

Abstract
Genetic variation and early adverse environmental events work together to increase risk for schizophrenia. γ-aminobutyric acid (GABA), the major inhibitory neurotransmitter in adult mammalian brain, plays a major role in normal brain development, and has been strongly implicated in the pathobiology of schizophrenia. GABA synthesis is controlled by two glutamic acid decarboxylase (GAD) genes, GAD1 and GAD2, both of which produce a number of alternative transcripts. Genetic variants in the GAD1 gene are associated with increased risk for schizophrenia, and reduced expression of its major transcript in the human dorsolateral prefrontal cortex (DLPFC). No consistent changes in GAD2 expression have been found in brains from patients with schizophrenia. In this work, with the use of RNA sequencing and PCR technologies, we confirmed and tracked the expression of an alternative truncated transcript of GAD2 (ENST00000428517) in human control DLPFC homogenates across lifespan besides the well-known full length transcript of GAD2. In addition, using quantitative RT-PCR, expression of GAD2 full length and truncated transcripts were measured in the DLPFC of patients with schizophrenia, bipolar disorder and major depression. The expression of GAD2 full length transcript is decreased in the DLPFC of schizophrenia and bipolar disorder patients, while GAD2 truncated transcript is increased in bipolar disorder patients but decreased in schizophrenia patients. Moreover, the patients with schizophrenia with completed suicide or positive nicotine exposure showed significantly higher expression of GAD2 full length transcript. Alternative transcripts of GAD2 may be important in the growth and development of GABA-synthesizing neurons as well as abnormal GABA signaling in the DLPFC of patients with schizophrenia and affective disorders.
Introduction
GABA, the major inhibitory neurotransmitter in mammalian brain, has been implicated in both brain development and schizophrenia [1–3]. Two genes, GAD1 and GAD2, control GABA synthesis, but only the former has been clearly implicated in schizophrenia [4,5]. GAD1 and GAD2 are located on different chromosomes in mammals and encode two major isoforms of the enzyme glutamate decarboxylase (GAD), GAD67 and GAD65 respectively [6]. Within cells, GAD2 full length protein (65kDa) is maintained in a largely inactive form, apoGAD, (approximately 93%), which is converted to an enzymatically active form through the binding of pyridoxal 5’-phosphate. In contrast, GAD1 full length protein (67kDa) is found mainly as holoGAD (approximately 72%), which is enzymatically active and continually produces GABA [7]. Studies of GAD2 in postmortem brains of patients with schizophrenia have been inconsistent and mostly negative. In prefrontal cortex (PFC), GAD2 expression has been reported as decreased [8], increased [9] and normal [10,11] in patients with schizophrenia.
The GAD2 and GAD1 full length proteins have quite different distributions and functions. Located predominantly in synapses, GAD2 full length protein is associated with synaptic vesicles and produces synaptic GABA during intense neuronal activity [12]. GAD1 full length protein, on the other hand, can be found in somatic-dendritic regions as well as the synaptic terminal [13]. Studies in transgenic mice suggest additional distinct roles of these two GAD isoforms. Homozygous GAD1 knockout mice die at birth and have 10% of the normal GABA content, whereas GAD2 knockout mice are viable and have normal GABA content at birth [14,15]. Although viable, GAD2 deficient mice are susceptible to seizures, show a reduction in GABA release during prolonged activation of inhibitory neurons, and decreased GABA release in the visual cortex with potassium stimulation [14,16,17]. Interestingly, GAD2 knockout mice exhibit deficits in prepulse inhibition, an abnormality involving defective modulation of the startle reflex, also associated with schizophrenia [18]. These findings support the notion that GAD2 plays a role in the synthesis of GABA for synaptic release.
Previous work from our group has demonstrated the importance of alternate transcripts from GAD1 in both early brain development and schizophrenia [19]. The characterization of GAD2 alternative transcripts has not been completed. In order to study GAD2 thoroughly in brains of patients with schizophrenia we have done the following experiments: 1. Identified alternate transcripts in postmortem human brain; 2. Measured the expression of GAD2 transcripts from prenatal week 14 through 80 years of age in human prefrontal cortex (PFC); and 3. Compared the expression of GAD2 transcripts in dorsolateral prefrontal cortex (DLPFC) of patients with schizophrenia, affective disorders and normal controls.
Materials and Methods
Human postmortem brain tissue collection, tissue retrieval and RNA extraction
Postmortem neonate, infant, child, adolescent and adult brains were collected at the Clinical Brain Disorders Branch (CBDB), National Institute of Mental Health (NIMH) through the Northern Virginia and District of Columbia Medical Examiners’ Office. Informed consent to study brain tissue was obtained from the legal next of kin for all cases, according to NIMH Protocol 90-M-0142 and processed approved by the NIMH/National Institutes of Health Institutional Review Board [20]. Additional fetal, child and adolescent brain tissue samples were provided by the National Institute of Child Health and Human Development Brain and Tissue Bank for Developmental Disorders under contacts N01-HD-4-3368 and NO1-HD-4_3383, approved by institutional review board of the University of Maryland at Baltimore and the State of Maryland, and the tissue was donated to the NIMH under the terms of a material transfer agreement. All samples were obtained with audio-taped informed consent from the legal next of kin to study brain tissue, as approved by the Institutional Review Board of the National Institutes of Health and University of Maryland, in lieu of a written consent form. Brains were hemisected, and cut into 1.0–1.5-cm-thick coronal slabs, flash frozen, and stored at -80°C. DLPFC gray matter was dissected using a dental drill. For CBDB cases, the DLPFC (Brodmann’s areas 9 and 46) were dissected from middle frontal gyrus from coronal slab immediately anterior to the genu of the corpus callosum. For fetal cases, the PFC was obtained from the frontal cortex dissected at the dorsal convexity, midway between the frontal pole and anterior temporal pole. To measure the pH, pulverized cerebellum was used from each sample. To assess the expression of GAD2 alternative transcripts in the DLPFC, 176 schizophrenia patients, 61 bipolar disorder patients, 138 major depressive disorder (MDD), and 364 non-neurological and non-psychiatric controls were studied. A subset of non-psychiatric non-neurological controls, spanning from gestational weeks 14 to 20 and from birth up to 85 years of age, were used to measure expression of selected GAD2 transcripts in the prefrontal cortex (Table 1). Toxicological analysis was performed on all samples. Positive toxicology was exclusionary for control subjects but not for patients with psychiatric disorders. The majority of positive toxicology reports in psychiatric cases were due to the presence of psychotropic medications. In a subset of psychiatric cases, toxicology studies also revealed the presence of a variety of illicit substances. Nonpsychiatric controls were excluded if toxicology was positive for psychotropic medications or illicit substances. 43.6% of psychiatric patients (63.8% in MDD, 65.6% in bipolar disorder, and 20% in schizophrenia) are suicides. Nicotine exposure was positive in 41.8% patients with psychiatric disorders (43.4% in schizophrenia, 43.7% in bipolar disorder and 35.1% in MDD) and 24.6% controls.
Table 1. Demographic information of our human postmortem samples.
| Cohort | Number | Race | Sex | Age | PMI(h) | pH | RIN |
|---|---|---|---|---|---|---|---|
| Controls | 326 | 117AA/107CAUC/6HISP/6AS | 167M/69F | 41±17.1 | 29.3±14.5 | 6.55±0.28 | 8.30±0.69 |
| SZ patients | 176 | 73AA/96CAUC/4HISP/3AS | 111M/65F | 50±15 | 38.6±24.1 | 6.40±0.25 | 7.84±0.96 |
| BP patients | 61 | 6AA/51CAUC/1HISP/3AS | 36M/25F | 45±14 | 32.9±18.4 | 6.36±0.28 | 7.99±0.86 |
| MDD patients | 138 | 14AA/119CAUC/3HISP/2AS | 79M/59F | 45±14 | 37.8±28.3 | 6.35±0.28 | 8.03±0.88 |
| Lifespan | |||||||
| Fetal Controls | 43 | 37AA/5CAUC/1HISP | 22M/21F | -0.42±0.07a | 2.58±2.14 | N/A | 8.81±1.29 |
| Postnatal Controls | 283 | 139AA/132CAUC/6HISP/6AS | 198M/85F | 34.99±20.84 | 28.9±14.4 | 6.52±0.30 | 8.22±0.79 |
AA, African American; CAUC, Caucasian; AS, Asian; HISP, Hispanic; F, Female; M, Male; Sz, Schizophrenia; BP, Bipolar Disorder; MDD, Major Depressive Disorder; PMI, Postmortem Interval; RIN, RNA Integrity Number.
aFetal cohort in gestational age in weeks.
Dissected tissue was pulverized and stored at -80°C; RNA extractions and reverse transcriptase reactions were performed as described previously [20]. Briefly, total RNA was extracted from 300mg of tissue with TRIZOL Reagent (Life Technologies, Grand Island, New York). The yield of total RNA was determined by measuring the absorbance at 260 nm. To generate a RNA Integrity Number (RIN), RNA quality was assessed by high-resolution capillary electrophoreses (Agilent Technologies, Palo Alto, California). Using SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen, Carlsbad, CA, USA) to synthesize cDNA, 4μg of total RNA was reverse transcribed in a 50μl reaction system.
RNA Sequencing
To identify the library of transcripts derived from the GAD2 gene, RNA sequencing was performed on pooled commercial fetal and adult whole human brain poly A+ RNAs (Clontech, Lot#7110099, Lot#110415). mRNA molecules were fragmented into small pieces using divalent cations under elevated temperatures. Reverse transcriptase and random hexamers were used to convert the cleaved RNA fragments into first strand cDNA. Second strand cDNA was synthesized using DNA Polymerase I and RNaseH. These cDNA fragments were then subjected to an end repair process using T4 DNA polymerase, T4 polynucleotide kinase (PNK) and Klenow DNA polymerase, the addition of a single ‘A’ base using Klenow exo (3’ to 5’ exo minus), and then ligation of the Illumina P adapters using T4 DNA Ligase. An index (up to 12) was inserted into Illumina adapters so that multiple samples could be sequenced in one lane of an 8-lane flow cell if necessary. These products were then purified and enriched by PCR to create the final cDNA library for high throughput DNA sequencing using HiSeq 2000 (Illumina, Inc.). To analyze the RNASeq data, sequencing reads were mapped against the whole reference genome by using TopHat software. The Cufflinks software was used to assemble transcripts and estimate their abundance.
Rapid amplification of cDNA (RACE) ends and end-to-end PCR
To identify the 3’ ends of GAD2 transcript in human brain, we performed 3’ RACE, using fetal and adult brain poly A+ RNA with GAD2 gene specific sense primers binding at exon 1. We used the SMART RACE cDNA Amplification Kit (Clontech) and Advantage 2 PCR Kit (Clonetech) for these assays. Commercial human fetal and adult brain poly A+ RNAs (Clontech) were reverse-transcribed to cDNA by MMLV reverse transcriptase (Clontech) according to the manufacturer’s protocol. The PCR amplification profile was 94°C for 2 min, 45 cycles of 94°C for 30 s, 68–72°C for 30s, 68°C for 2–6 min, and 68°C for 10min after the last cycle. Based on known GAD2 gene exons (NM_001134366.1) and 3r RACE results, we designed primer pairs to amplify full length and portion of GAD2 transcripts using Platinum TaqDNA polymerase (Invitrogen). The PCR conditions were 94°C for 3 minutes, 30 cycles of 94°C for 30 sec, 56°C for 30 sec, 72°C for 1–5 minute, and 72°C for 10 minutes after the last cycle. RACE and end-to-end PCR products were cloned into E. Coli by PCR-TOPO 4.0 vectors (InvitrogenTM) and sequenced. All PCR results were confirmed in separate PCR assays and Sanger sequencing.
DNA extraction and sequencing
For extraction of DNA from E. coli, the Qiagen QlAprep spin miniprep kit (QIAGEN Science, Maryland, USA) was used. DNA was sent for sequencing at the National Institute of Neurological Disorders and Strokes (NINDS), using Big Dye Terminators on an ABI Perkin Elmer 9700 Thermal Cycler according to the manufacturer’s protocol. DNA was then sequenced using an Applied Biosystem 3100 Genetic Analyzer. The final data was analyzed using the Applied Biosystems Sequence Scanner V.1.0.
Quantitative real-time PCR
mRNA expression levels of alternative GAD2 transcripts were measured in postmortem DLPFC samples, including 417 non-psychiatric non-neurological control subjects, 176 schizophrenia patients, 61 bipolar disorder patients, and 138 major depression patients by real-time quantitative (RT-PCR), using ABI Prism 7900 sequence detection system with 384-well format. TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA, USA) were used; for human GAD2 (spanning exons 15–16, Hs00609534_m1); for human GAD2 truncated transcript (spanning exons 4 a-intron4; Table 2). mRNA expression levels of two GAD2 alternative transcripts (GAD2 full length transcript and truncated transcript) were normalized to the geometric means of three constitutively-expressed genes: β-actin (ACTB), β2-microglobulin (B2M) and β-glucuronidase (GUSB). The geometric means of the three housekeeping genes did not show diagnostic group difference. Genotypes were generated as described previously by Straub et al [21].
Table 2. QPCR Primers and Probes.
| Transcript | Primers and Probes |
|---|---|
| ENST00000428517 | Forward: AATTGGGAATTGGCAGACCA |
| Reverse: ATTTTCGATGAGACAATACCTGTTT | |
| Probe: TTGATGCATTGCCAAACA |
Statistical analysis
Statistical analyses were performed using R package (Version3.2.2). The expression data was log2 transformed to make the data more symmetrical for ANCOVA/linear modeling. Levene's test showed that the assumption of homogeneity of variance was met. The lifespan curve was generated using LOESS fit (local polynomial regression fitting) by using an R package with default parameters. ANCOVA/linear model was used for all comparisons. Multiple regression analyses were carried out to evaluate the contributions of age, pH, postmortem interval (PMI), race, sex and RNA integrity number (RIN), ethanol exposure and nicotine exposure on mRNA expression. Multiple regression analyses were also performed to assess the effect of completed suicide, psychotropic medications (toxicology screen results about antidepressants, antipsychotics, anticonvulsants, benzodiazepines, and opiates), lifetime neuroleptic exposure, average daily neuroleptic dose, final neuroleptic dose, and illicit substances on mRNA expression in the patients with psychiatric disorders. Estimated of lifetime neuroleptic exposure, average daily dose and final neuroleptic dose were all converted to chlorpromazine equivalents for statistical comparisons. Comparisons between diagnostic groups were made by ANCOVA for mRNA expression with diagnosis as an independent variable. Covariates were chosen for each ANCOVA from multiple regression analyses. Comparisons between patients and controls following overall ANCOVA were conducted by post-hoc Tukey HSD tests. Comparisons within the different diagnostic groups for completed suicide, nicotine exposure, ethanol exposure and psychotropic medications were conducted by ANCOVA, followed by Bonferroni correction.
Results
Alternative transcripts of GAD2 in human brain
An analysis of pooled RNA from normal fetal or adult brains (acquired commercially) by RNA sequencing failed to identify any novel splicing junctions of GAD2 in the human DLPFC, but confirmed the production of a previously identified truncated transcript (ENST00000428517) composed of several 5’ exons of GAD2 (Fig 1).
Fig 1. Mapped Reads and Junction Plots from RNA sequencing of GAD2.

Using pooled whole brain, known and putative novel exons for GAD2 in both adult and fetal human brain were identified using RNA-Seq. The y-axis is the coverage information, while the x-axis shows the position of the reads. The red arc-shapes between exons represented the splicing junctions. The thickness of the red arc-shapes represented the abundance of the splicing events. We observed the continuously splicing events across all the known junction of the GAD2 gene in adult brain but not in fetal brain. These results suggested there isn’t a novel splicing junction for GAD2 in the human DLPFC, and potential truncated transcripts composed by several 5’ exons.
To validate the findings from the RNA-Seq data, we carried out 3’RACE and observed an alternative termination point with a poly A+ tail in intron 3. By end-to end PCR, we confirmed the well-known full length transcript encoding for the 65kDa GAD protein and the previously identified truncated transcript, ENST00000428517, consisting of the first four exons of GAD2 (Fig 2A). RNA sequencing data suggested that the expression of the GAD2 truncated transcript is about 78% of the expression of the full length GAD2 in the fetal brain, about 12% in the childhood (0~10 years old) and 4% after 10 years old [22]. The expression levels of the full length and truncated GAD2 transcripts in 20 pooled human tissues (acquired commercially from Clontech, Human Total RNA Master Panel II, Lot#1403130A) showed that both GAD2 transcripts are primarily expressed in brain (Fig 2B). The truncated transcript shares the same exon1 and translational start site with the full length transcript, but lacks an in-frame stop codon.
Fig 2. Alternative transcript map for GAD2.

(A) Alternative transcripts of GAD2 in human Brain. Full length GAD2 transcript (NM_001134366.1, encoding the 65kDa GAD protein) and a truncated transcript (ENST00000428517) were identified. Key: Black rectangles, known exons. (B) The expression of GAD2 transcripts in 20 human tissues. GAD2 were mainly expressed in human brain. These two transcripts had opposite developmental expression changes in human brain. All the expression levels were normalized by ACTB, B2M and GUSB.
Lifespan expression pattern of GAD2 alternative transcripts
In a cohort of 326 non-neurologic non-psychiatric control subjects ranging from the second trimester in the fetus through 85 years of age, prefrontal cortical samples were studied to determine the expression pattern of the two transcripts across the human lifespan. Unlike the lifespan pattern of the GAD2 full length transcript (Fig 3A), whose expression was low in the fetus and then gradually rose to its adult peak at about 20 years of age, the GAD2 truncated transcript, ENST00000428517, showed a mildly inverted expression pattern. ENST00000428517 expression peaked during fetal period, and then gradually decreased after birth (Fig 3B).
Fig 3. Lifespan expression of the GAD2 alternative transcripts.
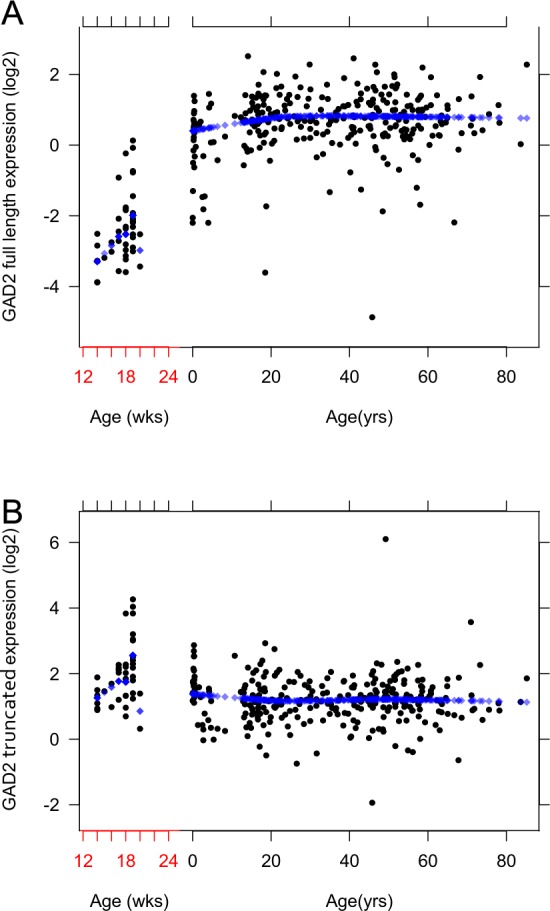
Each figure shows the expression patterns of the selected GAD2 alternative transcripts across the lifespan in the human DLPFC: (A) The expression of GAD2 full length across the lifespan in the human DLPFC. (B) The expression of ENST00000428517 across the lifespan in the human DLPFC. The expression of full length GAD2 transcript increases from 14 weeks of gestational age until the first decade of life and levels off throughout the rest of the lifespan. ENST00000428517 expression in the DLPFC is highest in the fetus and then declines gradually. The x-axis shows age: Before birth gestational age is in weeks (wks) and after birth in years (yrs). On the y-axis, the expressions levels were Log2 normalized. Each dot represents an individual subject.
Expression of GAD2 splice variants RNA in schizophrenia, bipolar disorder and major depression
Multiple regression analyses revealed that the mRNA expression level of GAD2 full length transcript positively correlated with age (p = 2.71E-05), sex (p = 1.96E-08), RIN (p<2E-16), pH (p = 1.19E-10) and PMI (P = 9.2E-03) in the DLPFC. The mRNA expression of ENST00000428517 was positively correlated with PMI (p = 1.60E-02) and sex (p = 1.94E-03) in the DLPFC. Each positive variable was included as covariates in ANCOVA. An ANCOVA revealed that in the DLPFC, the expression of GAD2 full length showed an overall effect of diagnosis (F(3,610) = 26.67,p = 3.22E-16; Fig 4A). Post-hoc analyses revealed that the expression of GAD2 full length was significantly decreased in schizophrenia patients (24% decrease, p = 7.05E-10) and bipolar disorder patients (22% decrease, p = 1.28E-02), with no changes in MDD patients (p = 5.92E-02) compared with controls. Regarding the truncated transcript ENST00000428517, there was also an overall effect of diagnosis (F(3,613) = 23.17,p = 3.21E-14; Fig 4B). Patients with bipolar disorder exhibited a 48% increase in ENST00000428517 mRNA expression (p = 5.76E-06), patients with schizophrenia (p = 5.76E-06) showed a 24% decrease, while MDD (p = 0.55) patients showed no difference.
Fig 4. Alternative GAD2 transcript expression in four diagnostic groups.

The expression of each alternative transcript was studied across four cohorts of subjects (non-psychiatric controls, schizophrenia, bipolar disorder, and major depressive disorder). Each panel shows the comparison of each transcript across the groups: (A) GAD2 full length in DLPFC; (B) ENST00000428517 in DLPFC. The x-axis shows the different diagnostic groups: Control, nonpsychiatric control group; Schizo, patients with schizophrenia; MDD, patients with major depressive disorder. The y-axis represents relative expression in the DLPFC. The y-axis is the least-squares means of expression with log2 normalization, computed for covariates at their means. Asterisks mark symbolizes statistically significance between psychiatric group and control group. Each dot represents an individual subject.
Expression of GAD2 full length transcript with nicotine exposure and suicide
The toxicology screen provided the positive or negative results for a large number of substances including nicotine, ethanol, opiates, anticonvulsants, antidepressants, and antipsychotics. The clinical and medical examiner records provided basic demographic information plus lifetime neuroleptic exposure, average daily dose, final neuroleptic dose, and suicide. The multiple regression analyses revealed that the expression of GAD2 full length transcript in DLPFC was significantly correlated with completed suicide (p = 9.61E-09) and nicotine exposure (p = 2.59E-02) in the psychiatric patients taken as a whole across diagnoses. The subjects with schizophrenia and completed suicide (p = 2.73E-03, Fig 5A) or positive nicotine exposure (p = 1.87E-03, Fig 5B) showed significantly higher expression of GAD2 full length transcript compared with natural deaths or nicotine free patients with schizophrenia. As there was a significant correlation between sex and the expression of GAD2 full length transcript, we ran further analyses to determine if sex might affect the expression of GAD2 full length transcript in the DLPFC within each diagnostic group. The female control subjects had significantly lower expression of GAD2 full length transcript compared to male controls (p = 1.73E-03). This was also seen independently in schizophrenia (P = 1.19E-02) and MDD (1.20E-03) patients (Fig 5C). There was no significant effect of psychotropic medications on the expression of GAD2 transcripts in the DLPFC.
Fig 5. Expression of GAD2 full length transcript correlated with suicide, nicotine or sex.

The y-axis represents relative expression in the DLPFC. The y-axis is the least-squares means of expression with log2 normalization, computed for covariates at their means. Each dot represents an individual subject. (A) GAD2 full length expression in DLPFC between completed suicide and death by natural causes in schizophrenia patients. The x-axis represents the schizophrenia patients with or without completed suicide. (B) GAD2 full length expression in DLPFC between nicotine positive and nicotine free schizophrenia patients on toxicology testing. The x-axis represents the schizophrenia patients with or without nicotine exposure based on toxicology testing. (C) GAD2 full length expression in DLPFC between female and male subjects. The x-axis shows the different diagnostic groups: Control, nonpsychiatric control group; Schizo, patients with schizophrenia; MDD, patients with major depressive disorder. M represent male, F represent female.
Discussion
Our results confirmed that GAD2 produces a truncated transcript in the human brain, in addition to the well-known full length transcript that encodes the 65kDa GAD enzyme. Characterization of the expression of these transcripts across the lifespan in the human DLPFC suggests that they are developmentally regulated. Finally, the expression of both GAD2 transcripts differed significantly between patients with psychiatric disorders compared to non-psychiatric controls.
Alternative splicing of GAD2
This study is the first to characterize alternative splicing of GAD2 in the human brain. We confirmed the presence of the full length transcript responsible of GAD2, and identified its developmental expression pattern in the prefrontal cortex. The truncated GAD2 transcript consisting of the first four exons of GAD2, ENST00000428517 lacks an in-frame stop codon. While the sequence for ENST00000428517 previously has been deposited into the UCSC Genome Browser, nothing has been published regarding its expression in human brain. Lacking of an in-frame stop codon, the truncated transcript is prone to nonstop decay, a cellular mRNA surveillance mechanism to prevent mRNA without a stop codon from translation [23,24]. As alternative splicing can lead to increased functional diversity in mRNA transcripts [25,26], this truncated GAD2 splice variant probably works as on-off regulation to adjust the level of the enzymatic protein produced by full length GAD2 transcript, which has a well established role in GABA synthesis. This truncated transcript might be critical to maintain a low level of full length GAD2 expression during fetal brain development since its lifespan expression pattern is the inverse of the full length GAD2 transcript. Additional experiments will be needed in order to define the function of this truncated transcript
GAD2 splice variants expression is developmentally regulated
Studies have shown that multiple transcripts derived from genes have expression patterns that differ across the lifespan [27,28]. Chan and colleagues detected the full length GAD2 mRNA in the human fetal frontal pole at 12 gestational weeks, with expression being highest at the beginning of the second trimester and decreasing rapidly and becoming undetectable by gestational week 19 [29]. However, in this study we show that the expression of the full length GAD2 mRNA increased with age, and had a particularly rapid rise during the first two decades of life, which mirrors (albeit on a different time scale) with expression studies in the rodent [30,31]. The lifetime expression trajectory of the ENST00000428517 is highest during fetal periods and gradually decreases after birth. Since the truncated transcript will be degraded quickly as it is an mRNA lacking a stop codon, its expression more likely acts as a component of the regulation of full length GAD2 expression levels during fetal brain development. During early CNS development, GABA levels are tightly controlled, as this neurotransmitter plays a major role in the regulation of cell proliferation, differentiation, and migration [32–34]. The role of GABA in early brain development may be mediated in part by the ENST00000428517 transcript since it is highly expressed during in the second trimester fetus, at a time before the rise in levels of synaptosomal full length GAD2.
Schizophrenia and GAD2
Recent studies indicate that genes involved in the etiology of schizophrenia may also be involved in the pathology of bipolar disorder and major depression [28,35–37]. Accordingly we examined the expression of GAD2 splice variants across these disorders. The full length GAD2 mRNA was significantly decreased in expression in the DLPFC of patients with schizophrenia and bipolar disorder compared with controls. While a decrease in full length GAD2 mRNA expression in bipolar disorder has been previously reported in the hippocampus, this same group did not see an effect in schizophrenia [38]. Another group has reported that GAD2 full length protein expression was decreased in the primary auditory cortex and cerebellum of schizophrenia patients [37,39] and cerebellum of bipolar disorder patients [37]. Our finding of decreased full length GAD2 mRNA potentially reflects an immature level of prefrontal development or regression after onset of illness, a supposition based on the developmental trajectory of GAD2 full length expression across normal development. Schizophrenia and bipolar disorder not only have some common clinical phenotypes, but appear to share some genetic risk factors [40,41]. It is possible that the decrease of full length GAD2 expression in both diagnostic groups might be involved in the molecular mechanism of the shared phenotypes. The truncated GAD2 transcript showed a significant decrease in expression level in patients with schizophrenia and an increase in patients with bipolar disorder, but no difference was detected in MDD patients. The expression level of ENST00000428517 suggests that this transcript behaves differently in the pathology associated with schizophrenia and bipolar disorder. The role of the truncated transcript ENST00000428517 is undefined, but it may be a component of the molecular dysfunction associated with these two psychiatric disorders.
It must be highlighted that any differences in mRNA expression between diagnostic groups are difficult to interpret. It is very hard to disambiguate changes related to the pathophysiology of illness from those secondary to the epiphenomena of illness, such as medications, cigarette smoking, environmental deprivation, and/or the stress of a lifetime combating chronic disability. Regardless, there appears to be diagnostic variation for alternative GAD2 transcripts in schizophrenia and affective disorders.
Our toxicology screen results suggest, unsurprisingly, that the psychiatric patients in this study have been exposed to multiple substances since positive toxicology was not an exclusion criterion for the patients with a psychiatric disorder. We studied the possibility that exposure to either prescription medications, nicotine, or illicit substances may contribute to expression changes seen in patients. Chronic administration of psychotropic medications, including clozapine, fluoxetine, haloperidol, lithium, olanzapine and valproic acid reportedly alters the level of full length GAD2 and full length GAD1 in rat, measured by both real-time PCR and western blotting [42]. In particular, the mood stabilizer valproic acid stimulated the proliferation of the GABAergic neurons with an associated dramatic increase in levels of GABA and full length GAD1/2 [43]. In contrast to animal studies, our analyses didn’t reveal positive correlations between the expression of GAD2 transcripts in the DLPFC and psychotropic medications in psychiatric patient cohorts.
The most common psychoactive substance on toxicological screening was nicotine, and this was not an exclusion criterion in any diagnostic group. The schizophrenia patients with nicotine exposure had increased expression of GAD2 full length transcript in the DLPFC comparing to nicotine-free patients with schizophrenia. There have been no previous studies on the impact of nicotine exposure on the expression of GAD2 in brain. Further animal studies are needed to confirm this finding and delineate the molecular mechanism. The correlation between GAD2 full length and nicotine is intriguing and suggests a previously unrecognized role for nicotine in cortical function in schizophrenia. This is especially important given the prevalence of cigarette smoking among schizophrenia patients [44].
43.6% psychiatric patients involved in this study were completed suicide. More than half of patients with bipolar disorder or MDD were suicides. We observed the up-regulated expression of GAD2 full length transcript in patients with schizophrenia who are suicides. Previously, suicidal patients with mood disorders had an increase in the relative density of GAD-immunoreactive neuropil [45] and abnormalities in the glutamate-glutamine and GABA-glutamine cycles had significant impact on suicidal behavior [46]. Our finding suggests that up-regulated 65kDa GAD, the translated protein produced by the GAD2 full length transcript, might in part be responsible for the increased level of GAD in suicidal patients with mood disorders, and provides additional and potentially important information regarding the role of GABA on suicidal behavior. If confirmed, this offers a new insight into the neurobiology of suicide.
Full length GAD2 expression is present in most classes of GABA neurons and is particularly prominent in axon terminals [47]. The data in this study is derived from homogenized tissue, and contains GAD2 transcripts from a variety of cell types and axon terminals. It is possible that the expression changes of GAD2 gene are restricted to a particular class of neurons or projections, which requires another level of investigation, which is necessary to understand the expression changes of GAD2 gene at cellular level in brain.
In general, the findings presented here confirm that splice variants of GAD2 are expressed differently in the human DLPFC across normal brain development and that these two transcripts may be involved in the pathophysiology of schizophrenia and affective disorders. While our results did show that ENST00000428517 is a fetal predominant transcript, its role in development is unknown. In order to address this issue, it would be insightful to conduct behavioral tests on transgenic ENST00000428517 knockout, knockdown and knock-up mice, followed by molecular biological experiments to observe if there are any changes in brain morphology and neurochemistry. It is known that GAD2 knockout mice are susceptible to seizures, have impairments in plasticity-related tasks, such as ocular dominance plasticity during the critical period and induction of early LTD in the visual cortex [14,16,17,48]. The identification of a fetal predominant novel truncated transcript produced by GAD2 suggests that this gene may play a more complex role in brain development and function than is widely appreciated.
Acknowledgments
We thank Amy Deep-Soboslay, M.Ed. of the Clinical Brain Disorders Branch, Genes, Cognition and Psychosis Program, Intramural Research Program, and NIMH for efforts in clinical diagnosis and demographic characterization, and Vesna Imamovic, Yeva Snitkovsky, Jewell King, Jonathan Sirovatka, and Liqin Wang, M.D., for their excellent technical assistance. Special gratitude also is extended to H. Ronald Zielke, Ph.D., Robert D. Vigorito, M.S., P.A., and Robert M. Johnson, B.S., of the National Institute of Child Health and Human Development Brain and Tissue Bank for Developmental Disorders at the University of Maryland for their provision of fetal, child, and adolescent brain specimens for this study.
Data Availability
All relevant data are within the paper.
Funding Statement
The authors have no support or funding to report.
References
- 1.Luján R, Shigemoto R, López-Bendito G. Glutamate and GABA receptor signalling in the developing brain. Neuroscience. 2005;130: 567–580. 10.1016/j.neuroscience.2004.09.042 [DOI] [PubMed] [Google Scholar]
- 2.Wassef A, Baker J, Kochan LD. GABA and schizophrenia: a review of basic science and clinical studies. J Clin Psychopharmacol. 2003;23: 601–640. 10.1097/01.jcp.0000095349.32154.a5 [DOI] [PubMed] [Google Scholar]
- 3.Taylor SF, Tso IF. GABA abnormalities in schizophrenia: a methodological review of in vivo studies. Schizophr Res. 2015;167: 84–90. 10.1016/j.schres.2014.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walls AB, Nilsen LH, Eyjolfsson EM, Vestergaard HT, Hansen SL, Schousboe A, et al. GAD65 is essential for synthesis of GABA destined for tonic inhibition regulating epileptiform activity. J Neurochem. 2010;115: 1398–1408. 10.1111/j.1471-4159.2010.07043.x [DOI] [PubMed] [Google Scholar]
- 5.Akbarian S, Huang H-S. Molecular and cellular mechanisms of altered GAD1/GAD67 expression in schizophrenia and related disorders. Brain Res Rev. 2006;52: 293–304. 10.1016/j.brainresrev.2006.04.001 [DOI] [PubMed] [Google Scholar]
- 6.Bu DF, Erlander MG, Hitz BC, Tillakaratne NJ, Kaufman DL, Wagner-McPherson CB, et al. Two human glutamate decarboxylases, 65-kDa GAD and 67-kDa GAD, are each encoded by a single gene. Proc Natl Acad Sci U S A. 1992;89: 2115–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Battaglioli G, Liu H, Martin DL. Kinetic differences between the isoforms of glutamate decarboxylase: implications for the regulation of GABA synthesis. J Neurochem. 2003;86: 879–887. [DOI] [PubMed] [Google Scholar]
- 8.Hashimoto T, Bazmi HH, Mirnics K, Wu Q, Sampson AR, Lewis DA. Conserved regional patterns of GABA-related transcript expression in the neocortex of subjects with schizophrenia. Am J Psychiatry. 2008;165: 479–489. 10.1176/appi.ajp.2007.07081223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dracheva S, Elhakem SL, McGurk SR, Davis KL, Haroutunian V. GAD67 and GAD65 mRNA and protein expression in cerebrocortical regions of elderly patients with schizophrenia. J Neurosci Res. 2004;76: 581–592. 10.1002/jnr.20122 [DOI] [PubMed] [Google Scholar]
- 10.Guidotti A, Auta J, Davis JM, Di-Giorgi-Gerevini V, Dwivedi Y, Grayson DR, et al. Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch Gen Psychiatry. 2000;57: 1061–1069. [DOI] [PubMed] [Google Scholar]
- 11.Glausier JR, Kimoto S, Fish KN, Lewis DA. Lower Glutamic Acid Decarboxylase 65-kDa Isoform Messenger RNA and Protein Levels in the Prefrontal Cortex in Schizoaffective Disorder but Not Schizophrenia. Biol Psychiatry. 2014; 10.1016/j.biopsych.2014.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patel AB, de Graaf RA, Martin DL, Battaglioli G, Behar KL. Evidence that GAD65 mediates increased GABA synthesis during intense neuronal activity in vivo. J Neurochem. 2006;97: 385–396. 10.1111/j.1471-4159.2006.03741.x [DOI] [PubMed] [Google Scholar]
- 13.Kanaani J, Kolibachuk J, Martinez H, Baekkeskov S. Two distinct mechanisms target GAD67 to vesicular pathways and presynaptic clusters. J Cell Biol. 2010;190: 911–925. 10.1083/jcb.200912101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Asada H, Kawamura Y, Maruyama K, Kume H, Ding R, Ji FY, et al. Mice lacking the 65 kDa isoform of glutamic acid decarboxylase (GAD65) maintain normal levels of GAD67 and GABA in their brains but are susceptible to seizures. Biochem Biophys Res Commun. 1996;229: 891–895. [DOI] [PubMed] [Google Scholar]
- 15.Asada H, Kawamura Y, Maruyama K, Kume H, Ding RG, Kanbara N, et al. Cleft palate and decreased brain gamma-aminobutyric acid in mice lacking the 67-kDa isoform of glutamic acid decarboxylase. Proc Natl Acad Sci U S A. 1997;94: 6496–6499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hensch TK, Fagiolini M, Mataga N, Stryker MP, Baekkeskov S, Kash SF. Local GABA circuit control of experience-dependent plasticity in developing visual cortex. Science. 1998;282: 1504–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tian N, Petersen C, Kash S, Baekkeskov S, Copenhagen D, Nicoll R. The role of the synthetic enzyme GAD65 in the control of neuronal gamma-aminobutyric acid release. Proc Natl Acad Sci U S A. 1999;96: 12911–12916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heldt SA, Green A, Ressler KJ. Prepulse inhibition deficits in GAD65 knockout mice and the effect of antipsychotic treatment. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol. 2004;29: 1610–1619. 10.1038/sj.npp.1300468 [DOI] [PubMed] [Google Scholar]
- 19.Hyde TM, Lipska BK, Ali T, Mathew SV, Law AJ, Metitiri OE, et al. Expression of GABA signaling molecules KCC2, NKCC1, and GAD1 in cortical development and schizophrenia. J Neurosci Off J Soc Neurosci. 2011;31: 11088–11095. 10.1523/JNEUROSCI.1234-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lipska BK, Deep-Soboslay A, Weickert CS, Hyde TM, Martin CE, Herman MM, et al. Critical factors in gene expression in postmortem human brain: Focus on studies in schizophrenia. Biol Psychiatry. 2006;60: 650–658. 10.1016/j.biopsych.2006.06.019 [DOI] [PubMed] [Google Scholar]
- 21.Straub RE, Lipska BK, Egan MF, Goldberg TE, Callicott JH, Mayhew MB, et al. Allelic variation in GAD1 (GAD67) is associated with schizophrenia and influences cortical function and gene expression. Mol Psychiatry. 2007;12: 854–869. 10.1038/sj.mp.4001988 [DOI] [PubMed] [Google Scholar]
- 22.Jaffe AE, Shin J, Collado-Torres L, Leek JT, Tao R, Li C, et al. Developmental regulation of human cortex transcription and its clinical relevance at single base resolution. Nat Neurosci. 2015;18: 154–161. 10.1038/nn.3898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frischmeyer PA, van Hoof A, O’Donnell K, Guerrerio AL, Parker R, Dietz HC. An mRNA surveillance mechanism that eliminates transcripts lacking termination codons. Science. 2002;295: 2258–2261. 10.1126/science.1067338 [DOI] [PubMed] [Google Scholar]
- 24.van Hoof A, Frischmeyer PA, Dietz HC, Parker R. Exosome-mediated recognition and degradation of mRNAs lacking a termination codon. Science. 2002;295: 2262–2264. 10.1126/science.1067272 [DOI] [PubMed] [Google Scholar]
- 25.Graveley BR. Alternative splicing: increasing diversity in the proteomic world. Trends Genet TIG. 2001;17: 100–107. [DOI] [PubMed] [Google Scholar]
- 26.Kelemen O, Convertini P, Zhang Z, Wen Y, Shen M, Falaleeva M, et al. Function of alternative splicing. Gene. 2013;514: 1–30. 10.1016/j.gene.2012.07.083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Colantuoni C, Lipska BK, Ye T, Hyde TM, Tao R, Leek JT, et al. Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature. 2011;478: 519–523. 10.1038/nature10524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tao R, Li C, Newburn EN, Ye T, Lipska BK, Herman MM, et al. Transcript-specific associations of SLC12A5 (KCC2) in human prefrontal cortex with development, schizophrenia, and affective disorders. J Neurosci. 2012;32: 5216–5222. 10.1523/JNEUROSCI.4626-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chan SO, Lyman WD, Chiu FC. Temporal and spatial expression of glutamic acid decarboxylases in human fetal brain. Brain Res Mol Brain Res. 1997;46: 318–320. [DOI] [PubMed] [Google Scholar]
- 30.Greif KF, Erlander MG, Tillakaratne NJ, Tobin AJ. Postnatal expression of glutamate decarboxylases in developing rat cerebellum. Neurochem Res. 1991;16: 235–242. [DOI] [PubMed] [Google Scholar]
- 31.Popp A, Urbach A, Witte OW, Frahm C. Adult and embryonic GAD transcripts are spatiotemporally regulated during postnatal development in the rat brain. PloS One. 2009;4: e4371 10.1371/journal.pone.0004371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.LoTurco JJ, Owens DF, Heath MJ, Davis MB, Kriegstein AR. GABA and glutamate depolarize cortical progenitor cells and inhibit DNA synthesis. Neuron. 1995;15: 1287–1298. [DOI] [PubMed] [Google Scholar]
- 33.Marty S, Berninger B, Carroll P, Thoenen H. GABAergic stimulation regulates the phenotype of hippocampal interneurons through the regulation of brain-derived neurotrophic factor. Neuron. 1996;16: 565–570. [DOI] [PubMed] [Google Scholar]
- 34.Manent J-B, Represa A. Neurotransmitters and brain maturation: early paracrine actions of GABA and glutamate modulate neuronal migration. Neurosci Rev J Bringing Neurobiol Neurol Psychiatry. 2007;13: 268–279. 10.1177/1073858406298918 [DOI] [PubMed] [Google Scholar]
- 35.Fatemi SH, Earle JA, McMenomy T. Reduction in Reelin immunoreactivity in hippocampus of subjects with schizophrenia, bipolar disorder and major depression. Mol Psychiatry. 2000;5: 654–663, 571. [DOI] [PubMed] [Google Scholar]
- 36.Costa E, Davis JM, Dong E, Grayson DR, Guidotti A, Tremolizzo L, et al. A GABAergic cortical deficit dominates schizophrenia pathophysiology. Crit Rev Neurobiol. 2004;16: 1–23. [DOI] [PubMed] [Google Scholar]
- 37.Fatemi SH, Hossein Fatemi S, Stary JM, Earle JA, Araghi-Niknam M, Eagan E. GABAergic dysfunction in schizophrenia and mood disorders as reflected by decreased levels of glutamic acid decarboxylase 65 and 67 kDa and Reelin proteins in cerebellum. Schizophr Res. 2005;72: 109–122. 10.1016/j.schres.2004.02.017 [DOI] [PubMed] [Google Scholar]
- 38.Heckers S, Stone D, Walsh J, Shick J, Koul P, Benes FM. Differential hippocampal expression of glutamic acid decarboxylase 65 and 67 messenger RNA in bipolar disorder and schizophrenia. Arch Gen Psychiatry. 2002;59: 521–529. [DOI] [PubMed] [Google Scholar]
- 39.Moyer CE, Delevich KM, Fish KN, Asafu-Adjei JK, Sampson AR, Dorph-Petersen K-A, et al. Reduced glutamate decarboxylase 65 protein within primary auditory cortex inhibitory boutons in schizophrenia. Biol Psychiatry. 2012;72: 734–743. 10.1016/j.biopsych.2012.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cross-Disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet Lond Engl. 2013;381: 1371–1379. 10.1016/S0140-6736(12)62129-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cross-Disorder Group of the Psychiatric Genomics Consortium, Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet. 2013;45: 984–994. 10.1038/ng.2711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fatemi SH, Reutiman TJ, Folsom TD. Chronic psychotropic drug treatment causes differential expression of Reelin signaling system in frontal cortex of rats. Schizophr Res. 2009;111: 138–152. 10.1016/j.schres.2009.03.002 [DOI] [PubMed] [Google Scholar]
- 43.Laeng P, Pitts RL, Lemire AL, Drabik CE, Weiner A, Tang H, et al. The mood stabilizer valproic acid stimulates GABA neurogenesis from rat forebrain stem cells. J Neurochem. 2004;91: 238–251. 10.1111/j.1471-4159.2004.02725.x [DOI] [PubMed] [Google Scholar]
- 44.Kelly DL, McMahon RP, Wehring HJ, Liu F, Mackowick KM, Boggs DL, et al. Cigarette smoking and mortality risk in people with schizophrenia. Schizophr Bull. 2011;37: 832–838. 10.1093/schbul/sbp152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gos T, Günther K, Bielau H, Dobrowolny H, Mawrin C, Trübner K, et al. Suicide and depression in the quantitative analysis of glutamic acid decarboxylase-Immunoreactive neuropil. J Affect Disord. 2009;113: 45–55. 10.1016/j.jad.2008.04.021 [DOI] [PubMed] [Google Scholar]
- 46.Bernstein H- G, Tausch A, Wagner R, Steiner J, Seeleke P, Walter M, et al. Disruption of glutamate-glutamine-GABA cycle significantly impacts on suicidal behaviour: survey of the literature and own findings on glutamine synthetase. CNS Neurol Disord Drug Targets. 2013;12: 900–913. [DOI] [PubMed] [Google Scholar]
- 47.Esclapez M, Tillakaratne NJ, Kaufman DL, Tobin AJ, Houser CR. Comparative localization of two forms of glutamic acid decarboxylase and their mRNAs in rat brain supports the concept of functional differences between the forms. J Neurosci Off J Soc Neurosci. 1994;14: 1834–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choi S- Y, Morales B, Lee H- K, Kirkwood A. Absence of long-term depression in the visual cortex of glutamic Acid decarboxylase-65 knock-out mice. J Neurosci Off J Soc Neurosci. 2002;22: 5271–5276. 20026507 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.