

Abstract
MicroRNAs (miRNAs) play an important role in the development and progression of human diseases. The identification of disease-associated miRNAs will be helpful for understanding the molecular mechanisms of diseases at the post-transcriptional level. Based on different types of genomic data sources, computational methods for miRNA-disease association prediction have been proposed. However, individual source of genomic data tends to be incomplete and noisy; therefore, the integration of various types of genomic data for inferring reliable miRNA-disease associations is urgently needed. In this study, we present a computational framework, CHNmiRD, for identifying miRNA-disease associations by integrating multiple genomic and phenotype data, including protein-protein interaction data, gene ontology data, experimentally verified miRNA-target relationships, disease phenotype information and known miRNA-disease connections. The performance of CHNmiRD was evaluated by experimentally verified miRNA-disease associations, which achieved an area under the ROC curve (AUC) of 0.834 for 5-fold cross-validation. In particular, CHNmiRD displayed excellent performance for diseases without any known related miRNAs. The results of case studies for three human diseases (glioblastoma, myocardial infarction and type 1 diabetes) showed that all of the top 10 ranked miRNAs having no known associations with these three diseases in existing miRNA-disease databases were directly or indirectly confirmed by our latest literature mining. All these results demonstrated the reliability and efficiency of CHNmiRD, and it is anticipated that CHNmiRD will serve as a powerful bioinformatics method for mining novel disease-related miRNAs and providing a new perspective into molecular mechanisms underlying human diseases at the post-transcriptional level. CHNmiRD is freely available at http://www.bio-bigdata.com/CHNmiRD.
Introduction
MicroRNAs (miRNAs) are endogenous small non-coding RNAs (~22nt) that function by binding to the 3’ untranslated regions (3’UTRs) of target mRNAs, and then inhibiting their expression [1, 2]. According to miRBase (Release 21) [3], more than 1800 human miRNAs have been discovered in the last few years. MiRNAs are known to participate in many important biological processes including cell proliferation, differentiation and apoptosis[4]. The dysregulation of miRNA expression is therefore associated with a broad range of diseases [5], such as cardiovascular diseases [6, 7], neurodevelopmental diseases [8–10] and cancers [5, 11, 12]. Identification of disease-related miRNAs will provide novel insights into the molecular mechanisms underlying human diseases at the post-transcriptional level.
Many miRNAs were found to be associated with certain diseases using various biological experiment methods. To provide a mechanism to comprehensively search for these experimentally verified miRNA-disease associations, researchers have constructed several publicly-available and manually-curated databases, such as HMDD [13] and miR2Disease [14]. However, the collection and inclusion of verified miRNA-disease associations in these databases is far from complete, and identifying disease-related miRNAs from the multitude of candidate miRNAs by biological experimentation is time consuming and labor-extensive. Therefore, the development of effective computational methods for inferring miRNA-disease associations at the systematic level is urgently needed.
Computational methods can produce statistically significant results from a large amount of biological data and serve as a powerful tool for guiding further biological experiments. Based on miRNA functional similarity network (MFSN), different algorithms (Jiang’s method [15], RWRMDA [16], NetCBI [17], HDMP [18], RLSMDA [19]) have been developed to predict disease-related miRNAs (S1 Table). For example, Jiang et al. [15] constructed a MFSN by establishing a relationship between two miRNAs based on their significantly shared common targets, and they then integrated the MFSN with a disease network to infer potential miRNA-disease associations. The MFSN they constructed considered the number of overlapping miRNA targets while neglecting the functional link between them, and only the direct neighbor information of each miRNA was utilized in their scoring system. Additionally, this method was not work for disease whose all neighbor diseases are not associated with any known miRNAs. In RWRMDA [16], NetCBI [17], HDMP [18] and RLSMDA [19], the MFSN they adopted was constructed based solely on the information of known miRNA-disease associations using Wang et al.’s method [20]. Moreover, in these methods, the same miRNA-disease relations were used to construct the MFSN and evaluate the performance, which might over-estimate the performance. In addition, RWRMDA and HDMP were not applicable to disease which did not have any known related miRNAs. Recently, based on protein-protein interaction (PPI) networks, Shi et al. [21] developed a computational framework to identify miRNA-disease associations by focusing on the functional link between miRNA targets and disease genes. Additionally, Mork et al. [22] presented a method in which miRNA-disease associations were inferred by integrating miRNA-protein associations and protein-disease associations. However, these two methods neglected to use information of known miRNA-disease associations, which could improve their predictive performance. In contrast, Xu et al. [23] and Jiang et al. [24] constructed different feature vectors and trained a support vector machine classifier for distinguishing positive miRNA-disease associations from negative ones, respectively. But, there were no verified negative microRNA-disease associations, which result in the difficulty or impossibility for collection of negative disease-related miRNAs. Hence, the low-quality negative samples used in these two studies might largely reduce the predictive accuracy. High-throughput technologies have produced huge amounts of genomic data, which can be used in many ways to predict miRNA-disease associations. However, individual sources of genomic data tend to be noisy and incomplete, which downgrades the prioritization algorithms. Therefore, the question of how to effectively integrate different types of genomic data to improve predictive performance is a major challenge.
In this study, we constructed a complex heterogeneous network (CHN) by integrated PPI data, gene ontology (GO) data, miRNA-target relationships, disease phenotype data and known miRNA-disease associations. Based on the CHN, a computational model, CHNmiRD, was developed to identify miRNA-disease associations by performing random walk analysis. The results of cross validation and case studies suggested that CHNmiRD was effective for uncovering unknown miRNA-disease associations.
Materials and Methods
Human miRNA-disease associations and miRNA targets
Human miRNA-disease associations were retrieved from HMDD (version 2.0) [13]. This version of HMDD, released in 2013, has recorded 10,368 high-quality, experimentally verified miRNA-disease associations from 3,511 papers. Repeating miRNA-disease entries were removed, miRNA precursors were mapped to mature miRNAs using miRBase, and disease names were curated based on Online Mendelian Inheritance in Man (OMIM) [25] disease ID. Finally, 3,536 miRNA-disease associations involving 370 miRNAs and 105 diseases were obtained (S1 File). These miRNA-disease associations were used to construct a disease phenotype-miRNA network and used as the gold standard dataset for evaluating performance.
The miRNA targets were chosen from three widely used and experimentally validated miRNA target databases: TarBase (version 6.0) [26], miRTarBase (version 4.5) [27] and miRecords (version 4) [28]. We merged these three databases, and after removing miRNAs that have only one target and unifying the name of mature miRNAs based on miRBase, 37,659 targeting pairs involving 402 miRNAs and 12,360 target genes were obtained (S2 File).
Disease phenotype network and disease phenotype-miRNA network
Disease phenotype similarity scores were calculated by MimMiner [29] which computed a disease phenotype similarity score for two disease phenotypes based on the text mining analysis of their disease phenotype descriptions contained in the OMIM database. For each disease phenotype, the similarity between it and any other disease phenotypes in the OMIM database was computed using MimMiner, and the K most similar disease phenotypes, called K-nearest neighbors (KNN), were identified. The disease phenotype was connected with its KNNs and weighted using the similarity measure calculated by MimMiner. The network constructed by this method was called the KNN graph. In this study, we constructed a disease phenotype network (DPN) using 5-NN network (S3 File) followed some previous studies [30, 31].
As described above, the disease phenotype-miRNA relationships were extracted from the HMDD database (version 2.0) [32]. These relationships can be viewed as a bipartite disease phenotype-miRNA network (DPMN) in which one node is the miRNA, and the other is the disease phenotype, and the edges are the disease phenotype-miRNA relationships. This network can be used as a bridge to construct a CHN (described later).
MiRNA functional similarity network based on PPI and GO
MiRNA performs its regulatory function primarily through its target mRNA(s), and miRNAs with similar functions tend to target functionally related genes [33]. Therefore, for a given pair of miRNAs, their functional similarity score could be obtained by calculating the functional similarity of their target mRNA set. Firstly, the functional similarity score of two miRNA target sets was calculated based on PPI considering the functional communication and physical interaction between gene sets by using GsNetCom [34]. Secondly, we adopted GSFS [35] to compute the functional similarity score of two miRNA target sets based on three sub-ontologies (biological process, BP; molecular function, MF; and cellular component, CC) of GO. Finally, four miRNA functional similarity matrices were obtained by using different data sources. In order to make use of the global network similarity information, four weighted MFSNs were constructed according to the above miRNA functional similarity matrices, in which the edges were assigned different functional similarity scores between miRNAs.
Random walk with restart algorithm
Random walk with restart (RWR) is a global network ranking algorithm [36]. The random walker starts from a seed node (or a set of seed nodes, simultaneously) and proceeds to randomly selected neighbors based on the probabilities of the edges between the two nodes. Formally, RWR is an iterative algorithm and defined as follows:
| (1) |
where P0 is the initial probability vector, constructed such that equal probabilities are assigned to all of the seed nodes, with the sum of the probabilities equal to 1. Pt is a vector in which the i-th element holds the probability of finding the random walker at node i at step t. M is the transition matrix of the network, in which (i, j)-th element of M denotes the transition probability from node i to node j, and it is computed as the row-normalized adjacency matrix of the network. α is the restart probability of the walker returning to the seed node, the closer the value of α is to 0, the more global the view observed.
We performed the algorithm until the probability of all of the nodes reached a steady state, measured by the change between Pt and Pt+1 (measured by the L1 norm) falling below 10−10. The stable probability is defined as P∞, which gives a measure of similarity between non-seed nodes and seed nodes.
Ranking algorithm based on random walk with restart on complex heterogeneous networks
In this study, we presented a complex heterogeneous network computational model, CHNmiRD, to infer potential miRNA-disease associations by combining an integrated multigraph MFSN and DPN. Our method was an expansion of a previous method for predicting disease-related protein-coding genes [31]. The strategy to identify miRNA-disease associations using CHNmiRD is shown in Fig 1. The main flow of CHNmiRD consists of four steps: (1) constructing an integrated multigraph MFSN; (2) generating the CHN; (3) deciding the transition matrix of the CHN and (4) deciding the initial probability vector of the RWR algorithm to rank candidate disease miRNAs.
Fig 1. An overview of the CHNmiRD method.

Firstly, four MFSNs were constructed based on different genomic data by means of miRNA-target relationships and a disease phenotype network was constructed using the information of disease phenotype similarity. Then the complex heterogeneous network was generated by connecting the disease phenotype network and the integrated multigraph MFSN using the known miRNA-disease relationship information. Finally, the predicting miRNA-disease associations were obtained by implementing RWR algorithm on the complex heterogeneous network.
As mentioned above, four MFSNs were obtained based on different types of genomic data and were merged into a single multigraph MFSN. On the merged multigraph MFSN, the transition probability from node i to node j was computed as the expected value of the transition probabilities corresponding to four types of links between node i to node j. Suppose Ak is the transition matrix of the network k (k = 1, 2, 3, 4), and the corresponding (i, j)-th element of the matrix is Ak(i, j) denoting the transition probability from node miRNA i to node miRNA j. The transition probability from node i to node j on the integrated multigraph MFSN can then be computed as
| (2) |
Where Ni is the number of networks to which node miRNA i is associated. ωk is the probability of choosing the k-th network. Here, we set denoting the selection of any network with equal probability. Thus, an integrated multigraph MFSN could be obtained (S4 File).
A CHN was constructed by connecting a DPN and an integrated multigraph MFSN through the use of the human miRNA-disease associations from the HMDD database. Suppose A(m×m), B(n×n) and C(m×n) denote adjacency matrices for the integrated multigraph MFSN, DPN and DPMN, respectively. The adjacency matrix of the CHN can then be represented as , where CT is the transpose of C.
Next, we computed the transition matrix of the CHN. Suppose the transition matrix of the CHN is , where MmiRmiR and MDD are transition matrices indicating the probability from one miRNA (disease) to another miRNA (disease) in the random walk, respectively; MmiRD is the transition matrix from the integrated multigraph MFSN to the DPN, and MDmiR is the transition matrix from the DPN to the integrated multigraph MFSN. Let λ be the jumping probability, that is, the probability of jumping from the integrated multigraph MFSN to the DPN or vice versa. Let miRi denote the i-th miRNA in the integrated multigraph MFSN and di represents the i-th disease phenotype in the DPN. The transition matrix can thus be defined as follows:
The transition probability from miRi to miRj is defined as
| (3) |
The transition probability from di to dj is defined as
| (4) |
The transition probability from miRi to dj is defined as
| (5) |
The transition probability from di to miRj is defined as
| (6) |
Let u0 and v0 be the initial probability vectors of the integrated multigraph MFSN and DPN, respectively. The initial probability vector of the CHN can then be represented as . The parameter η ∈ (0,1) weighs the importance of the integrated multigraph MFSN and DPN. The initial probability of the integrated multigraph MFSN u0 is constructed such that equal probabilities are assigned to all of the seed nodes with the sum of the probabilities equal to 1. Similarly, the initial probability of the DPN v0 can be obtained.
Finally, we substituted the transition matrix M and initial probability P0 into the iterative equation (Eq 1). After a few steps, a stable probability vector can be obtained. All candidate miRNAs can now be ranked according to u∞, and the top ranked miRNAs can be considered as having a high probability of being associated with the disease of interest.
Results
Performance of CHNmiRD
For simplicity, we chose the following parameters to assess the performance of CHNmiRD in identifying potential miRNA-disease associations: α = 0.7 and λ = η = 0.5. The effect of these parameters was examined in the next section. 5-fold cross validation analysis of 3,462 known experimentally verified miRNA-disease associations, including 69 diseases associated with no less than 5 miRNAs, was used for this assessment. For a given disease d, the known experimentally verified miRNAs associated with disease d were randomly divided into 5 subsets. One subset was used as testing case, while the known disease d-related miRNAs in the rest sets and disease d were used as seed nodes in the multigraph MFSN and DPN, respectively. The candidate miRNAs included all of the miRNAs without known associations with disease d. We tested how well this testing case ranked relative to the candidate miRNA set for the given disease d. If the ranking of the testing miRNA exceeded a given threshold, this experimentally verified miRNA-disease association was considered to be successfully predicted by CHNmiRD.
The ROC curve is a plot of the true positive rate (sensitivity) against the false positive rate (1-specificity) for different thresholds. Suppose TP denotes true positive, TN denotes true negative, FN denotes false negative, and FP denotes false positive, then the sensitivity is calculated as TP/(TP+FN), and specificity is calculated as TN/(TN+FP). Sensitivity refers to the proportion of the testing miRNAs ranked higher than a given threshold, and specificity refers to the proportion of the testing miRNAs ranked lower than this given threshold. We plotted an ROC curve by varying the threshold and calculated the value of the area under the ROC curve (AUC). AUC values range from 0 to 1, with 0.5 and 1.0 indicating random and perfect predictive performance, respectively. CHNmiRD achieved an AUC value of 0.834 when testing 3,462 known experimentally verified miRNA-disease associations (Fig 2). To examine whether the result generated by chance, the seed miRNAs were randomly selected from candidate miRNAs for each disease and the AUC value was calculated (Fig 2). The results indicated that the real AUC value was much higher than that in randomization tests. 19 human diseases which are associated with at least 50 miRNAs were also evaluated. As shown in Table 1, lung cancer achieved the highest AUC value while systemic lupus erythematosus had the lowest one. The average AUC value of these 19 diseases was 0.844. These results demonstrated that CHNmiRD was effective in recovering known experimentally verified miRNA-disease associations.
Fig 2. ROC curves and AUC values of CHNmiRD and other similar methods for 5-fold cross validation.
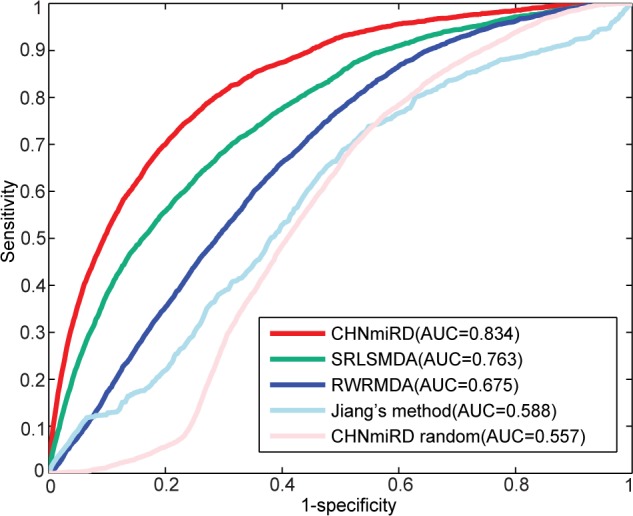
Table 1. AUC values of CHNmiRD and other similar methods for 19 human diseases using 5-fold cross validation.
| Disease name | MIM ID | No. miR | CHNmiRD | Jiang’s | RWRMDA | SRLSMDA |
|---|---|---|---|---|---|---|
| Lung cancer | 211980 | 208 | 0.920 | 0.589 | 0.777 | 0.832 |
| Breast cancer | 114480 | 229 | 0.911 | 0.573 | 0.777 | 0.913 |
| Colorectal cancer | 114500 | 239 | 0.904 | 0.557 | 0.807 | 0.909 |
| osteosarcoma | 259500 | 54 | 0.900 | 0.664 | 0.685 | 0.810 |
| Hepatocellular cancer | 114550 | 243 | 0.895 | - | 0.779 | 0.819 |
| Pancreatic cancer | 260350 | 127 | 0.876 | 0.648 | 0.691 | 0.844 |
| Bladder cancer | 109800 | 106 | 0.875 | 0.567 | 0.701 | 0.817 |
| Esophageal cancer | 133239 | 171 | 0.873 | 0.568 | 0.737 | 0.868 |
| Glioblastoma | 137800 | 155 | 0.872 | 0.563 | 0.744 | 0.857 |
| Melanoma | 155600 | 175 | 0.858 | 0.590 | 0.709 | 0.840 |
| Prostate cancer | 176807 | 148 | 0.857 | 0.576 | 0.725 | 0.854 |
| nasopharyngeal cancer | 607107 | 51 | 0.848 | 0.711 | 0.627 | 0.697 |
| kidney cancer | 144700 | 125 | 0.833 | 0.579 | 0.735 | 0.820 |
| Thyroid cancer | 188550 | 58 | 0.828 | 0.622 | 0.628 | 0.785 |
| Acute myeloid leukemia | 601626 | 86 | 0.822 | - | 0.575 | 0.611 |
| Cervical cancer | 603956 | 64 | 0.820 | 0.583 | 0.630 | 0.785 |
| Medulloblastoma | 155255 | 76 | 0.786 | 0.556 | 0.669 | 0.780 |
| Adrenal cortical carcinoma | 202300 | 67 | 0.777 | 0.625 | 0.617 | 0.677 |
| Systemic lupus erythematosus | 152700 | 83 | 0.711 | - | 0.594 | 0.622 |
Note: ‘No.miR’ indicates the number of miRNAs associated with a disease. ‘-’ denotes the disease- miRNA associations could not be predicted by Jiang’s method because of the lack of data.
To further evaluate the performance of individual data sources, we performed the same prediction framework by substituting the MFSN based on individual data sources for the integrated MFSN. The results are shown in Table 2. Although the PPI obtained the highest AUC value (0.817) among the four data sources, it was lower than that of the integrated method (0.834). The results showed that prediction performance improved upon integration of different genomic data sources. In addition, the coverage of miRNAs was different and biased for individual data sources. Therefore, some known disease-related miRNAs were ignored in the prediction process when using individual data sources. For example, 8 of 370 disease-related miRNAs were absent in the MFSN constructed based on BP ontology, and 36 of 370 disease-related miRNAs could not be prioritized when using CC ontology. Compared with individual data sets, the combined algorithm produced a higher coverage of miRNAs, which could be preferable for searching for novel disease-related miRNAs.
Table 2. Performance of individual data source.
| Data source | PPI | BP | MF | CC |
|---|---|---|---|---|
| AUC | 0.817 | 0.771 | 0.765 | 0.751 |
| No. of missing disease-related miRNAs | 0 | 8 | 17 | 36 |
Robustness of CHNmiRD
To evaluate the robustness of CHNmiRD, we considered different miRNA targets, miRNA-disease associations, DPNs and parameters. The predicted targets of 402 miRNAs were obtained from TargetScan (version 6.2) [37], miRDB (version 5.0) [38] and TargetMiner (May 2012) [39] (see S5 File). CHNmiRD was implemented for 5-fold cross validation. As a result, an AUC value of 0.832 was achieved (S1 Fig), which was comparable with that of the experimentally verified targets. To examine whether CHNmiRD was sensitive to the miRNA-disease associations, we randomly removed the miRNA-disease associations from 5% to 30% with a step of 5%. The results showed that the number of miRNA-disease associations had slight effect on the results (S2 Table). Additionally, we constructed DPNs using 3-NN network and 7-NN network, and CHNmiRD was then performed. As a result, the AUC values of 0.833 and 0.834 were obtained for 3-NN network and 7-NN network using 5-fold cross validation (S2 Fig). This was comparable with that of 5-NN network (0.834), demonstrating that CHNmiRD was robust to the selection of K for the KNN network. CHNmiRD included three parameters: (1) the restart probability α; (2) the jumping probability λ; and (3) the parameter η which controlled the effect of the two seed nodes, seed miRNAs and seed diseases. Based on previous studies demonstrating that the predictive result was robust to the restart probability, parameter α was selected to be 0.7 [40–42]. To investigate the possible effects of parameters λ and η on the performance of CHNmiRD, various values were used for these two parameters, and 5-fold cross validation was performed. The AUC values for different combinations of these two parameters are shown in Table 3. The results of the validation showed that parameter η had only a slight effect on the performance, while an increase of parameter λ improved performance. Specifically, when parameter λ was in the range of 0.5 to 0.9, performance became stable and performed better. Thus, the dependence of this method on these two parameters is minimal, particularly when the value of λ is above 0.5.
Table 3. AUC values for different combinations of the two parameters.
| η | |||||
|---|---|---|---|---|---|
| λ | 0.1 | 0.3 | 0.5 | 0.7 | 0.9 |
| 0.1 | 0.706 | 0.714 | 0.723 | 0.732 | 0.743 |
| 0.3 | 0.800 | 0.800 | 0.800 | 0.799 | 0.801 |
| 0.5 | 0.835 | 0.834 | 0.834 | 0.832 | 0.831 |
| 0.7 | 0.850 | 0.849 | 0.849 | 0.848 | 0.846 |
| 0.9 | 0.856 | 0.856 | 0.857 | 0.856 | 0.855 |
CHNmiRD versus similar existing methods
To further demonstrate the advantages of CHNmiRD in identifying miRNA-disease associations, we compared our model with the following similar existing methods: Jiang’s method [15], RWRMDA [16] and SRLSMDA [19]. Jiang et al. [15] adopted a hypergeometric distribution model for inferring potential miRNA-disease associations based on the human phenome-miRNAnome network. Chen et al. proposed two methods to uncover the relationships between miRNAs and diseases: RWRMDA and SRLSMDA. RWRMDA [16] used random walk method on the MFSN, while SRLSMDA [19] combined the optimal classifiers in disease space and miRNA space using regularized least squares method. We applied the RWRMDA to the integrated multigraph MFSN and applied Jiang’s method and SRLSMDA to the CHN, respectively. 5-fold cross validation was then performed using the same dataset. The best parameters were selected for other prediction methods (see S6 File) and the AUC values were obtained (see Fig 2 and Table 1). As the results indicated, CHNmiRD (AUC = 0.834) performed better than Jiang’s method (AUC = 0.588), RWRMDA (AUC = 0.675) and SRLSMDA (AUC = 0.763).
Additionally, there were some diseases without known related miRNAs and the pathological mechanism of these diseases at the miRNA level was completely unknown. A recent study indicated that SRLSMDA showed a better performance for this kind of disease [19]. We therefore tested the efficacy of CHNmiRD in searching miRNA-disease associations for these diseases. In the DPMN, 105 diseases were connected to 370 miRNAs. For each of these 105 diseases, we removed all of the relationships of this disease to miRNAs and used this disease as a seed node to implement CHNmiRD and RLSMDA. If one of the known disease-related miRNAs was ranked in the top N of the ranked list, we considered it to be a successful prediction. Here, we set N as 1, 5, 10, 20 and 50. As indicated in Table 4, CHNmiRD successfully ranked 40 miRNAs as top 1, while SRLSMDA only ranked 14 miRNAs as top 1. Moreover, CHNmiRD performed better than SRLSMDA as N varied.
Table 4. The number of successfully predicted miRNAs with different Ns.
| Top N | Top 1 | Top 5 | Top 10 | Top 20 | Top 50 |
|---|---|---|---|---|---|
| SRLSMDA | 14 | 80 | 140 | 280 | 779 |
| CHNmiRD | 40 | 138 | 249 | 434 | 987 |
Case studies
To illustrate the application of CHNmiRD in identifying novel disease-related miRNAs, case studies of glioblastoma (GBM), myocardial infarction (MI) and type 1 diabetes (T1D) considering different available numbers of seed miRNAs were examined. For a given disease, the known miRNAs associated with that disease were referred to as seed miRNAs. Based on the aforementioned known miRNA-disease associations, GBM had 155 seed miRNAs, MI had 40 seed miRNAs, and T1D had 1 seed.
For each of these three diseases, all of the candidate miRNAs (non-seed miRNAs) were ranked based on CHNmiRD (S3 Table), and the top 10 predicted miRNAs in the ranked list were examined. Because the known miRNA-disease associations were collected from the HMDD database, which was last updated in 2013, we manually verified these miRNA-disease associations by checking more recently published literatures. The results are illustrated in Table 5. Ten, 8 and 3 of the top 10 predicted miRNAs were confirmed in GBM, MI and T1D, respectively, according to recently reported biological experiments, and almost all of these had high ranks in the predicted miRNA lists. Although the remaining 9 predicted miRNA-disease associations had not yet been validated directly, these associations could be interpreted indirectly by recent studies. For instance, gene expression profile analysis of patient whole blood revealed that hsa-miR-182-5p was deregulated in patients with coronary artery disease [43]. Additionally, hsa-miR-19b-3p was reported to be a potential anti-thrombotic protector in patients with unstable angina [44], which has a high probability of developing into acute myocardial infarction. The remaining 7 predicted miRNAs which were not validated to be associated with T1D directly were found to be associated with diabetes [45, 46] and type 2 diabetes (T2D) [47–49]. It is worth noting that T1D had only one seed miRNA, but CHNmiRD achieved excellent performance. Collectively, these results not only indicated the reliability of CHNmiRD in identifying novel disease-associated miRNAs, but also demonstrated its potential application value in biomedical research.
Table 5. Literature evidence for top 10 miRNAs of glioblastoma, myocardial infarction and type 1 diabetes.
| miRNA | Rank | Literature validation | PubMed ID | Year |
|---|---|---|---|---|
| Glioblastoma | ||||
| hsa-miR-200a-3p | 1 | Yes/directly | 24755707 | 2014 |
| hsa-miR-190a-5p | 2 | Yes/directly | 23863200 | 2013 |
| hsa-miR-126-3p | 3 | Yes/directly | 21713760 | 2012 |
| hsa-miR-126-5p | 4 | Yes/directly | 21713760 | 2012 |
| hsa-miR-223-3p | 5 | Yes/directly | 24438238 | 2014 |
| hsa-miR-29b-3p | 6 | Yes/directly | 24155920 | 2013 |
| hsa-miR-34c-5p | 7 | Yes/directly | 24140020 | 2013 |
| hsa-miR-34b-5p | 8 | Yes/directly | 24213470 | 2012 |
| hsa-miR-1-3p | 9 | Yes/directly | 24310399 | 2014 |
| hsa-miR-34b-3p | 10 | Yes/directly | 24213470 | 2012 |
| Myocardial infarction | ||||
| hsa-miR-146a-5p | 1 | Yes/directly | 23208587 | 2013 |
| hsa-miR-17-5p | 2 | Yes/directly | 24900964 | 2014 |
| hsa-miR-17-3p | 3 | Yes/directly | 24900964 | 2014 |
| hsa-miR-125b-2-3p | 4 | Yes/directly | 24627568 | 2014 |
| hsa-miR-125b-5p | 5 | Yes/directly | 24627568 | 2014 |
| hsa-miR-182-3p | 6 | No/ indirectly | - | - |
| hsa-miR-19b-3p | 7 | No/ indirectly | - | - |
| hsa-miR-34c-5p | 8 | Yes/directly | 23047694 | 2012 |
| hsa-miR-29c-3p | 9 | Yes/directly | 20164119 | 2010 |
| hsa-miR-29c-5p | 10 | Yes/directly | 24900964 | 2014 |
| Type 1 diabetes | ||||
| hsa-miR-155-5p | 1 | Yes/directly | 24223694 | 2013 |
| hsa-miR-16-5p | 2 | No/ indirectly | 23233752 | 2013 |
| hsa-miR-146a-5p | 3 | Yes/directly | 24796653 | 2014 |
| hsa-miR-15a-5p | 4 | No/ indirectly | 24397367 | 2014 |
| hsa-miR-21-5p | 5 | Yes/directly | 24937532 | 2014 |
| hsa-miR-15a-3p | 6 | No/ indirectly | 24397367 | 2014 |
| hsa-miR-17-5p | 7 | No/ indirectly | 22960330 | 2012 |
| hsa-miR-16-1-3p | 8 | No/ indirectly | 23233752 | 2013 |
| hsa-miR-96-5p | 9 | No/ indirectly | 24981880 | 2014 |
| hsa-miR-128-3p | 10 | No/ indirectly | 24944010 | 2014 |
Discussion
In this work, a computational framework, CHNmiRD, was presented for the prediction of novel miRNA-disease associations by integrating multiple genomic and phenotype data. Based on PPI data and GO data (three sub-ontologies: BP, MF and CC), four MFSNs were constructed using miRNA-target relationships and were further merged in to an integrated multigraph MFSN. A CHN was then constructed by connecting the integrated multigraph MFSN and DPN using the known miRNA-disease relationship information. Finally, novel miRNA-disease associations were predicted by implementing a global network distance measure-based random walk analysis on the CHN.
Comparing the integrated data with the individual data sources using the same method, we found that PPI data was the most effective in prioritizing candidate miRNAs among the four data sets. However, the performance of PPI data was inferior to the combined method, because individual data tend to be incomplete and noisy. In addition, the combined method covered more miRNAs, which was favorable for uncovering novel disease-related miRNAs.
The results of cross validation indicated the improved performance of CHNmiRD over other similar existing methods, especially for diseases without any known associated miRNAs. In addition, CHNmiRD did not need negative samples and the performance became stable and performed better when parameter λ was in the range of 0.5 to 0.9. Furthermore, case studies demonstrated the reliability and effectiveness of this method in revealing novel disease-related miRNAs. Each of the top 10 miRNAs in the three case studies was either directly or indirectly validated by recently published research. It worth noting that we did not compare CHNmiRD with our previously described method [21] because of different data sources used in the two methods. Moreover, the known disease-miRNA associations were not used in our previous method, thus the cross validation could not be implemented.
The CHNmiRD is based on the CHN, and thus the efficacy of CHNmiRD is affected by the quality of the CHN. For future studies, more bioinformatics data should be integrated to improve the quality of the CHN. For example, expression profile and/or pathway data can be added into the integrated MFSN, and the similarity of disease phenotypes based on ontological descriptions can also be added into the DPN. We anticipate that our algorithm may be more comprehensive and effective with the increasing amount of available miRNA-related biological data. In summary, CHNmiRD could potentially provide an improved tool for predicting novel miRNA-disease associations and play an important role in deciphering the pathogenesis of complex human diseases at the post-transcriptional level.
Supporting Information
(TIF)
(A). The DPN constructed based on 3-NN network. (B). The DPN constructed based on 7-NN network.
(TIF)
(XLS)
(XLS)
(XLS)
(XLSX)
(DOC)
AUC values of RWRMDA for 5-fold cross validation with variation of the parameter (Table A). AUC values of SRLSMDA for 5-fold cross validation with variation of the parameter (Table B).
(DOC)
(DOC)
(DOC)
(XLS)
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported in part by the National Natural Science Foundation of China (Grant Nos. 31500675), the Postdoctoral Foundation of Heilongjiang Province (Grant No. LBH–Z15162), the Natural Science Foundation of Heilongjiang Province (Grant No. QC2013C019) and the Science Foundation of Heilongjiang Provincial Health Department (Grant Nos. 2013126). There was no additional external funding received for this study.
References
- 1.Ambros V. The functions of animal microRNAs. Nature. 2004;431(7006):350–5. . [DOI] [PubMed] [Google Scholar]
- 2.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–33. 10.1016/j.cell.2009.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34(Database issue):D140–4. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19(1):92–105. 10.1101/gr.082701.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sumazin P, Yang X, Chiu HS, Chung WJ, Iyer A, Llobet-Navas D, et al. An extensive microRNA-mediated network of RNA-RNA interactions regulates established oncogenic pathways in glioblastoma. Cell. 2011;147(2):370–81. 10.1016/j.cell.2011.09.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Small EM, Olson EN. Pervasive roles of microRNAs in cardiovascular biology. Nature. 2011;469(7330):336–42. 10.1038/nature09783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thum T, Catalucci D, Bauersachs J. MicroRNAs: novel regulators in cardiac development and disease. Cardiovasc Res. 2008;79(4):562–70. 10.1093/cvr/cvn137 [DOI] [PubMed] [Google Scholar]
- 8.Hebert SS, Horre K, Nicolai L, Papadopoulou AS, Mandemakers W, Silahtaroglu AN, et al. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer's disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci U S A. 2008;105(17):6415–20. 10.1073/pnas.0710263105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parekh V. miR-34b-a novel plasma marker for Huntington disease? Nat Rev Neurol. 2011;7(6):304 . [DOI] [PubMed] [Google Scholar]
- 10.Kim AH, Parker EK, Williamson V, McMichael GO, Fanous AH, Vladimirov VI. Experimental validation of candidate schizophrenia gene ZNF804A as target for hsa-miR-137. Schizophr Res. 2012;141(1):60–4. 10.1016/j.schres.2012.06.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gee HE, Camps C, Buffa FM, Colella S, Sheldon H, Gleadle JM, et al. MicroRNA-10b and breast cancer metastasis. Nature. 2008;455(7216):E8–9; author reply E. 10.1038/nature07362 [DOI] [PubMed] [Google Scholar]
- 13.Lu M, Zhang Q, Deng M, Miao J, Guo Y, Gao W, et al. An analysis of human microRNA and disease associations. PLoS One. 2008;3(10):e3420 10.1371/journal.pone.0003420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang Q, Wang Y, Hao Y, Juan L, Teng M, Zhang X, et al. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009;37(Database issue):D98–104. 10.1093/nar/gkn714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang Q, Hao Y, Wang G, Juan L, Zhang T, Teng M, et al. Prioritization of disease microRNAs through a human phenome-microRNAome network. BMC Syst Biol. 2010;4 Suppl 1:S2 10.1186/1752-0509-4-S1-S2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen X, Liu MX, Yan GY. RWRMDA: predicting novel human microRNA-disease associations. Mol Biosyst. 2012;8(10):2792–8. 10.1039/c2mb25180a [DOI] [PubMed] [Google Scholar]
- 17.Chen H, Zhang Z. Similarity-based methods for potential human microRNA-disease association prediction. BMC Med Genomics. 2013;6:12 10.1186/1755-8794-6-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xuan P, Han K, Guo M, Guo Y, Li J, Ding J, et al. Prediction of microRNAs associated with human diseases based on weighted k most similar neighbors. PLoS One. 2013;8(8):e70204 10.1371/journal.pone.0070204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen X, Yan GY. Semi-supervised learning for potential human microRNA-disease associations inference. Sci Rep. 2014;4:5501 10.1038/srep05501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang D, Wang J, Lu M, Song F, Cui Q. Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics. 2010;26(13):1644–50. 10.1093/bioinformatics/btq241 [DOI] [PubMed] [Google Scholar]
- 21.Shi H, Xu J, Zhang G, Xu L, Li C, Wang L, et al. Walking the interactome to identify human miRNA-disease associations through the functional link between miRNA targets and disease genes. BMC Syst Biol. 2013;7:101 10.1186/1752-0509-7-101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mork S, Pletscher-Frankild S, Palleja Caro A, Gorodkin J, Jensen LJ. Protein-driven inference of miRNA-disease associations. Bioinformatics. 2013;30(3):392–7. 10.1093/bioinformatics/btt677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu J, Li CX, Lv JY, Li YS, Xiao Y, Shao TT, et al. Prioritizing candidate disease miRNAs by topological features in the miRNA target-dysregulated network: case study of prostate cancer. Mol Cancer Ther. 2011;10(10):1857–66. 10.1158/1535-7163.MCT-11-0055 [DOI] [PubMed] [Google Scholar]
- 24.Jiang Q, Wang G, Jin S, Li Y, Wang Y. Predicting human microRNA-disease associations based on support vector machine. Int J Data Min Bioinform. 2013;8(3):282–93. . [DOI] [PubMed] [Google Scholar]
- 25.Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33(Database issue):D514–7. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vergoulis T, Vlachos IS, Alexiou P, Georgakilas G, Maragkakis M, Reczko M, et al. TarBase 6.0: capturing the exponential growth of miRNA targets with experimental support. Nucleic Acids Res. 2011;40(Database issue):D222–9. 10.1093/nar/gkr1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsu SD, Tseng YT, Shrestha S, Lin YL, Khaleel A, Chou CH, et al. miRTarBase update 2014: an information resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2014;42(Database issue):D78–85. 10.1093/nar/gkt1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xiao F, Zuo Z, Cai G, Kang S, Gao X, Li T. miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res. 2009;37(Database issue):D105–10. 10.1093/nar/gkn851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Driel MA, Bruggeman J, Vriend G, Brunner HG, Leunissen JA. A text-mining analysis of the human phenome. Eur J Hum Genet. 2006;14(5):535–42. . [DOI] [PubMed] [Google Scholar]
- 30.Li Y, Patra JC. Integration of multiple data sources to prioritize candidate genes using discounted rating system. BMC Bioinformatics. 2010;11 Suppl 1:S20 10.1186/1471-2105-11-S1-S20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y, Li J. Disease gene identification by random walk on multigraphs merging heterogeneous genomic and phenotype data. BMC Genomics. 2012;13 Suppl 7:S27 10.1186/1471-2164-13-S7-S27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, Qiu C, Tu J, Geng B, Yang J, Jiang T, et al. HMDD v2.0: a database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2014;42(Database issue):D1070-4. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun J, Zhou M, Yang H, Deng J, Wang L, Wang Q. Inferring potential microRNA-microRNA associations based on targeting propensity and connectivity in the context of protein interaction network. PLoS One. 2013;8(7):e69719 10.1371/journal.pone.0069719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Q, Sun J, Zhou M, Yang H, Li Y, Li X, et al. A novel network-based method for measuring the functional relationship between gene sets. Bioinformatics. 2011;27(11):1521–8. 10.1093/bioinformatics/btr154 [DOI] [PubMed] [Google Scholar]
- 35.Lv S, Li Y, Wang Q, Ning S, Huang T, Wang P, et al. A novel method to quantify gene set functional association based on gene ontology. J R Soc Interface. 2011;9(70):1063–72. 10.1098/rsif.2011.0551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kohler S, Bauer S, Horn D, Robinson PN. Walking the interactome for prioritization of candidate disease genes. Am J Hum Genet. 2008;82(4):949–58. 10.1016/j.ajhg.2008.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27(1):91–105. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X. miRDB: a microRNA target prediction and functional annotation database with a wiki interface. Rna. 2008;14(6):1012–7. 10.1261/rna.965408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bandyopadhyay S, Mitra R. TargetMiner: microRNA target prediction with systematic identification of tissue-specific negative examples. Bioinformatics. 2009;25(20):2625–31. 10.1093/bioinformatics/btp503 [DOI] [PubMed] [Google Scholar]
- 40.Li Y, Patra JC. Genome-wide inferring gene-phenotype relationship by walking on the heterogeneous network. Bioinformatics. 2010;26(9):1219–24. 10.1093/bioinformatics/btq108 [DOI] [PubMed] [Google Scholar]
- 41.Jiang R, Gan M, He P. Constructing a gene semantic similarity network for the inference of disease genes. BMC Syst Biol. 2011;5 Suppl 2:S2 10.1186/1752-0509-5-S2-S2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Macropol K, Can T, Singh AK. RRW: repeated random walks on genome-scale protein networks for local cluster discovery. BMC Bioinformatics. 2009;10:283 10.1186/1471-2105-10-283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taurino C, Miller WH, McBride MW, McClure JD, Khanin R, Moreno MU, et al. Gene expression profiling in whole blood of patients with coronary artery disease. Clin Sci (Lond). 2010;119(8):335–43. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li S, Ren J, Xu N, Zhang J, Geng Q, Cao C, et al. MicroRNA-19b functions as potential anti-thrombotic protector in patients with unstable angina by targeting tissue factor. J Mol Cell Cardiol. 2014;75C:49–57. . [DOI] [PubMed] [Google Scholar]
- 45.Zitman-Gal T, Green J, Pasmanik-Chor M, Golan E, Bernheim J, Benchetrit S. Vitamin D manipulates miR-181c, miR-20b and miR-15a in human umbilical vein endothelial cells exposed to a diabetic-like environment. Cardiovasc Diabetol. 2014;13:8 10.1186/1475-2840-13-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Szeto CC, Ching-Ha KB, Ka-Bik L, Mac-Moune LF, Cheung-Lung CP, Gang W, et al. Micro-RNA expression in the urinary sediment of patients with chronic kidney diseases. Dis Markers. 2012;33(3):137–44. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spinetti G, Fortunato O, Caporali A, Shantikumar S, Marchetti M, Meloni M, et al. MicroRNA-15a and microRNA-16 impair human circulating proangiogenic cell functions and are increased in the proangiogenic cells and serum of patients with critical limb ischemia. Circ Res. 2013;112(2):335–46. 10.1161/CIRCRESAHA.111.300418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang Z, Chen H, Si H, Li X, Ding X, Sheng Q, et al. Serum miR-23a, a potential biomarker for diagnosis of pre-diabetes and type 2 diabetes. Acta Diabetol. 2014. . [DOI] [PubMed] [Google Scholar]
- 49.Chakraborty C, Doss CG, Bandyopadhyay S, Agoramoorthy G. Influence of miRNA in insulin signaling pathway and insulin resistance: micro-molecules with a major role in type-2 diabetes. Wiley Interdiscip Rev RNA. 2014;5(5):697–712. 10.1002/wrna.1240 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TIF)
(A). The DPN constructed based on 3-NN network. (B). The DPN constructed based on 7-NN network.
(TIF)
(XLS)
(XLS)
(XLS)
(XLSX)
(DOC)
AUC values of RWRMDA for 5-fold cross validation with variation of the parameter (Table A). AUC values of SRLSMDA for 5-fold cross validation with variation of the parameter (Table B).
(DOC)
(DOC)
(DOC)
(XLS)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.