

Abstract
High levels of aneuploidy have been observed in disease-free tissues, including post-mitotic tissues such as the brain. Using a quantitative interphase-fluorescence in situ hybridization approach, we previously reported a chromosome-specific, age-related increase in aneuploidy in the mouse cerebral cortex. Increased aneuploidy has been associated with defects in DNA repair and the spindle assembly checkpoint, which in turn can lead to premature aging. Here, we quantified the frequency of aneuploidy of three autosomes in the cerebral cortex and cerebellum of adult and developing brain of Bub1bH/H mice, which have a faulty mitotic checkpoint, and Ercc1−/Δ7 mice, defective in nucleotide excision repair and inter-strand cross-link repair. Surprisingly, the level of aneuploidy in the brain of these murine models of accelerated aging remains as low as in the young adult brains from control animals, i.e. <1% in the cerebral cortex and ∼0.1% in the cerebellum. Therefore, based on aneuploidy, these adult mice with reduced life span and accelerated progeroid features are indistinguishable from age-matched, normal controls. Yet, during embryonic development, we found that Bub1bH/H, but not Ercc1−/Δ7 mice, have a significantly higher frequency of aneuploid nuclei relative to wild-type controls in the cerebral cortex, reaching a frequency as high as 40.3% for each chromosome tested. Aneuploid cells in these mutant mice are likely eliminated early in development through apoptosis and/or immune-mediated clearance mechanisms, which would explain the low levels of aneuploidy during adulthood in the cerebral cortex of Bub1bH/H mice. These results shed light on the mechanisms of removal of aneuploidy cells in vivo.
Introduction
Genomic stability and maintenance of the correct chromosome number are important for the functioning of cells under normal physiological conditions. Defects in proteins responsible for chromosomal stability and genome maintenance have been associated with a large array of pathologies (1,2). Mitotic errors due to defects in genes involved in chromosome segregation result in aneuploidy, a karyotype characterized by whole chromosome gains and losses. Aneuploidy is typically associated with spontaneous abortions, birth defects, cancer and premature aging. Indeed, germ-line aneuploidy is incompatible with life, apart from few exceptions (trisomy 13, 18 and 21) (3). Patients with mosaic variegated aneuploidy syndrome, a rare autosomal disorder associated with chromosome mis-segregation and aneuploidy, show predisposition to cancer and progeroid features (4). Aneuploidy is now considered a hallmark of both cancer and aging, and mice models prone to its accumulation manifest various phenotypes (5). For instance, deficiency of mitotic arrest deficient-like 1 (Mad1l1) (6) and centromere protein E (Cenpe) (7) increases tumor incidence in mice, while heterozygosis of mitotic arrest deficient-like 2 (Mad2l1) causes chromosome mis-segregation and apoptosis. Interestingly, susceptibility to tumors in mice with altered expression of the Bub1 mitotic checkpoint serine/threonine kinase (Bub1) gene is dependent to its levels of expression. The dual outcome may be due to a Bub1 role in mediating cell death (8). These findings suggest that the consequences of aneuploidy could vary based on the severity of ploidy changes, and/or the frequency of affected cells. Furthermore, proteins involved in chromosome stability may have functions other than the control of mitosis.
Therefore, it is emerging that the consequences of aneuploidy, especially in disease-free tissues, are complex and poorly understood. Yet, aneuploidy caused by defects in some spindle assembly checkpoint (SAC) components or nucleotide excision repair, double-strand break repair and inter-strand cross-link repair in mice have been associated with multiple symptoms of premature aging. For example, mice expressing the SAC protein Bub1b) at 11% of normal levels (Bub1bH/H; H for hypomorphic) have a median life span of only 6 months, present with a variety of premature aging phenotypes, and elevated levels of aneuploidy in the spleen from 2 months onward (9). Likewise, mice heterozygous for two other SAC proteins, Bub3 mitotic checkpoint protein (Bub3) and ribonucleic acid export 1 (Rae1) also show increased aneuploidy and progeroid features (10). Excision repair cross-complementation group 1 (Ercc1) knockout mice show polyploidy in perinatal liver, progressing to severe aneuploidy by 3 weeks of age (11). A more benign variant of this mouse model, the Ercc1−/Δ7 mutant, carrying a null mutation in one allele and a 7-amino acid truncation in the second allele, displays several aging-associated phenotypes, a life span of only 6 months, accelerated age-related cognitive decline, and neurodegeneration (12,13). Mutant mice models, such as the Bub1b and Bub3/Rae1, suggest that accumulation of aneuploidy can be tolerated in adult tissues, albeit at a severe cost of fitness or the appearance of premature aging features. Indeed, sustained high-level expression of Bub1b preserves genomic integrity by correcting SAC signaling and has the ultimate effect of extending life span (14).
While it has been hypothesized since the 1960s that aneuploidy in disease-free somatic cells increases with age (15), it has been difficult to quantify chromosome numbers in non-dividing post-mitotic tissues during the aging process. Using a quantitative, dual-labeling interphase-fluorescence in situ hybridization (FISH) approach we previously reported that aneuploidy increases with age in the cerebral cortex of mice, with Chromosomes 7, 18 and Y most severely affected (i.e. up to 9.8% of non-neuronal brain nuclei of old mice being aneuploid for these chromosomes combined). While at old age aneuploidy was specific to non-neuronal nuclei, at early age both neuronal and non-neuronal cells were equally affected (16).
To test if the increased aneuploidy observed in mice during normal brain aging would be reinforced in Bub1b and Ercc1 mutant progeroid mice, we measured the levels of single-chromosome aneuploidy during adult and embryonic life. We focused our analysis on three autosomes, MMU7 (Mus Musculus Chromosome 7) and MMU18, as the most affected in our previous analysis, and MMU1 being the least affected.
Surprisingly, we found no evidence for accelerated accumulation of aneuploid nuclei in either cortex or cerebellum of the mutant mice. Conversely significantly high levels of aneuploidy were detected during embryonic development in Bub1bH/H mice, which agreed with a significant increase of terminal deoxynucleotidyl transferase (TdT) 2′-deoxyuridine 5′-triphosphate (dUTP) nick-end labeling (TUNEL)-positive cells. Our data suggests that the aneuploid progeny is eliminated early during development in Bub1bH/H mice, providing an explanation for the lack of aneuploidy accumulation in the adult brain.
Results
Low levels of aneuploidy are detected in both cortex and cerebellum of adult Bub1bH/H and Ercc1−/Δ7 mice
We sought to quantify the levels of whole chromosome aneuploidy in interphase nuclei from adult Bub1bH/H and Ercc1−/Δ7 defective mice at 6 and 3.5 months of age, respectively. Based on the median lifespan described for these mutant mice these time points should resemble ploidy changes observed in wild-type (WT) mice at old age (28 months) (9,12). Adult WT mice of the same age and breeding backgrounds were used as controls. For this purpose, we applied a previously described custom designed two-color FISH approach that provides highly quantitative and reproducible aneuploidy analysis for individual chromosomes in interphase nuclei by assessing gain and loss based on the enumeration at two separate loci on the same chromosome (Fig. 1A) (16,17). For each nucleus, aneuploidy was only scored when both loci showed a loss or gain of signal (Fig. 1B). We manually scored a total of 6559 nuclei (∼300 per mouse genotype) from the cerebral cortex and the cerebellum of adult Bub1bH/H, Ercc1−/Δ7 and age-matched control mice (Supplementary Material, Table S1).
Figure 1.

Probes used for FISH analysis and aneuploidy levels in adult mice. (A) Four differently labeled BAC clones (spectrum aqua, far red, spectrum orange and spectrum green) mapping to mouse chromosomes 1 and 18 at two distinct genomic loci enable the identification of diploid from aneuploid cells. (B) Two-color interphase FISH: the identification of whole chromosome loss or gain is determined by the numerical correspondence between the two colors. Representative hybridizations of cortical nuclei. Left panel shows a nucleus with two copies for MMU1 (2n) and right panel shows an aneuploid nucleus that contains three copies (gain). (C) Aneuploidy levels measured in the cortex of 6-month-old Bub1bH/H mice and age-matched controls. (D) Aneuploidy levels measured in the cortex of 14-week-old Ercc1−/Δ7 mice and age-matched controls. (E) Additional analysis of aneuploidy levels for MMU7 measured in the cortex of 6-month-old Bub1bH/H mice and age-matched controls. (F) Aneuploidy levels measured in the cerebellum of 14-week-old Ercc1−/Δ7 mice and age-matched controls. No statistically significant increase in aneuploidy was observed for the three chromosomes tested in these models at the selected time points.
We initially focused our analysis on the cerebral cortex because of the age-related accumulation of aneuploidy previously observed in this specific area of the brain in WT C57BL/6 mice when compared with the cerebellum. During normative aging, aneuploidy in 28-month-old cortices reached 9.8% for Chromosomes 7, 18 and Y combined (16). Because the reported median life span of WT controls ranges between 30 and 22 months (9,12,18), we predicted the levels of aneuploidy in the cortex of Bub1bH/H and Ercc1−/Δ7 adult progeroid mice (6 and 3.5 months, respectively) to be comparable with the overall levels previously measured WT mice at old age (28 months) when using the same FISH probes (16).
Surprisingly, both Bub1bH/H and Ercc1−/Δ7 models exhibited a low percentage of aneuploid cells in the cerebral cortex with no statistically significant difference when compared with their respective strain and age-matched controls. Cortical nuclei of Bub1bH/H mice showed aneuploidy levels <1% for all chromosomes (Fig. 1C), i.e. 0.84% (SD ± 0.8) for Chromosome 1, 0.55% (SD ± 0.95) for Chromosome 7 and 0.55% (SD ± 0.48) for Chromosome 18. The aneuploidy levels of Ercc1−/Δ7 cortical nuclei were <0.5% for all chromosomes (Fig. 1D), i.e. 0.33% (SD ± 0.57) for Chromosome 1, 0.3% (SD ± 0.53) for Chromosome 7 and 0.31% (SD ± 0.54) for Chromosome 18. Both chromosome gains (red) and losses (green) contributed to aneuploidy (Supplementary Material, Fig. S1). To confirm these results, we performed additional analysis on three independent Bub1bH/H mice and respective controls using FISH probes for MMU7. Although the overall aneuploidy levels for this data set are higher (average 2.6% SD ± 0.57 for the Bub1bH/H), the results confirmed the lack of significant accumulation of aneuploidy in the cortex of adult Bub1bH/H mice (Fig. 1E). These findings suggest that age-related aneuploidization of the brain is not recapitulated in Bub1bH/H and Ercc1−/Δ7 mice with segmentally progeroid features.
We next quantified, as an additional control, aneuploidy levels in the cerebellum of Ercc1−/Δ7 mice and age-matched controls (16). The cerebellum was selected based on our previous observation that aneuploidy does not accumulate with age within this tissue in WT C57BL/6 mice. Out of 977 nuclei inspected for ploidy changes of Chromosomes 1, 7 and 18 in the Ercc1−/Δ7 mice, only one was found to carry a whole chromosome loss (MMU1), resulting in overall aneuploidy levels of only 0.1% (Fig. 1F). This low frequency was identical to what observed in age-matched controls, in which also only one aneuploid cell was identified (gain of MMU1) out of 969 analyzed.
Interestingly, aneuploidy in the cerebellum shows a trend toward an extremely low level (0.1% in the cerebellum of both controls and Ercc1−/Δ7 mice versus 0.89% in controls, and 0.31% in the cortex of Ercc1−/Δ7 mice) confirming our previous results (16).
Increased aneuploidy is observed during embryonic brain development of Bub1bH/H and Ercc1−/Δ7 mice
The extremely low aneuploidy found in adult cerebral cortex of Bub1bH/H mice was unexpected since splenocytes and mouse embryonic fibroblasts derived from Bub1bH/H mice are known to have increased aneuploidy and to undergo premature sister chromatid separation more frequently than their WT counterparts (9). In addition, toward the end of their lifespan, Ercc1−/Δ7 mice present higher levels of anisokaryosis and cytoplasmic inclusions in their liver, both of which can occur in aneuploid or polyploid nuclei (12). Knowing that the mammalian developing brain contains a high frequency of aneuploid neural progenitor cells (19,20), we sought to investigate the levels of aneuploidy in embryos at E13.5 in both Bub1bH/H and Ercc1−/Δ7 mice. We focused our analysis to the developing cortex (i.e. the tissue surrounding the lateral ventricle that develops into the adult cerebral cortex) (Fig. 2A and B, red and blue boxes) by analyzing frozen sections from Bub1bH/H, Ercc1−/Δ7 and age-matched control mice (Supplementary Material, Table S1). Both chromosome gains and losses contributed to aneuploidy (Supplementary Material, Fig. S2), but the percentage of gains was >2-fold higher than the percentage of loss (Supplementary Material, Table S2), and this difference was statistically significant for all groups analyzed (P < 0.0001 in WT, Bub1bH/H and Ercc1−/Δ7). The percentage of aneuploidy for Chromosomes 1 and 18 analyzed individually was ∼31% (SD ± 0.82) in the control WT mice. This is in agreement with levels found previously in E11–E15 murine embryonic cerebral cortex (19). We found a statistically significant increase of ∼25% in aneuploidy in Bub1bH/H mice (40.3% of aneuploid nuclei, SD ± 0.16), relative to WT controls for both chromosomes (P < 0.05 for Chromosome 1 and P < 0.01 for Chromosome 18) (Fig. 2D). We also found a 19% increase in aneuploidy in Ercc1−/Δ7 mice (36.5% of aneuploid nuclei, SD ± 0.84) when compared with WT, but this difference did not reach statistical significance for either chromosome (P > 0.05 for both chromosomes) (Fig. 2E). These results confirm that aneuploidy is present during normal mammalian brain development and it is elevated in both Bub1bH/H and Ercc1−/Δ7 developing cortices relative to WT controls, although this increase is statistically significant only for Bub1bH/H mice.
Figure 2.

Analysis of aneuploidy during the embryonic brain development. (A) Representative H&E staining of E13.5 head section of WT, Bub1bH/H and Ercc1−/Δ7 mice. The red square depicted in the 2.4× magnification box is indicative of the embryonic brain area. (B) Higher magnification of the same sections highlighting the developing cortex in which aneuploidy analysis was performed. The blue square depicted in the 10× magnification indicates the specific area where the FISH analysis was performed. (C) Representative FISH images for MMU1 in each mouse strain highlighting diploid and aneuploid cells (contours) and respective ploidy (the numbers of signals are indicated with the correspondent color of the fluorophore used for staining). (D) Aneuploidy levels measured in the developing cortex of E13.5 Bub1bH/H mice and age-matched controls. The increase in aneuploidy is statistically significant for both chromosomes (*P = 0.0159 for MMU1 and *P = 0.0072 for MMU18). (E) Aneuploidy levels measured in the developing cortex of E13.5 Ercc1−/Δ7 mice and age-matched controls. The increase in aneuploidy is not statistically significant for either chromosome (P = 0.1149 for MMU1 and P = 0.3997 for MMU18).
SA-β-gal staining of the developing cortex does not differ between controls and Bub1bH/H or Ercc1−/Δ7 mice
The findings that aneuploidy is elevated during embryogenesis in the murine developing cortex but is extremely low during adulthood suggests that aneuploid cells are being eliminated, and/or overgrown by more fit diploid cells. Because mammalian aneuploid cells were shown to undergo premature senescence to prevent the propagation of cells with abnormal DNA content (9,10,21–23), we performed senescence-associated β-galactosidase (SA-β-gal) staining in frozen sections from all samples to measure differences in the number of senescent cells. SA-β-gal staining was evident in normal embryonic tissues, confirming findings by others (24,25), particularly in the choroid plexus, ependymal cells and lens (Supplementary Material, Fig. S3). Bub1b mice (both Bub1bH/H and controls) had scattered positive cells in ventricular zones (third and fourth ventricles) that were rare in Ercc1 mice (both Ercc1−/Δ7 and controls) (Supplementary Material, Table S3). We could not identify positive staining within the developing cerebral cortex in both genotypes. These results suggest that aneuploid cells that are present during embryonic development in Bub1bH/H and Ercc1−/Δ7 mice do not undergo senescence.
Aneuploid cells present at E13.5 in the developing cortex are likely removed by apoptotic mechanisms and immune-mediated clearance
The low level of aneuploidy observed in the adult brain relative to the embryonic cortex suggests that, during nervous system development, aneuploid cells may be preferentially prone to cell death (19). We, therefore, performed in the developing cortex (E13.5) a quantification of apoptotic cells.
To test whether aneuploid cells undergo caspase-dependent apoptosis, we performed immunohistochemistry (IHC) staining for cleaved-Casp3 (CC3) in frozen head sections from E13.5 Bub1bH/H and Ercc1−/Δ7 mice versus controls. We found that positive cells were rare in all samples, even when multiple sections were analyzed for each mouse (n = 4). Positive CC3 cells were often found associated with ganglia and at the interface between the third and fourth ventricles, but they were absent from the region analyzed by FISH (except for only one positive cell in one WT mouse) (Supplementary Material, Table S4). The observed lack of CC3 staining within the developing cortex in both genotypes argues against caspase-mediated apoptosis being the main mechanism for elimination of aneuploid cells under these experimental conditions.
Because very small number of apoptotic cells is detected by CC3 staining, it is likely that other mechanisms compensate for developmental apoptosis in the absence of Casp3 (26,27). We, therefore, investigated if other cell death markers would reveal differential staining within the developing cortex. We focused this analysis on Bub1bH/H mice and age-matched controls being that the increase in aneuploidy of Ercc1−/Δ7 mice was not statistically significant different from WT. TUNEL assay was performed to evaluate the extent of apoptosis based on the extent of DNA fragmentation. We found that the developing cortex of Bub1bH/H mice contained significantly more TUNEL-positive cells per field relative to WT (P < 0.01) (Fig. 3). These results suggest that the aneuploid cells that are present at E13.5 in Bub1bH/H mice are removed by nuclear fragmentation in a Casp3 independent cell death pathway.
Figure 3.
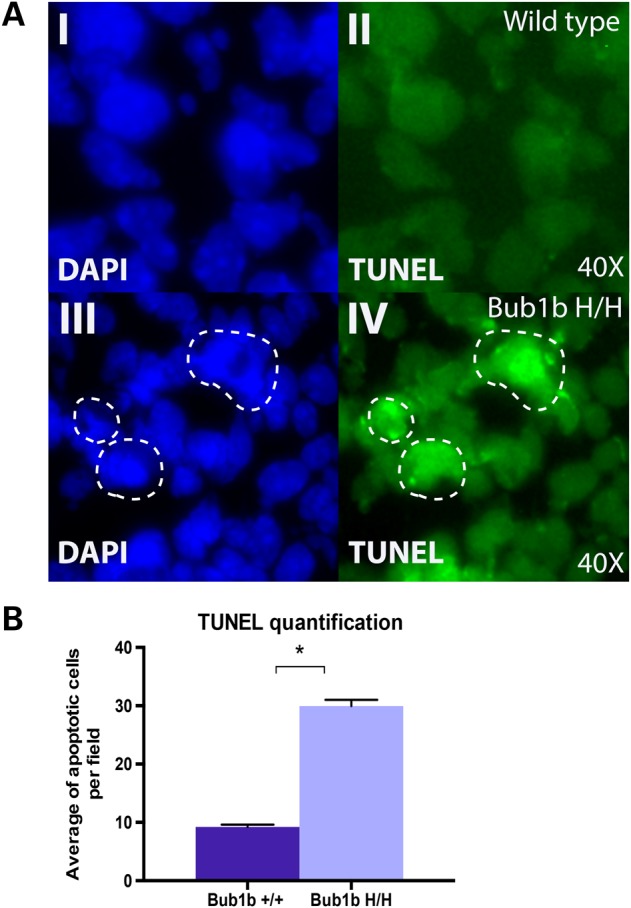
Analysis of cell death by TUNEL staining in the developing cortex: (A) Representative images from TUNEL staining in E13.5 WT (top) and Bub1bH/H (bottom) mice. (I and III) DAPI nuclei staining only; (II and IV) fluorescein TUNEL staining only. (B) Quantification of TUNEL staining images represented as the average of apoptotic cells per 40× field per genotype. The difference is statistically significant (*P = 0.00148).
To verify that the number of degenerating neural progenitor cells in Bub1bH/H mice is also increased relative to age-matched controls we used Fluoro-jade B, a fluorochrome commonly used to label degenerating neurons (28). As a confirmation of specific labeling of our embryonic samples, we detected Fluoro-jade B-stained cells in both WT and mutant mice within the ganglia, the same region in which CC3-positive cells were often observed (Supplementary Material, Fig. S4). Fluoro-jade B staining often coincided with nuclei that had a pyknotic or fragmented appearance, suggestive of cell death (Fig. 4A). We found scattered Fluoro-jade B-stained cells in the developing cortex of both Bub1bH/H and WT mice and a trend toward more positive cells in Bub1bH/H mice, but this difference was not statistically significant (Fig. 4B). This observation suggests that neural progenitor cells may be the main components of the aneuploid pool, which is in agreement with previous reports (19).
Figure 4.

Analysis of degenerating neurons by Fluoro-jade B labeling in the developing cortex. (A) Representative images from Fluoro-jade B staining in E13.5 WT and Bub1bH/H mice at 40× and 100× magnifications. (I and IV) 40× Fluoro-jade B staining only; (II and V) 40× DAPI nuclei staining only; (III and VI) 40× merged image; (VII and X) 100× Fluoro-jade B staining only; (VIII and XI) 100× DAPI nuclei staining only; (IX and XII) 100× merged image. (B) Quantification of Fluoro-jade B staining images represented as average of positively stained cells per 40× field per genotype. The difference is not statistically significant (P = 0.05612).
Microglia are key immune effectors that populate the central nervous system (CNS) and are important for its maturation and maintenance, acting as sensors of pathological events and caretakers of brain homeostasis (29,30). We, therefore, hypothesized that dying or dead aneuploid cells are eliminated from the developing brain by immune-mediated clearance. We performed immunofluorescence (IF) staining in frozen embryonic head sections for the epidermal growth factor-like module-containing mucin-like hormone receptor-like 1 (Adgre1, i.e. F4/80), a common marker found in microglia, perivascular, meningeal and choroid plexus macrophages (29). We found a trend for more macrophages infiltrated within the developing cortex of Bub1bH/H mice when compared with WT, suggesting that macrophages may be involved in this process. However, this difference was not statistically significant (Fig. 5).
Figure 5.

Analysis of macrophage infiltration in the developing cortex (Adgre1 marker, i.e. F4/80): (A) Representative images from Adgre1 IF staining in E13.5 WT and Bub1bH/H mice at low (10×) and higher magnifications (40×). (I and IV) 10× Adgre1 staining only; (II and V) 10× DAPI nuclei staining only; (III and VI) 10× merged image; (VII and X) 40× Adgre1 staining only; (VIII and XI) 40× DAPI nuclei staining only; (IX and XII) 40× merged image. (B) Quantification of Adgre1 IF staining represented as average of positively stained cells per 40× field per genotype. The difference is not statistically significant (P = 0.29294).
Discussion
By using a custom two—color FISH approach for the sensitive quantification of whole chromosome gains and losses, we measured the frequency of aneuploidy for autosomes 1, 7 and 18 in the brain of the short-lived, progeroid Bub1bH/H and Ercc1−/Δ7 mice. Our analysis revealed that aneuploidy levels in these two murine models of accelerated aging are low during adulthood (∼1%), a frequency similar to their respective aged-matched WT controls and comparable with the levels routinely observed in the brain of young (4 months old) C57BL/6 mice (16). However, contrary to normative aging where chromosome-specific aneuploidy accumulates up to 9.8% in 28-month-old animals, premature aging mice defective for the SAC component Bub1b or for the DNA repair component Ercc1 maintain aneuploidy at very low levels. This lack of increased aneuploidy in the adult cortex and cerebellum of these progeroid models reinforces the notion that aging is a complex process that cannot be easily ascribed to one molecular defect and therefore, not all aspects of normative aging are recapitulated.
On the other hand, we observed high frequency of aneuploid nuclei during normal embryonic development (31% of total nuclei on average), confirming findings by others (19,20). We used 12 μm tissue sections for FISH to minimize the possibility of analyzing incomplete nuclei and, since we found chromosome gains >2-fold higher than losses, we are confident that our results reflects the ploidy of the tissue and are not confounded by artifacts. Our analysis revealed increased aneuploidy at E13.5 in mutant mice relative to WT; however, only Bub1bH/H mice reached statistical significance and, for this reason, they were analyzed in more depth with the goal to elucidate the mechanisms that result in the low aneuploidy observed during adulthood.
Aneuploidy induce changes in cellular functions and metabolism, that together with high levels of genomic instability could trigger cell-cycle arrest and/or apoptosis in order to maintain tissue homeostasis and prevent the proliferation of genetically modified cells that could be detrimental. Aneuploid cells generated by down-regulation of SAC components Bub1 mitotic checkpoint serine/threonine kinase (Bub1) and Mad2l1 or centromere protein A (Cenpa) were shown to undergo premature senescence (21–23). Also, Musio et al. (31) showed that cells with defects in chromosome segregation undergo elimination by programmed cell death (PCD). Furthermore, it has been demonstrated that the proliferation of aneuploid cells is limited by a Trp53-dependent cell cycle delay or apoptosis (32,33). All these findings support the idea that aneuploid cells have reduced fitness, and their removal or prevention of their propagation is important to maintain normal physiological tissue functions.
High levels of SA-β-gal activity were detected in kidney sections of Bub1bH/H mice (9) and in various cell types in Ercc1−/Δ7 mice (34–36), suggesting that cellular senescence could play a role in the clearance of aneuploid cells. However, we did not observe SA-β-gal-stained cells in the developing cortex of mutant or WT mice. This cannot be explained by the loss of β-gal activity in stored frozen sections, because some staining was often evident in the choroid plexus and lens from the majority of sections. Senescent cells can also be identified by their lack of proliferation and activation of tumor suppressor networks (37,38); thus, to rule out the possibility that aneuploid cells enter premature senescence, it would be interesting to the follow-up our findings and examine the levels of expression of senescent markers such as Cdkn1a (p21), Cdkn2a (p16) and Cdkn2a (p19ARF) during embryonic brain development of Bub1bH/H and Ercc1−/Δ7 mice.
The brains of Bub1bH/H and Ercc1−/Δ7mice develop normally, suggesting that these genes do not have an essential function for brain development (9,12). However, both mice models present markers of age-related neurodegeneration such as increased glial fibrillary acidic protein and microglial staining (13,39). In addition, Bub1bH/H mice exhibit age-related accelerated gliosis in the cortex (39), and Ercc1−/Δ7 mice undergo age-related accelerated brain mass reduction, decrease in neuronal plasticity and contain CC3-positive cells throughout their brains (12,13,40). The finding that aneuploidy is significantly elevated in the embryonic brain of Bub1bH/H mice relative to WT is consistent with severely compromised SAC functions in cells deficient for Bub1b, which often display lagging chromosomes and premature sister chromatid separation (9). Previous studies suggested susceptibility for numerical aneuploidy accumulation in Ercc1−/Δ7mice (12,41). Given the function of this protein for DNA inter-strand cross-links repair (42,43), it is possible that these mice contain structural chromosomal rearrangements that are not detected by our FISH approach. It is tempting to speculate that both the age-related gliosis in Bub1bH/H mice and the reduction in brain mass in Ercc1−/Δ7 mice are associated with the lack of aneuploid cells during adulthood, and that they could all be consequences of increased apoptosis. The higher generation of cells with genomic instability in these mutant genotypes could trigger mechanisms for their ablation, aiming to guard tissue homeostasis. However, cell death induction might be too high and consequently detrimental for the organism, resulting in the age-related phenotypes observed in these mice. This hypothesis is supported by our observation of Fluoro-jade B-positive cluster of cells throughout the developing cortex in one Ercc1−/Δ7 mouse, which was absent from the WT mouse inspected (data not shown). The significant increase in TUNEL-positive cells and the observed trend toward more degenerating neurons and macrophages in the embryonic brain of Bub1bH/H mice also support the hypothesis that aneuploid cells are being eliminated from the brain.
The finding that WT E13.5 mice contain ∼31% of aneuploid cells confirms levels reported previously, and the drastic reduction in their percentage during adulthood suggests that aneuploid cells may be prone to preferential cell death during nervous system development (19). The observed basal level of TUNEL and Fluoro-jade B-positive cells in WT mice is in agreement with the phenomenon of PCD, the widespread occurrence of natural neuronal cell death in the central and peripheral nervous system (26,27,44–47). Depending on the brain region, it is estimated that 20–50% of neurons die due to PCD (26,45), which interestingly coincides with the percentage of aneuploid cells present during normal embryonic development. These observations suggest that one of the main functions of PCD is clearance of aneuploid cells (20). Although Casp3 is the main apoptotic pathway in the CNS (48), CC3 staining was rare throughout the sections analyzed, which is in agreement with others (26,27). However, because TUNEL and in situ end-labeling methods are good tools for detecting cell death in embryos (26,27,46,47), we believe that it represents a reliable assessment of apoptosis. Because it has been previously shown that the aneuploid pool at E11–E15 is composed by cerebral cortical neuroblasts (19), the presence of Fluoro-jade B-positive cells in the developing cortex of all genotypes possibly represent aneuploid neural progenitor cells that are undergoing degeneration.
In animal studies, macrophage migration coincides with peaks of apoptotic cells (27,44). Microglia, macrophages of the CNS, are important for brain homeostasis as they monitor the functionality of neurons, cell death, debris phagocytosis and clearance (29,30). The Adgre1 receptor, one of the most specific cell-surface markers for murine macrophages (29,49,50), was chosen to verify microglia infiltration in the developing cortex. Adgre1-stained cells were observed in both WT and Bub1bH/H mice at E13.5, in agreement with their presence in the brain as early as E8.5 (51). Since a trend for higher infiltration was seen in Bub1bH/H mice, it suggests that elevated aneuploidy activates immune responses to aid in their clearance.
The trend toward increased numbers of Fluoro-jade B-positive cells and microglia in the embryonic brain of Bub1bH/H mice is of particular interest because it pinpoints to a mechanism for the elimination of aneuploid cells in this tissue. However, more mice needs to be analyzed to determine if statistically significance can be achieved.
In this study, we analyzed only the cortex and the cerebellum; therefore, we cannot exclude that aneuploidy in these mutant mice occurs with variable frequency in other areas of the brain at advanced age. Our previous work on normative aging (16) and the data generated in this study suggest that susceptibility to aneuploidy in the adult brain may vary between anatomical regions (i.e. the cortex versus the cerebellum). Our data also suggest that the brain uniquely has the ability to regulate levels of aneuploidy to maintain tissue homeostasis. Future studies are required to better elucidate the mechanisms leading to elimination of unfit aneuploid cells in this tissue.
Materials and Methods
Tissue isolation
Ercc1−/Δ7 and Bub1bH/H mice have been previously described (9,12). The cerebral cortex and the cerebellum were dissected from 3.5 months (Ercc1−/Δ7) and 6 months (Bub1bH/H) male murine models and age-matched littermate controls as indicated in Supplementary Material, Table S1. Freshly isolated heads of E13.5 animals were snap frozen in liquid nitrogen, embedded in optimal cutting temperature compound, sectioned at 12 µm and mounted on glass slides that were stored in −80°C until analysis. All experiments concerning Ercc1−/Δ7 and control mice, as described in the local protocol number 20070013, were reviewed and approved by the Committee on Animal Experimentation of the Antonie van Leeuwennhoek terrain in Bilthoven, the Netherlands. Experiments performed on Bub1bH/H and control mice were carried on under protocol A48013, approved by the Institutional Animal Care and Use Committee of Mayo Clinic, MN, USA. Both protocols adhere to all applicable national and institutional regulations.
Extraction of adult cortical nuclei for direct FISH analysis
The cortex and the cerebellum from adult mice were processed using the same protocol. The entire procedure was performed on ice using pre-cooled buffers to preserve the integrity of the nuclei. The cortex and the cerebellum were homogenized for 45 s in 5 ml of lysis buffer. The suspension was transferred to a clear ultra-centrifuge tube (14 × 89 mm, Beckman Coulter, Inc.) and layered on 9 ml of sucrose solution. The samples were placed into SWiT40 rotor buckets and ultra-centrifuged at 4°C for 2.5 h at 25,000 rpm. After the centrifugation, 1 ml of phosphate buffered saline (PBS) was added to the pellet and kept on ice for 20 min. The nuclei were then dissolved through pipetting for 1 min, centrifuged at 4°C for 10 min at 800 rpm and then fixed with ice-cold methanol/acetic acid (Fisher) (3:1) for 10 min on ice. The nuclei were stored in fixative solution at −20°C until they were dropped onto a clean slide in 48% humidity at 24°C for FISH analysis.
Localization of the developing cortex in E13.5 embryos for direct FISH analysis
The region of the future cerebral cortex in embryonic E13.5 sections was identified by H&E staining of 12 µm thick slides for each embryo. Brain regions were verified using an atlas of mouse development (52). FISH experiments were performed on 12 µm whole head cryosections to preserve tissue integrity and morphology. Images for analysis were taken specifically within the developing cortex region (i.e. tissue surrounding the lateral ventricle).
Fluorescence in situ hybridization
All hybridizations were performed blind, de-identified keys were assigned by an independent individual not involved in this study and revealed only after data acquisition and analysis. FISH was performed using the two locus-specific probes as previously described: for Chromosome 1, we used the bacterial artificial chromosome (BAC) clones RP23-34K7 (1qA1) and RP23-102B11 (1qA2); for Chromosome 7, we used the BAC clones RP23-336P12 (7qA1) and RP23-316N16 (7qF5); for Chromosome 18, we used the BAC clones RP23-332K12 (18qA2) and RP23-16K15 (18qE1) as depicted in Figure 1A.
The probes were labeled by nick translation using, DY-415-dUTP, DY-495-dUTP, DY-590-dUTP and DY-647P1-dUTP (Dyomics, Jena, GE). The slides with nuclei from adult cerebellum or cortex or containing 12 µm sections from the E13.5 head were denatured with 50% formamide (FA)/2× saline sodium citrate (SSC) at 80°C for 1.5 min and then dehydrated with serial ethanol washing steps (ice-cold 70, 90 and 100% for 3 min each). Probes were denatured in the hybridization solution (50% dextran sulfate/2× SSC) at 85°C for 5 min, applied to the slides, and incubated overnight at 37°C in a humidified chamber. The slides were then washed three times for 5 min with 50% FA/2× SSC, 1× SSC and 4× SSC/0.1%Tween. Slides were dehydrated with serial ethanol washing steps (see above) and mounted with ProLong Gold antifade reagent with DAPI (Invitrogen) for imaging.
Image acquisition
FISH images were acquired with a manual inverted fluorescence microscope (Axiovert 200, Zeiss) with fine focusing oil immersion lens (×40, NA 1.3 oil). Multiple focal planes were obtained for each channel to ensure that signals on different focal planes were included. The resulting fluorescence emissions were collected using 350–470 nm (for DAPI), 436–480 nm (for DY-415-dUTP), 470–540 nm (for DY-495-dUTP), 546–600 nm (for DY-590-dUTP) and 620–700 nm (for DY-647P1-dUTP) filters. The microscope was equipped with a Camera Hall 100 and the Applied Spectral Imaging software.
Data analysis of interphase nuclei
Images from cortex and cerebellum isolated nuclei and from the developing cortex for each hybridization were randomly acquired and saved as .tiff composite files. An average of 330 cells were visually inspected for each chromosome and FISH signals manually counted for both adult cortex and cerebellum. At least 500 nuclei located in the developing cortex region were analyzed per mouse and per chromosome in E13.5 samples. Nuclei that seemed broken or lacking signals from both probes in a chromosome were not considered. A total of 19,513 nuclei were inspected in this study. The detailed number of nuclei scored for each chromosome tested and for each mouse analyzed is listed in Supplementary Material, Tables S1 and S2.
Detection of apoptotic cells by CC3 IHC
Frozen 12 µm E13.5 head sections were air dried for 30 min and then fixed in 10% neutral buffered formalin for 10 min. Endogenous peroxidase activity was quenched by incubating the slides with 0.15% hydrogen peroxide in methanol for 10 min. Blocking was performed in 5% donkey serum supplemented with 2% bovine serum albumin (Fisher) dissolved in PBS for 1 h. The sections were stained by routine IHC methods, using horseradish peroxidase (HRP) polymer conjugate to localize the antibody bound to antigen, with diaminobenzidine as the final chromogen [SignalStain® Boost IHC Detection Reagent (HRP, Rabbit) #8114; Cell Signalling]. All immune-stained sections were lightly counterstained with hematoxylin. The primary antibody to CC3, (ASP 175, #9661, Cell Signalling), was used at 1:50 diluted in antibody diluent (Cell Signalling, #8112), for 2 h at room temperature. Histological evaluation was performed blindly [histopathology core (RSS)].
The assessment of cellular senescence in murine embryonic brain using the senescence-associated β-sgalactosidase staining (SA-ningdase stain. Frozen 12 µm E13.5 head sections were fixed with 0.5% glutaraldehyde (Sigma-Aldrich) for 15 min at room temperature and then incubated in staining solution for 24 h at 37°C, as previously described (53,54). Slides were then washed in PBS, lightly counterstained with hematoxylin (Fisher) and mounted with Permount (Fisher). Visual inspection was performed with Axio Observer Z1 (Zeiss). The slides were scored for the presence of SA-β-gal staining in different regions of the brain and then classified according to amount of blue-stained cells. Histological evaluation was performed blindly (RSS).
Analysis of apoptosis by TUNEL staining
The TUNEL reaction was performed using the ‘In Situ Cell Death Detection Kit, Fluorescein’ (Roche) according to manufacturer's instructions. Briefly, frozen 12 µm E13.5 head sections were fixed with 4% paraformaldehyde for 20 min, washed, permeabilized and incubated for 1 h with the TUNEL reaction mixture. Negative (no enzyme TdT) and positive controls (DNase I treated) were included. Slides were mounted with ProLong Gold antifade reagent with DAPI (Invitrogen, Carlsbad, CA, USA) for imaging and analyzed with a manual inverted fluorescence microscope (Axiovert 200, Zeiss) using 350–470 nm (for DAPI) and 470–540 nm (for TUNEL) filters. The number of positively stained cells per 40× field was manually scored in 20 randomly acquired images of the developing cortex from 2 WT and 2 Bub1bH/H mice. The average of stained cells per field per mouse was then averaged again to generate the values for each group (WT littermates and HH).
Fluoro-jade B staining for detecting neuronal degeneration
The staining was performed using Fluoro-Jade® B (EMD Millipore) following manufacturer's instructions. Briefly, frozen 12 µm E13.5 head sections were fixed with 4% paraformaldehyde for 20 min, washed and then immersed in a solution containing 1% sodium hydroxide (Fisher) in 80% ethanol, followed by 70% ethanol, distilled water and 0.06% potassium permanganate (Fisher) to reduce tissue background. Sections were then stained in 0.0004% Fluoro-jade B solution with 0.1% acetic acid for 20 min, followed by serial washes, DAPI staining and Permount (Fisher) mounting. The sections were analyzed with a manual inverted fluorescence microscope (Axiovert 200, Zeiss) using 350–470 nm (for DAPI) and 470–540 nm (for Fluoro-jade B) filters. The number of positively stained cells per 40× field was manually scored in 25 randomly acquired images of the lateral ventricle from 2 WT and 2 Bub1bH/H mice. The average number of stained cells per field per mouse was then averaged again to generate the values for each group (WT littermates and HH).
Detection of macrophages by ADGRE1 (i.e. F4/80) IF
Frozen 12 µm E13.5 head sections were air dried for 1 h and then fixed in cold acetone for 10 min. Slides were hydrated in PBS, blocked in 5% goat serum (Gemini) for 1 h and incubated with 10 µg/ml of a rat monoclonal antibody raised against the macrophage marker Adgre1 (CI:A3-1) (Abcam—ab6640) overnight at 4°C. The following day sections were washed and incubated with an Alexa Fluor® 488 goat anti-rat secondary antibody at 1:300 dilution for 1 h at room temperature, washed and mounted with ProLong Gold antifade reagent with DAPI (Invitrogen) for imaging. Images were obtained with a manual inverted fluorescence microscope (Axiovert 200, Zeiss) using 350–470 nm (for DAPI) and 470–540 nm (for Adgre1) filters. The number of positively stained cells per 40× field was manually scored in 15 randomly acquired images of the lateral ventricle from 3 WT and 2 Bub1bH/H mice. The average of stained cells per field per mouse was then averaged again to generate the values for each group (WT and HH).
Statistical analysis
The statistical significance was calculated using t-test with 95% confidence interval with the software GraphPad Prism 6.0.
Supplementary Material
Funding
This work was supported by grants from the National Institutes of Health [AG17242 to J.V., MH070596 and NS088943 to J.H., P30CA013330 to R.S.S. and C.M. (Albert Einstein Cancer Center grant)]. Part of this work was supported by funds from the Glenn Foundation for Medical Research.
Supplementary Material
Acknowledgements
We thank the Molecular Cytogenetic Core at Albert Einstein College of Medicine for help with the FISH analysis and the Histology and Comparative Pathology Facility at Albert Einstein College of Medicine for help with the localization of the developing cortex in E13.5 embryo sections, IHC and analysis for cleaved-Casp3 and SA-β-gal staining scoring.
Conflict of Interest statement. None declared.
References
- 1.Maslov A.Y., Vijg J. (2009) Genome instability, cancer and aging. Biochim. Biophys. Acta, 1790, 963–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vijg J., Suh Y. (2013) Genome instability and aging. Ann. Rev. Physiol., 75, 645–668. [DOI] [PubMed] [Google Scholar]
- 3.Faggioli F., Vijg J., Montagna C. (2011) Chromosomal aneuploidy in the aging brain. Mech. Ageing Dev., 132, 429–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanks S., Coleman K., Reid S., Plaja A., Firth H., Fitzpatrick D., Kidd A., Mehes K., Nash R., Robin N. et al. (2004) Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat. Genetics, 36, 1159–1161. [DOI] [PubMed] [Google Scholar]
- 5.Ricke R.M., van Deursen J.M. (2013) Aneuploidy in health, disease, and aging. J. Cell Biol., 201, 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iwanaga Y., Chi Y.H., Miyazato A., Sheleg S., Haller K., Peloponese J.M. Jr., Li Y., Ward J.M., Benezra R., Jeang K.T. (2007) Heterozygous deletion of mitotic arrest-deficient protein 1 (MAD1) increases the incidence of tumors in mice. Cancer Res., 67, 160–166. [DOI] [PubMed] [Google Scholar]
- 7.Weaver B.A., Silk A.D., Montagna C., Verdier-Pinard P., Cleveland D.W. (2007) Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell, 11, 25–36. [DOI] [PubMed] [Google Scholar]
- 8.Jeganathan K., Malureanu L., Baker D.J., Abraham S.C., van Deursen J.M. (2007) Bub1 mediates cell death in response to chromosome missegregation and acts to suppress spontaneous tumorigenesis. J. Cell Biol., 179, 255–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baker D.J., Jeganathan K.B., Cameron J.D., Thompson M., Juneja S., Kopecka A., Kumar R., Jenkins R.B., de Groen P.C., Roche P. et al. (2004) BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat. Genet, 36, 744–749. [DOI] [PubMed] [Google Scholar]
- 10.Baker D.J., Jeganathan K.B., Malureanu L., Perez-Terzic C., Terzic A., van Deursen J.M. (2006) Early aging-associated phenotypes in Bub3/Rae1 haploinsufficient mice. J. Cell Biol., 172, 529–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McWhir J., Selfridge J., Harrison D.J., Squires S., Melton D.W. (1993) Mice with DNA repair gene (ERCC-1) deficiency have elevated levels of p53, liver nuclear abnormalities and die before weaning. Nat. Genet., 5, 217–224. [DOI] [PubMed] [Google Scholar]
- 12.Dolle M.E., Kuiper R.V., Roodbergen M., Robinson J., de Vlugt S., Wijnhoven S.W., Beems R.B., de la Fonteyne L., de With P., van der Pluijm I. et al. (2011) Broad segmental progeroid changes in short-lived Ercc1(-/Delta7) mice. Pathobiol. Aging Age Relat. Dis., 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borgesius N.Z., de Waard M.C., van der Pluijm I., Omrani A., Zondag G.C., van der Horst G.T., Melton D.W., Hoeijmakers J.H., Jaarsma D., Elgersma Y. (2011) Accelerated age-related cognitive decline and neurodegeneration, caused by deficient DNA repair. J. Neurosci., 31, 12543–12553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baker D.J., Dawlaty M.M., Wijshake T., Jeganathan K.B., Malureanu L., van Ree J.H., Crespo-Diaz R., Reyes S., Seaburg L., Shapiro V. et al. (2013) Increased expression of BubR1 protects against aneuploidy and cancer and extends healthy lifespan. Nat. Cell Biol., 15, 96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jacobs P.A., Court Brown W.M., Doll R. (1961) Distribution of human chromosome counts in relation to age. Nature, 191, 1178–1180. [DOI] [PubMed] [Google Scholar]
- 16.Faggioli F., Wang T., Vijg J., Montagna C. (2012) Chromosome-specific accumulation of aneuploidy in the aging mouse brain. Hum. Mol. Genet., 21, 5246–5253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Faggioli F., Vijg J., Montagna C. (2014) Four-color FISH for the detection of low-level aneuploidy in interphase cells. Methods Mol. Biol., 1136, 291–305. [DOI] [PubMed] [Google Scholar]
- 18.Dolle M.E., Busuttil R.A., Garcia A.M., Wijnhoven S., van Drunen E., Niedernhofer L.J., van der Horst G., Hoeijmakers J.H., van Steeg H., Vijg J. (2006) Increased genomic instability is not a prerequisite for shortened lifespan in DNA repair deficient mice. Mutat. Res., 596, 22–35. [DOI] [PubMed] [Google Scholar]
- 19.Rehen S.K., McConnell M.J., Kaushal D., Kingsbury M.A., Yang A.H., Chun J. (2001) Chromosomal variation in neurons of the developing and adult mammalian nervous system. Proc. Natl Acad. Sci.USA, 98, 13361–13366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yurov Y.B., Iourov I.Y., Vorsanova S.G., Liehr T., Kolotii A.D., Kutsev S.I., Pellestor F., Beresheva A.K., Demidova I.A., Kravets V.S. et al. (2007) Aneuploidy and confined chromosomal mosaicism in the developing human brain. PLoS One, 2, e558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lentini L B.V., Schillaci T., Di Leonardo A. (2011) MAD2 depletion triggers premature cellular senescence in human primary fibroblasts by activating a P53 pathway preventing aneuploid cells propagation. [DOI] [PubMed]
- 22.Maehara K., Takahashi K., Saitoh S. (2010) CENP-A reduction induces a p53-dependent cellular senescence response to protect cells from executing defective mitoses. Mol. Cell. Biol., 30, 2090–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gjoerup O.V., Wu J., Chandler-Militello D., Williams G.L., Zhao J., Schaffhausen B., Jat P.S., Roberts T.M. (2007) Surveillance mechanism linking Bub1 loss to the p53 pathway. Proc.Natl Acad. Sci. USA, 104, 8334–8339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Storer M., Mas A., Robert-Moreno A., Pecoraro M., Ortells M.C., Di Giacomo V., Yosef R., Pilpel N., Krizhanovsky V., Sharpe J. et al. (2013) Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell, 155, 1119–1130. [DOI] [PubMed] [Google Scholar]
- 25.Munoz-Espin D., Canamero M., Maraver A., Gomez-Lopez G., Contreras J., Murillo-Cuesta S., Rodriguez-Baeza A., Varela-Nieto I., Ruberte J., Collado M. et al. (2013) Programmed cell senescence during mammalian embryonic development. Cell, 155, 1104–1118. [DOI] [PubMed] [Google Scholar]
- 26.Kuan C.Y., Roth K.A., Flavell R.A., Rakic P. (2000) Mechanisms of programmed cell death in the developing brain. Trends Neurosci., 23, 291–297. [DOI] [PubMed] [Google Scholar]
- 27.Rakic S., Zecevic N. (2000) Programmed cell death in the developing human telencephalon. Eur. J. Neurosci., 12, 2721–2734. [DOI] [PubMed] [Google Scholar]
- 28.Schmued L.C., Albertson C., Slikker W. Jr. (1997) Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res., 751, 37–46. [DOI] [PubMed] [Google Scholar]
- 29.Prinz M., Priller J., Sisodia S.S., Ransohoff R.M. (2011) Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat. Neurosci., 14, 1227–1235. [DOI] [PubMed] [Google Scholar]
- 30.Gomez Perdiguero E., Schulz C., Geissmann F. (2013) Development and homeostasis of ‘resident’ myeloid cells: the case of the microglia. Glia, 61, 112–120. [DOI] [PubMed] [Google Scholar]
- 31.Musio A., Montagna C., Zambroni D., Indino E., Barbieri O., Citti L., Villa A., Ried T., Vezzoni P. (2003) Inhibition of BUB1 results in genomic instability and anchorage-independent growth of normal human fibroblasts. Cancer Res., 63, 2855–2863. [PubMed] [Google Scholar]
- 32.Thompson S.L., Compton D.A. (2010) Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J. Cell Biol., 188, 369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li M., Fang X., Baker D.J., Guo L., Gao X., Wei Z., Han S., van Deursen J.M., Zhang P. (2010) The ATM-p53 pathway suppresses aneuploidy-induced tumorigenesis. Proc. Natl Acad. Sci. USA, 107, 14188–14193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Durik M., Kavousi M., van der Pluijm I., Isaacs A., Cheng C., Verdonk K., Loot A.E., Oeseburg H., Bhaggoe U.M., Leijten F. et al. (2012) Nucleotide excision DNA repair is associated with age-related vascular dysfunction. Circulation, 126, 468–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gregg S.Q., Gutierrez V., Robinson A.R., Woodell T., Nakao A., Ross M.A., Michalopoulos G.K., Rigatti L., Rothermel C.E., Kamileri I. et al. (2012) A mouse model of accelerated liver aging caused by a defect in DNA repair. Hepatology, 55, 609–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen Q., Liu K., Robinson A.R., Clauson C.L., Blair H.C., Robbins P.D., Niedernhofer L.J., Ouyang H. (2013) DNA damage drives accelerated bone aging via an NF-kappaB-dependent mechanism. J. Bone Miner. Res., 28, 1214–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuilman T., Michaloglou C., Mooi W.J., Peeper D.S. (2010) The essence of senescence. Genes Dev., 24, 2463–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rodier F., Campisi J. (2011) Four faces of cellular senescence. J. Cell Biol., 192, 547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hartman T.K., Wengenack T.M., Poduslo J.F., van Deursen J.M. (2007) Mutant mice with small amounts of BubR1 display accelerated age-related gliosis. Neurobiol. Aging, 28, 921–927. [DOI] [PubMed] [Google Scholar]
- 40.de Waard M.C., van der Pluijm I., Zuiderveen Borgesius N., Comley L.H., Haasdijk E.D., Rijksen Y., Ridwan Y., Zondag G., Hoeijmakers J.H., Elgersma Y. et al. (2010) Age-related motor neuron degeneration in DNA repair-deficient Ercc1 mice. Acta Neuropathol., 120, 461–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weeda G., Donker I., de Wit J., Morreau H., Janssens R., Vissers C.J., Nigg A., van Steeg H., Bootsma D., Hoeijmakers J.H. (1997) Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Curr. Biol., 7, 427–439. [DOI] [PubMed] [Google Scholar]
- 42.Niedernhofer L.J., Odijk H., Budzowska M., van Drunen E., Maas A., Theil A.F., de Wit J., Jaspers N.G., Beverloo H.B., Hoeijmakers J.H. et al. (2004) The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol. Cell. Biol., 24, 5776–5787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ahmad A., Robinson A.R., Duensing A., van Drunen E., Beverloo H.B., Weisberg D.B., Hasty P., Hoeijmakers J.H., Niedernhofer L.J. (2008) ERCC1-XPF endonuclease facilitates DNA double-strand break repair. Mol. Cell. Biol., 28, 5082–5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ferrer I., Bernet E., Soriano E., del Rio T., Fonseca M. (1990) Naturally occurring cell death in the cerebral cortex of the rat and removal of dead cells by transitory phagocytes. Neuroscience, 39, 451–458. [DOI] [PubMed] [Google Scholar]
- 45.Oppenheim R.W. (1991) Cell death during development of the nervous system. Ann. Rev. Neurosci., 14, 453–501. [DOI] [PubMed] [Google Scholar]
- 46.Blaschke A.J., Staley K., Chun J. (1996) Widespread programmed cell death in proliferative and postmitotic regions of the fetal cerebral cortex. Development, 122, 1165–1174. [DOI] [PubMed] [Google Scholar]
- 47.Blaschke A.J., Weiner J.A., Chun J. (1998) Programmed cell death is a universal feature of embryonic and postnatal neuroproliferative regions throughout the central nervous system. J. Comp. Neurol., 396, 39–50. [DOI] [PubMed] [Google Scholar]
- 48.Srinivasan A., Roth K.A., Sayers R.O., Shindler K.S., Wong A.M., Fritz L.C., Tomaselli K.J. (1998) In situ immunodetection of activated caspase-3 in apoptotic neurons in the developing nervous system. Cell Death Differ., 5, 1004–1016. [DOI] [PubMed] [Google Scholar]
- 49.Austyn J.M., Gordon S. (1981) F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur. J. Immunol., 11, 805–815. [DOI] [PubMed] [Google Scholar]
- 50.Morris L., Graham C.F., Gordon S. (1991) Macrophages in haemopoietic and other tissues of the developing mouse detected by the monoclonal antibody F4/80. Development, 112, 517–526. [DOI] [PubMed] [Google Scholar]
- 51.Alliot F., Godin I., Pessac B. (1999) Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res. Dev. Brain Res., 117, 145–152. [DOI] [PubMed] [Google Scholar]
- 52.Kaufman M.H. (1999) The Atlas of Mouse Development. Harcourt Brace, Co. Publishers, Cambridge, UK. [Google Scholar]
- 53.Dimri G.P., Lee X., Basile G., Acosta M., Scott G., Roskelley C., Medrano E.E., Linskens M., Rubelj I., Pereira-Smith O. et al. (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl Acad. Sci. USA, 92, 9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bandyopadhyay D., Gatza C., Donehower L.A., Medrano E.E. (2005) Analysis of cellular senescence in culture in vivo: the senescence-associated beta-galactosidase assay. Current protocols in cell biology/editorial board, Juan S. Bonifacino … [et al.], Chapter 18, Unit 18 19. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.