] [Google Scholar]
- Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol. 2010;87:779–89. doi: 10.1189/jlb.1109766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing L, et al. Temporal profile of astrocytes and changes of oligodendrocyte-based myelin following middle cerebral artery occlusion in diabetic and non-diabetic rats. Int J Biol Sci. 2013;9:190–9. doi: 10.7150/ijbs.5844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jojart I, et al. Immunoelectronhistochemical evidence for innervation of brain microvessels by vasopressin-immunoreactive neurons in the rat. Neurosci Lett. 1984;51:259–64. doi: 10.1016/0304-3940(84)90561-5. [DOI] [PubMed] [Google Scholar]
- Joyal JS, et al. Ischemic neurons prevent vascular regeneration of neural tissue by secreting semaphorin 3A. Blood. 2011;117:6024–35. doi: 10.1182/blood-2010-10-311589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karima M, et al. Enhanced superoxide release and elevated protein kinase C activity in neutrophils from diabetic patients: association with periodontitis. J Leukoc Biol. 2005;78:862–70. doi: 10.1189/jlb.1004583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney JB, et al. The VEGF receptor flt-1 (VEGFR-1) is a positive modulator of vascular sprout formation and branching morphogenesis. Blood. 2004;103:4527–35. doi: 10.1182/blood-2003-07-2315. [DOI] [PubMed] [Google Scholar]
- Kermani P, et al. Neurotrophins promote revascularization by local recruitment of TrkB+ endothelial cells and systemic mobilization of hematopoietic progenitors. J Clin Invest. 2005;115:653–63. doi: 10.1172/JCI200522655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaibullina AA, Rosenstein JM, Krum JM. Vascular endothelial growth factor promotes neurite maturation in primary CNS neuronal cultures. Brain Res Dev Brain Res. 2004;148:59–68. doi: 10.1016/j.devbrainres.2003.09.022. [DOI] [PubMed] [Google Scholar]
- Kim E, Tolhurst AT, Cho S. Deregulation of inflammatory response in the diabetic condition is associated with increased ischemic brain injury. J Neuroinflammation. 2014;11:83. doi: 10.1186/1742-2094-11-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, et al. Paracrine and autocrine functions of brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF) in brain-derived endothelial cells. J Biol Chem. 2004;279:33538–46. doi: 10.1074/jbc.M404115200. [DOI] [PubMed] [Google Scholar]
- Kim J, et al. Different prognostic value of white blood cell subtypes in patients with acute cerebral infarction. Atherosclerosis. 2012;222:464–7. doi: 10.1016/j.atherosclerosis.2012.02.042. [DOI] [PubMed] [Google Scholar]
- Klagsbrun M, Eichmann A. A role for axon guidance receptors and ligands in blood vessel development and tumor angiogenesis. Cytokine Growth Factor Rev. 2005;16:535–48. doi: 10.1016/j.cytogfr.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Kumari R, et al. Impaired wound healing after cerebral hypoxia-ischemia in the diabetic mouse. J Cereb Blood Flow Metab. 2007;27:710–8. doi: 10.1038/sj.jcbfm.9600382. [DOI] [PubMed] [Google Scholar]
- Kummer U, et al. Elevated Glucose Concentrations Promote Receptor-Independent Activation of Adherent Human Neutrophils: An Experimental and Computational Approach. Biophysical Journal. 2007;92:2597–2607. doi: 10.1529/biophysj.106.086769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriyama N, et al. Evaluation of factors associated with elevated levels of platelet-derived microparticles in the acute phase of cerebral infarction. Clin Appl Thromb Hemost. 2010;16:26–32. doi: 10.1177/1076029609338047. [DOI] [PubMed] [Google Scholar]
- Lalancette-Hebert M, et al. Galectin-3 is required for resident microglia activation and proliferation in response to ischemic injury. J Neurosci. 2012;32:10383–95. doi: 10.1523/JNEUROSCI.1498-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HS, et al. Brain angiogenesis in developmental and pathological processes: regulation, molecular and cellular communication at the neurovascular interface. FEBS J. 2009;276:4622–35. doi: 10.1111/j.1742-4658.2009.07174.x. [DOI] [PubMed] [Google Scholar]
- Lee SW, et al. SSeCKS regulates angiogenesis and tight junction formation in blood-brain barrier. Nat Med. 2003;9:900–6. doi: 10.1038/nm889. [DOI] [PubMed] [Google Scholar]
- Lee Y, et al. Therapeutically targeting neuroinflammation and microglia after acute ischemic stroke. Biomed Res Int. 2014;2014:297241. doi: 10.1155/2014/297241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennmyr F, et al. Expression of vascular endothelial growth factor (VEGF) and its receptors (Flt-1 and Flk-1) following permanent and transient occlusion of the middle cerebral artery in the rat. J Neuropathol Exp Neurol. 1998;57:874–82. doi: 10.1097/00005072-199809000-00009. [DOI] [PubMed] [Google Scholar]
- Lennmyr F, et al. Vascular endothelial growth factor gene expression in middle cerebral artery occlusion in the rat. Acta Anaesthesiol Scand. 2005;49:488–93. doi: 10.1111/j.1399-6576.2005.00646.x. [DOI] [PubMed] [Google Scholar]
- Li AF, et al. High glucose alters connexin 43 expression and gap junction intercellular communication activity in retinal pericytes. Invest Ophthalmol Vis Sci. 2003;44:5376–82. doi: 10.1167/iovs.03-0360. [DOI] [PubMed] [Google Scholar]
- Li J, et al. SMAD4-mediated WNT signaling controls the fate of cranial neural crest cells during tooth morphogenesis. Development. 2011;138:1977–89. doi: 10.1242/dev.061341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, et al. Neurovascular recovery via co-transplanted neural and vascular progenitors leads to improved functional restoration after ischemic stroke in rats. Stem Cell Reports. 2014a;3:101–14. doi: 10.1016/j.stemcr.2014.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SF, et al. Recombinant adeno-associated virus serotype 1-vascular endothelial growth factor promotes neurogenesis and neuromigration in the subventricular zone and rescues neuronal function in ischemic rats. Neurosurgery. 2009;65:771–9. doi: 10.1227/01.NEU.0000349931.61771.52. discussion 779. [DOI] [PubMed] [Google Scholar]
- Li W, et al. Adaptive cerebral neovascularization in a model of type 2 diabetes: relevance to focal cerebral ischemia. Diabetes. 2010;59:228–35. doi: 10.2337/db09-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, et al. Comparative analysis of the neurovascular injury and functional outcomes in experimental stroke models in diabetic Goto-Kakizaki rats. Brain Res. 2013;1541:106–14. doi: 10.1016/j.brainres.2013.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, et al. Ephrin-A3 and ephrin-A4 contribute to microglia-induced angiogenesis in brain endothelial cells. Anat Rec (Hoboken) 2014b;297:1908–18. doi: 10.1002/ar.22998. [DOI] [PubMed] [Google Scholar]
- Li Y, et al. The role of astrocytes in mediating exogenous cell-based restorative therapy for stroke. Glia. 2014c;62:1–16. doi: 10.1002/glia.22585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YN, et al. Ischemic neurons activate astrocytes to disrupt endothelial barrier via increasing VEGF expression. J Neurochem. 2014d;129:120–9. doi: 10.1111/jnc.12611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liauw J, et al. Thrombospondins 1 and 2 are necessary for synaptic plasticity and functional recovery after stroke. J Cereb Blood Flow Metab. 2008;28:1722–32. doi: 10.1038/jcbfm.2008.65. [DOI] [PubMed] [Google Scholar]
- Liebner S, Czupalla CJ, Wolburg H. Current concepts of blood-brain barrier development. Int J Dev Biol. 2011;55:467–76. doi: 10.1387/ijdb.103224sl. [DOI] [PubMed] [Google Scholar]
- Lin TN, et al. Differential regulation of thrombospondin-1 and thrombospondin-2 after focal cerebral ischemia/reperfusion. Stroke. 2003;34:177–86. doi: 10.1161/01.str.0000047100.84604.ba. [DOI] [PubMed] [Google Scholar]
- Lindberg OR, Brederlau A, Kuhn HG. Epidermal growth factor treatment of the adult brain subventricular zone leads to focal microglia/macrophage accumulation and angiogenesis. Stem Cell Reports. 2014;2:440–8. doi: 10.1016/j.stemcr.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, et al. Higher numbers of circulating endothelial progenitor cells in stroke patients with intracranial arterial stenosis. BMC Neurol. 2013;13:161. doi: 10.1186/1471-2377-13-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, et al. Beneficial effects of gfap/vimentin reactive astrocytes for axonal remodeling and motor behavioral recovery in mice after stroke. Glia. 2014;62:2022–33. doi: 10.1002/glia.22723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Lopez C, LeRoith D, Torres-Aleman I. Insulin-like growth factor I is required for vessel remodeling in the adult brain. Proc Natl Acad Sci U S A. 2004;101:9833–8. doi: 10.1073/pnas.0400337101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louissaint A, Jr, et al. Coordinated interaction of neurogenesis and angiogenesis in the adult songbird brain. Neuron. 2002;34:945–60. doi: 10.1016/s0896-6273(02)00722-5. [DOI] [PubMed] [Google Scholar]
- Lu H, et al. Overexpression of netrin-1 improves neurological outcomes in mice following transient middle cerebral artery occlusion. Front Med. 2011;5:86–93. doi: 10.1007/s11684-011-0118-x. [DOI] [PubMed] [Google Scholar]
- Lu H, et al. Netrin-1 hyperexpression in mouse brain promotes angiogenesis and long-term neurological recovery after transient focal ischemia. Stroke. 2012;43:838–43. doi: 10.1161/STROKEAHA.111.635235. [DOI] [PubMed] [Google Scholar]
- Mamluk R, et al. Neuropilin-1 binds vascular endothelial growth factor 165, placenta growth factor-2, and heparin via its b1b2 domain. J Biol Chem. 2002;277:24818–25. doi: 10.1074/jbc.M200730200. [DOI] [PubMed] [Google Scholar]
- Marti-Fabregas J, et al. Endothelial progenitor cells in acute ischemic stroke. Brain Behav. 2013;3:649–55. doi: 10.1002/brb3.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda M, et al. Decreased fluidity of polymorphonuclear leukocyte membrane in streptozocin-induced diabetic rats. Diabetes. 1990;39:466–70. doi: 10.2337/diab.39.4.466. [DOI] [PubMed] [Google Scholar]
- Matsuo Y, et al. Correlation between myeloperoxidase-quantified neutrophil accumulation and ischemic brain injury in the rat. Effects of neutrophil depletion. Stroke. 1994;25:1469–75. doi: 10.1161/01.str.25.7.1469. [DOI] [PubMed] [Google Scholar]
- Mause SF, Weber C. Microparticles: Protagonists of a Novel Communication Network for Intercellular Information Exchange. Circulation Research. 2010;107:1047–1057. doi: 10.1161/CIRCRESAHA.110.226456. [DOI] [PubMed] [Google Scholar]
- Mazzanti M, Sul JY, Haydon PG. Glutamate on demand: astrocytes as a ready source. Neuroscientist. 2001;7:396–405. doi: 10.1177/107385840100700509. [DOI] [PubMed] [Google Scholar]
- Miao HQ, et al. Neuropilin-1 mediates collapsin-1/semaphorin III inhibition of endothelial cell motility: functional competition of collapsin-1 and vascular endothelial growth factor-165. J Cell Biol. 1999;146:233–42. doi: 10.1083/jcb.146.1.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micu I, et al. NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature. 2006;439:988–92. doi: 10.1038/nature04474. [DOI] [PubMed] [Google Scholar]
- Morel O, et al. Sustained elevated amounts of circulating procoagulant membrane microparticles and soluble GPV after acute myocardial infarction in diabetes mellitus. Thromb Haemost. 2004;91:345–53. doi: 10.1160/TH03-05-0294. [DOI] [PubMed] [Google Scholar]
- Navaratna D, et al. Mechanisms and targets for angiogenic therapy after stroke. Cell Adh Migr. 2009;3:216–23. doi: 10.4161/cam.3.2.8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navaratna D, et al. Cerebrovascular degradation of TRKB by MMP9 in the diabetic brain. J Clin Invest. 2013;123:3373–7. doi: 10.1172/JCI65767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura S, et al. Platelet-derived microparticles may influence the development of atherosclerosis in diabetes mellitus. Atherosclerosis. 1995;116:235–40. doi: 10.1016/0021-9150(95)05551-7. [DOI] [PubMed] [Google Scholar]
- Nomura S. Dynamic role of microparticles in type 2 diabetes mellitus. Curr Diabetes Rev. 2009;5:245–51. doi: 10.2174/157339909789804404. [DOI] [PubMed] [Google Scholar]
- Ogata N, et al. Increased levels of platelet-derived microparticles in patients with diabetic retinopathy. Diabetes Res Clin Pract. 2005;68:193–201. doi: 10.1016/j.diabres.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Ogata N, et al. Elevation of monocyte-derived microparticles in patients with diabetic retinopathy. Diabetes Res Clin Pract. 2006;73:241–8. doi: 10.1016/j.diabres.2006.01.014. [DOI] [PubMed] [Google Scholar]
- Omoto S, et al. Significance of platelet-derived microparticles and activated platelets in diabetic nephropathy. Nephron. 1999;81:271–7. doi: 10.1159/000045292. [DOI] [PubMed] [Google Scholar]
- Ozerdem U, Stallcup WB. Early contribution of pericytes to angiogenic sprouting and tube formation. Angiogenesis. 2003;6:241–9. doi: 10.1023/B:AGEN.0000021401.58039.a9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panes J, et al. Diabetes exacerbates inflammatory responses to ischemia-reperfusion. Circulation. 1996;93:161–7. doi: 10.1161/01.cir.93.1.161. [DOI] [PubMed] [Google Scholar]
- Park JA, et al. Meteorin regulates angiogenesis at the gliovascular interface. Glia. 2008;56:247–58. doi: 10.1002/glia.20600. [DOI] [PubMed] [Google Scholar]
- Park KW, et al. The axonal attractant Netrin-1 is an angiogenic factor. Proc Natl Acad Sci U S A. 2004;101:16210–5. doi: 10.1073/pnas.0405984101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SW, et al. Angiopoietin 2 induces pericyte apoptosis via alpha3beta1 integrin signaling in diabetic retinopathy. Diabetes. 2014;63:3057–68. doi: 10.2337/db13-1942. [DOI] [PubMed] [Google Scholar]
- Poittevin M, et al. Diabetic microangiopathy: impact of impaired cerebral vasoreactivity and delayed angiogenesis after permanent middle cerebral artery occlusion on stroke damage and cerebral repair in mice. Diabetes. 2015;64(3):999–1010. doi: 10.2337/db14-0759. [DOI] [PubMed] [Google Scholar]
- Prakash R, et al. Enhanced cerebral but not peripheral angiogenesis in the Goto-Kakizaki model of type 2 diabetes involves VEGF and peroxynitrite signaling. Diabetes. 2012;61:1533–42. doi: 10.2337/db11-1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash R, et al. Cerebral Neovascularization and Remodeling Patterns in Two Different Models of Type 2 Diabetes. PLoS One. 2013a;8:e56264. doi: 10.1371/journal.pone.0056264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash R, et al. Vascularization pattern after ischemic stroke is different in control versus diabetic rats: relevance to stroke recovery. Stroke. 2013b;44:2875–82. doi: 10.1161/STROKEAHA.113.001660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price CJ, et al. Cerebral neutrophil recruitment, histology, and outcome in acute ischemic stroke: an imaging-based study. Stroke. 2004;35:1659–64. doi: 10.1161/01.STR.0000130592.71028.92. [DOI] [PubMed] [Google Scholar]
- Ransom BR, Ransom CB. Astrocytes: multitalented stars of the central nervous system. Methods Mol Biol. 2012;814:3–7. doi: 10.1007/978-1-61779-452-0_1. [DOI] [PubMed] [Google Scholar]
- Rasika S, Nottebohm F, Alvarez-Buylla A. Testosterone increases the recruitment and/or survival of new high vocal center neurons in adult female canaries. Proc Natl Acad Sci U S A. 1994;91:7854–8. doi: 10.1073/pnas.91.17.7854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieckmann P, Engelhardt B. Building up the blood-brain barrier. Nat Med. 2003;9:828–9. doi: 10.1038/nm0703-828. [DOI] [PubMed] [Google Scholar]
- Ritter L, et al. Exaggerated neutrophil-mediated reperfusion injury after ischemic stroke in a rodent model of type 2 diabetes. Microcirculation. 2011;18:552–61. doi: 10.1111/j.1549-8719.2011.00115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosell A, et al. Factors secreted by endothelial progenitor cells enhance neurorepair responses after cerebral ischemia in mice. PLoS One. 2013;8:e73244. doi: 10.1371/journal.pone.0073244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenstein JM, et al. Neurotrophic effects of vascular endothelial growth factor on organotypic cortical explants and primary cortical neurons. J Neurosci. 2003;23:11036–44. doi: 10.1523/JNEUROSCI.23-35-11036.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin LL, et al. A cell culture model of the blood-brain barrier. J Cell Biol. 1991;115:1725–35. doi: 10.1083/jcb.115.6.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhrberg C. Growing and shaping the vascular tree: multiple roles for VEGF. Bioessays. 2003;25:1052–60. doi: 10.1002/bies.10351. [DOI] [PubMed] [Google Scholar]
- Ruiz de Almodovar C, et al. Matrix-binding vascular endothelial growth factor (VEGF) isoforms guide granule cell migration in the cerebellum via VEGF receptor Flk1. J Neurosci. 2010;30:15052–66. doi: 10.1523/JNEUROSCI.0477-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz de Almodovar C, et al. VEGF mediates commissural axon chemoattraction through its receptor Flk1. Neuron. 2011;70:966–78. doi: 10.1016/j.neuron.2011.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sa-Pereira I, Brites D, Brito MA. Neurovascular unit: a focus on pericytes. Mol Neurobiol. 2012;45:327–47. doi: 10.1007/s12035-012-8244-2. [DOI] [PubMed] [Google Scholar]
- Schmidt-Lucke C, et al. Reduced number of circulating endothelial progenitor cells predicts future cardiovascular events: proof of concept for the clinical importance of endogenous vascular repair. Circulation. 2005;111:2981–7. doi: 10.1161/CIRCULATIONAHA.104.504340. [DOI] [PubMed] [Google Scholar]
- Shalaby F, et al. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–6. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- Shan LY, et al. Platelet-derived microparticles are implicated in remote ischemia conditioning in a rat model of cerebral infarction. CNS Neurosci Ther. 2013;19:917–25. doi: 10.1111/cns.12199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiga Y, et al. Neutrophil as a mediator of ischemic edema formation in the brain. Neurosci Lett. 1991;125:110–2. doi: 10.1016/0304-3940(91)90003-c. [DOI] [PubMed] [Google Scholar]
- Shin JW, Huggenberger R, Detmar M. Transcriptional profiling of VEGF-A and VEGF-C target genes in lymphatic endothelium reveals endothelial-specific molecule-1 as a novel mediator of lymphangiogenesis. Blood. 2008;112:2318–26. doi: 10.1182/blood-2008-05-156331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman WF, et al. Vascular, glial and neuronal effects of vascular endothelial growth factor in mesencephalic explant cultures. Neuroscience. 1999;90:1529–41. doi: 10.1016/s0306-4522(98)00540-5. [DOI] [PubMed] [Google Scholar]
- Sobrino T, et al. The increase of circulating endothelial progenitor cells after acute ischemic stroke is associated with good outcome. Stroke. 2007;38:2759–64. doi: 10.1161/STROKEAHA.107.484386. [DOI] [PubMed] [Google Scholar]
- Sobrino T, et al. Increased levels of circulating endothelial progenitor cells in patients with ischaemic stroke treated with statins during acute phase. Eur J Neurol. 2012;19:1539–46. doi: 10.1111/j.1468-1331.2012.03770.x. [DOI] [PubMed] [Google Scholar]
- Sonntag WE, et al. Alterations in insulin-like growth factor-1 gene and protein expression and type 1 insulin-like growth factor receptors in the brains of ageing rats. Neuroscience. 1999;88:269–79. doi: 10.1016/s0306-4522(98)00192-4. [DOI] [PubMed] [Google Scholar]
- Stratman AN, et al. Pericyte recruitment during vasculogenic tube assembly stimulates endothelial basement membrane matrix formation. Blood. 2009;114:5091–101. doi: 10.1182/blood-2009-05-222364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, et al. AAV-mediated netrin-1 overexpression increases peri-infarct blood vessel density and improves motor function recovery after experimental stroke. Neurobiol Dis. 2011;44:73–83. doi: 10.1016/j.nbd.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchaikovski V, et al. Diabetes mellitus activates signal transduction pathways resulting in vascular endothelial growth factor resistance of human monocytes. Circulation. 2009;120:150–9. doi: 10.1161/CIRCULATIONAHA.108.817528. [DOI] [PubMed] [Google Scholar]
- Todo K, et al. Granulocyte-macrophage colony-stimulating factor enhances leptomeningeal collateral growth induced by common carotid artery occlusion. Stroke. 2008;39:1875–82. doi: 10.1161/STROKEAHA.107.503433. [DOI] [PubMed] [Google Scholar]
- Tolosa L, et al. Vascular endothelial growth factor protects spinal cord motoneurons against glutamate-induced excitotoxicity via phosphatidylinositol 3-kinase. J Neurochem. 2008;105:1080–90. doi: 10.1111/j.1471-4159.2007.05206.x. [DOI] [PubMed] [Google Scholar]
- Tsai NW, et al. The association between circulating endothelial progenitor cells and outcome in different subtypes of acute ischemic stroke. Clin Chim Acta. 2014;427:6–10. doi: 10.1016/j.cca.2013.09.029. [DOI] [PubMed] [Google Scholar]
- Tsimerman G, et al. Involvement of microparticles in diabetic vascular complications. Thromb Haemost. 2011;106:310–21. doi: 10.1160/TH10-11-0712. [DOI] [PubMed] [Google Scholar]
- Ullian EM, et al. Schwann cells and astrocytes induce synapse formation by spinal motor neurons in culture. Mol Cell Neurosci. 2004;25:241–51. doi: 10.1016/j.mcn.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Urra X, et al. Monocyte subtypes predict clinical course and prognosis in human stroke. J Cereb Blood Flow Metab. 2009;29:994–1002. doi: 10.1038/jcbfm.2009.25. [DOI] [PubMed] [Google Scholar]
- Virgintino D, et al. An intimate interplay between precocious, migrating pericytes and endothelial cells governs human fetal brain angiogenesis. Angiogenesis. 2007;10:35–45. doi: 10.1007/s10456-006-9061-x. [DOI] [PubMed] [Google Scholar]
- Waltenberger J. Impaired collateral vessel development in diabetes: potential cellular mechanisms and therapeutic implications. Cardiovasc Res. 2001;49:554–60. doi: 10.1016/s0008-6363(00)00228-5. [DOI] [PubMed] [Google Scholar]
- Wang L, et al. Treatment of stroke with erythropoietin enhances neurogenesis and angiogenesis and improves neurological function in rats. Stroke. 2004;35:1732–7. doi: 10.1161/01.STR.0000132196.49028.a4. [DOI] [PubMed] [Google Scholar]
- Wang YQ, et al. VEGF overexpression enhances striatal neurogenesis in brain of adult rat after a transient middle cerebral artery occlusion. J Neurosci Res. 2007;85:73–82. doi: 10.1002/jnr.21091. [DOI] [PubMed] [Google Scholar]
- Werner N, et al. Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med. 2005;353:999–1007. doi: 10.1056/NEJMoa043814. [DOI] [PubMed] [Google Scholar]
- Wesley UV, et al. Galectin-3 enhances angiogenic and migratory potential of microglial cells via modulation of integrin linked kinase signaling. Brain Res. 2013;1496:1–9. doi: 10.1016/j.brainres.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierusz-Wysocka B, et al. Evidence of polymorphonuclear neutrophils (PMN) activation in patients with insulin-dependent diabetes mellitus. J Leukoc Biol. 1987;42:519–23. doi: 10.1002/jlb.42.5.519. [DOI] [PubMed] [Google Scholar]
- Winkler EA, Bell RD, Zlokovic BV. Central nervous system pericytes in health and disease. Nat Neurosci. 2011;14:1398–405. doi: 10.1038/nn.2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TW, Li WW, Li H. Netrin-1 attenuates ischemic stroke-induced apoptosis. Neuroscience. 2008;156:475–82. doi: 10.1016/j.neuroscience.2008.08.015. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Mahmood A, Chopp M. Angiogenesis, neurogenesis and brain recovery of function following injury. Curr Opin Investig Drugs. 2010;11:298–308. [PMC free article] [PubMed] [Google Scholar]
- Yan T, et al. Niaspan attenuates the adverse effects of bone marrow stromal cell treatment of stroke in type one diabetic rats. PLoS One. 2013;8:e81199. doi: 10.1371/journal.pone.0081199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan T, et al. HUCBCs Increase Angiopoietin 1 and Induce Neurorestorative Effects after Stroke in T1DM Rats. CNS Neurosci Ther. 2014;20:935–44. doi: 10.1111/cns.12307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan YP, et al. Galectin-3 mediates post-ischemic tissue remodeling. Brain Res. 2009;1288:116–24. doi: 10.1016/j.brainres.2009.06.073. [DOI] [PubMed] [Google Scholar]
- Yang J, et al. proBDNF negatively regulates neuronal remodeling, synaptic transmission, and synaptic plasticity in hippocampus. Cell Rep. 2014;7:796–806. doi: 10.1016/j.celrep.2014.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, et al. Niaspan enhances vascular remodeling after stroke in type 1 diabetic rats. Exp Neurol. 2011;232:299–308. doi: 10.1016/j.expneurol.2011.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, et al. Combination BMSC and Niaspan treatment of stroke enhances white matter remodeling and synaptic protein expression in diabetic rats. Int J Mol Sci. 2013;14:22221–32. doi: 10.3390/ijms141122221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yemisci M, et al. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat Med. 2009;15:1031–7. doi: 10.1038/nm.2022. [DOI] [PubMed] [Google Scholar]
- Yoder MC. Human endothelial progenitor cells. Cold Spring Harb Perspect Med. 2012;2:a006692. doi: 10.1101/cshperspect.a006692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharek A, et al. Angiopoietin1/Tie2 and VEGF/Flk1 induced by MSC treatment amplifies angiogenesis and vascular stabilization after stroke. J Cereb Blood Flow Metab. 2007;27:1684–91. doi: 10.1038/sj.jcbfm.9600475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Xu Q. Stem/Progenitor cells in vascular regeneration. Arterioscler Thromb Vasc Biol. 2014;34:1114–9. doi: 10.1161/ATVBAHA.114.303809. [DOI] [PubMed] [Google Scholar]
- Zhang ZG, et al. Bone marrow-derived endothelial progenitor cells participate in cerebral neovascularization after focal cerebral ischemia in the adult mouse. Circ Res. 2002;90:284–8. doi: 10.1161/hh0302.104460. [DOI] [PubMed] [Google Scholar]
- Zhao YH, et al. Endothelial progenitor cells: therapeutic perspective for ischemic stroke. CNS Neurosci Ther. 2013;19:67–75. doi: 10.1111/cns.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]