

Abstract
Nonsense-mediated mRNA decay (NMD) is a cellular quality-control mechanism that is thought to exacerbate the phenotype of certain pathogenic nonsense mutations by preventing the expression of semi-functional proteins. NMD also limits the efficacy of read-through compound (RTC)-based therapies. Here, we report a gene-specific method of NMD inhibition using antisense oligonucleotides (ASOs), and combine this approach with an RTC to effectively restore the expression of full-length protein from a nonsense-mutant allele.
RNA-based therapeutic approaches have become increasingly promising, as exemplified by the recent progress in the development of ASOs and stop-codon RTCs as therapeutics for various genetic diseases1,2. NMD is a quality-control process that degrades mRNAs with premature termination codons (PTCs) during translation3. mRNAs targeted by small molecules that allow the ribosome to read-through stop codons are often subject to NMD, reducing the therapeutic potential of such RTCs4. NMD suppression has thus gained attention as a potential RNA-based therapy, but the proposed approaches so far have relied on general inhibition of the NMD machinery5–7. Transcript-specific, as opposed to global, inhibition of NMD is desirable, because in addition to its protective role from the potential toxicity of certain truncated proteins, NMD plays an important role in global post-transcriptional gene regulation, for example, by alternative splicing coupled with NMD8,9. Indeed, NMD plays essential roles in development, such as in myogenesis, neurogenesis and lymphocyte maturation1. For example, UPF3B, a key regulator of NMD, is necessary for neural-progenitor cell differentiation and is mutated in various cognitive disorders10.
NMD relies on the presence of at least one exon-junction complex (EJC) bound to the mRNA downstream of a PTC. During translation, when a ribosome reaches a PTC, it associates with the release factors eRF1 and eRF3, and interacts with the downstream EJC through UPF1, UPF2, and UPF3A/3B proteins1. This physical interaction provides a platform to recruit additional NMD factors, targeting the mRNA for destruction. Given that most EJCs are assembled 20–24 nucleotides upstream of each exon-exon junction during splicing, and that the EJC is not required for splicing of most exons, we have developed an antisense approach that takes advantage of the predictable nature of EJC deposition11.
ASOs can be designed to block the binding sites of RNA-binding proteins, resulting in effects such as splicing modulation and inhibition of translation initiation, among others12. We tested whether ASOs can be used for targeted NMD inhibition by blocking EJC deposition on mRNA, downstream of a PTC. Although EJCs appear to be dispensable for most splicing events, they have roles in mRNP export from the nucleus and in translation13. Our approach is not expected to affect such processes, because we only target EJCs downstream of a PTC on a given mRNA, leaving the remaining upstream EJCs unaffected.
We first tested a well-characterized NMD substrate, the full-length three-exon HBB (β-globin) gene with the Q39X nonsense mutation (CAG→TAG) in exon 2 (HBB-T39)14. We stably expressed a single copy of this gene, following Flp-recombinase-mediated integration at a defined site in U2OS T-REx cells. In this context, HBB-T39 mRNA expression was reduced to about 5% compared to wild type HBB (Supplementary Fig. 1). We designed a set of 2′-O-(2-methoxyethyl) ribose (MOE) and phosphorothioate modified 15mer ASOs that together span the region of the HBB-T39 mRNA predicted to harbor an EJC, and screened them individually for NMD-inhibiting activity (Fig. 1a, b). After transfection, many of the ASOs caused skipping of HBB exon 2; however, several ASOs resulted in minimal exon skipping, and markedly increased the level of full-length spliced mRNA (Fig. 1c and Supplementary Fig. 2). We chose H-24 as the lead ASO, and confirmed its dose-dependent activity, which resulted in a 3-fold increase in HBB-T39 mRNA at the highest dose tested (Fig. 1d). Consistent with NMD inhibition, H-24 did not affect the levels of wild-type HBB mRNA (Supplementary Fig. 3). To verify the transcript-specific effect of NMD-inhibiting ASOs, we confirmed that the expression of several NMD-targeted endogenous mRNAs remained unchanged following ASO treatment (Supplementary Fig. 4)15.
Figure 1.
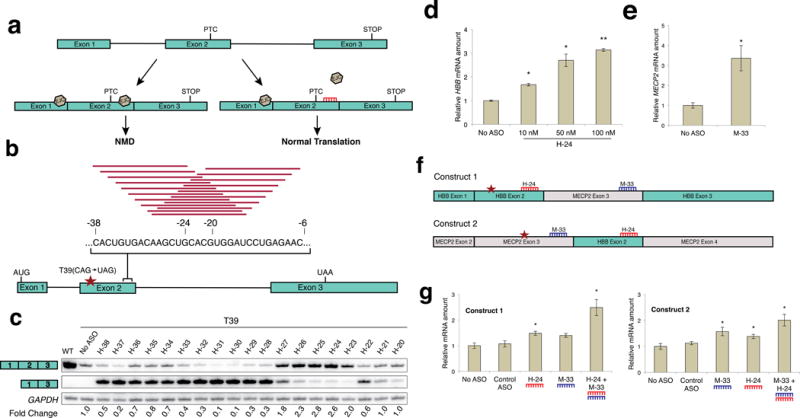
Design and testing of EJC-targeting ASOs. (a) Schematic of the antisense approach to inhibit NMD of a target mRNA. An ASO (red) complementary to the predicted EJC-deposition site downstream of the PTC interferes with EJC assembly, inhibiting NMD. (b) An example of the ASO screening strategy, using HBB with a PTC in codon 39, stably expressed in U2OS cells. A set of 19 uniformly modified MOE phosphorothioate 15-mer ASOs was designed to cover the presumptive EJC-deposition site at 1-nt resolution. (c) ASOs targeting the HBB exon 2 EJC region were individually transfected at a nominal concentration of 100 nM. HBB mRNA was measured by radioactive RT-PCR using primers in exons 1 and 3. Some of the ASOs caused skipping of exon 2. The fold change in full-length mRNA with the T39 mutation, relative to the no-ASO control in lane 2, is indicated below each lane. The uncropped autoradiograph is shown in Supplementary Figure 2. (d) The optimal ASO for HBB (H-24, based on three experiments as in (c)) was tested at the indicated nominal concentrations (n = 3, *P < 0.05, **P < 0.001 versus no-ASO control). (e) The same screening approach was used to identify an ASO (M-33) that effectively inhibits NMD of MECP2 mRNA with a PTC in its penultimate exon (n = 3). See also Supplementary Figure 3. (f) Constructs to test the feasibility of targeted antisense inhibition of NMD in the case of targets with more than one downstream EJC. (g) 50 nM of individual ASOs and 25 nM each of the combined ASOs were tested with each chimeric construct (n = 4). Error bars represent standard deviation (s.d.). The corresponding autoradiographs are shown in Supplementary Figure 7.
To test the generality of our approach, we constructed a three-exon NMD minigene reporter based on the MECP2 gene with the S65X nonsense mutation (TCA→TGA) in the middle exon (exon 3 in the natural gene), similarly integrated at a defined location in U2OS T-REx cells. MECP2-S65X mRNA was efficiently degraded by NMD (Supplementary Fig. 5). We screened a corresponding set of 19 ASOs spanning the presumptive EJC binding site near the 3′ end of this exon and identified multiple ASOs that increased the amount of full-length spliced mRNA (Supplementary Fig. 6). One of several effective ASOs for MECP2 (M-33) resulted in a ~3-fold increase in mRNA (Fig. 1e).
The above NMD substrates are simple three-exon genes or minigenes with a single predicted EJC downstream of a PTC. To test the broader applicability of our approach, we designed a pair of four-exon hybrid genes, consisting in one case of HBB exons 1 through 3, with exon 3 and portions of the flanking introns from MECP2 inserted within HBB intron 2, and in the other case of MECP2 exons 2 through 4, with HBB exon 2 and portions of the flanking introns inserted within MECP2 intron 3 (Fig. 1f and Methods). Using our optimal ASOs for these two genes (H-24 for HBB and M-33 for MECP2), we compared the effects of single and combined ASOs in inhibiting NMD of these target substrates with two presumptive downstream EJCs. In both cases, the combined treatment with both ASOs was more effective than with either ASO alone (Fig. 1g, Supplementary Fig. 7).
NMD is sensitive to the cellular state and environment, and can be inhibited by stress, e.g., hypoxia and amino-acid deprivation16. Furthermore, ASOs can have various off-target effects, although none has been linked to NMD efficiency17. To test the presumptive mechanism of action, we performed in vitro splicing followed by GST-eIF4A3 pull-down to confirm that an effective ASO blocks the interaction between the spliced mRNA and eIF4A3, the anchor component of the EJC18. Consistent with its predicted activity, H-24 ASO blocked the interaction between HBB mRNA and eIF4A3 in a dose-dependent manner (Fig. 2a).
Figure 2.
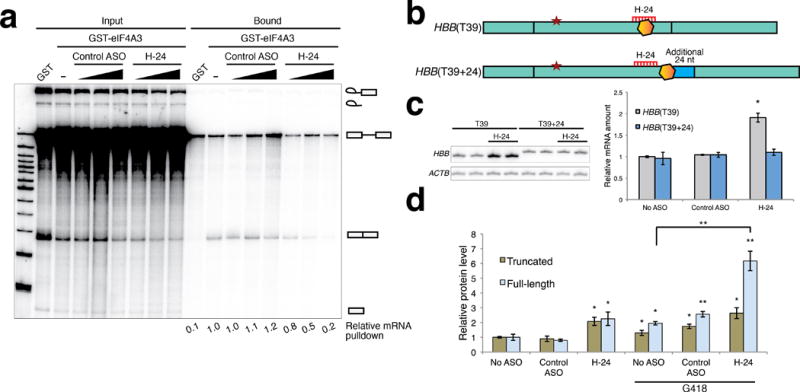
Validation of the mechanism of action of NMD inhibition. (a) In vitro splicing of a radiolabeled pre-mRNA comprising exon 2, intron 2, and exon 3 of HBB was carried out in HeLa nuclear extract in the presence of GST-eIF4A3 (or GST) and varying concentrations of control or targeting ASOs. Spliced mRNA was specifically pulled down by GST-eIF4A3 in the absence of ASO or in the presence of control ASO in the splicing reaction. H-24 ASO prevented the association of eIF4A3 with the spliced mRNA in a dose-dependent manner. The relative changes in mRNA pull-down efficiency, normalized to the spliced mRNA level of the corresponding input, are indicated below each lane. 5% of the input RNA is shown on the left lanes. [ASO] = 100 nM, 200 nM, or 500 nM. (b) Diagram of constructs to test the dependence of the ASO effect on the position of the EJC. Addition of extra sequence (24 nt) at the end of exon 2 shifts the ASO hybridization site farther upstream from the exon-exon junction and from the site of EJC deposition. (c) H-24 ASO at 50 nM did not affect the NMD efficiency of the modified construct. n = 3, *P < 0.01. (d) Antisense-inhibition of NMD increased expression of both the truncated protein and the full-length read-through product translated from a modified HBB-T39 mRNA that allows more efficient read-through. Expression of the full-length protein was highest when ASO (50 nM) and G-418 (1 mg/ml) were combined. The expression levels of truncated and full-length proteins are normalized to their levels in the ‘no ASO’ samples. See also Supplementary Figure 11 for a representative blot. Student’s t-test, * P < 0.05, ** P < 0.001; n = 3. Error bars represent ± s.d.
To further establish the mechanism of action, we modified the HBB-T39 construct by adding 24 nucleotides at the 3′ end of exon 2. Due to the position-specific nature of EJC binding, this modification shifts the predicted EJC-deposition site downstream of the sequence targeted by H-24 ASO (Fig. 2b)1, 18. The mRNA expressed from this construct (HBB-T39+24) was not affected by H-24, indicating that the ASO target sequence has to overlap with the predicted EJC-assembly site to inhibit NMD (Fig. 2c).
Additionally, to rule out the possibility that the increase in steady-state mRNA level reflects an increased rate of transcription, we performed an actinomycin D chase assay to measure the mRNA decay rate. As expected, cells treated with H-24 ASO expressed HBB-T39 mRNA with greater stability (Supplementary Fig. 8). HBB-T39 mRNA was reported to undergo biphasic decay, likely due to rapid decay of the NMD-sensitive mRNA molecules, occurring alongside slower decay of a subset of mRNA molecules that evade NMD19. Analysis of the half-lives in two phases revealed that the stability of HBB-T39 mRNA in ASO-treated cells was higher during the rapid-decay phase (estimated t1/2 = 6.8 min untreated vs. 16.5 min ASO-treated), whereas the decay rate during the slow phase remained unchanged (estimated t1/2 > 12 hr for both treatment groups).
To determine if the target-mRNA increase results in an increase in the encoded protein after ASO treatment, we monitored the effect of ASO on protein synthesis, via a GFP tag placed at the N-terminus of HBB-T39. We observed a robust increase in the amount of truncated protein in response to ASO treatment (Supplementary Fig. 9). Notably, ASO treatment did not reduce the amount of full-length protein expressed from the wild-type GFP fusion construct (Supplementary Fig. 10). We conclude that protein synthesis is not inhibited by an exon-targeting MOE phosphorothioate ASO, presumably because it is displaced from the mRNA upon passage of a translating ribosome.
RTCs are diverse classes of molecules that cause the ribosome to misrecognize a PTC and incorporate an amino acid, terminating at the natural stop codon downstream, and thereby restoring synthesis of full-length protein from mRNA with a nonsense mutation20. Although this is a promising strategy currently being tested in clinical trials, NMD diminishes the efficacy of read-through therapy by degrading the mRNAs targeted by such compounds4,5.
To test the utility of our approach in improving the efficacy of RTCs, we designed a read-through reporter, HBB-T39(UGAC), based on the above GFP-tagged HBB-T39 construct, by replacing the TAG PTC and the following four nucleotides with TGACTAG. This stop codon and the downstream sequence weaken translation termination, allowing some spontaneous read-through, and thereby increasing the sensitivity of detection of full-length protein21. Translational read-through suppresses NMD to some extent, as is evident by the 2–3 fold increase in HBB-T39(UGAC) mRNA, relative to HBB-T39 (Supplementary Fig. 1). Nevertheless, we observed strong increases in both the truncated protein and the read-through product upon further NMD suppression by H-24 ASO treatment (Fig. 2d and Supplementary Fig. 11).
G418 is an aminoglycoside with read-through activity4. Co-treatment of U2OS cells expressing HBB-T39(UGAC) with H-24 ASO and G418 increased the full-length protein level by 6-fold, compared to a 2-fold increase with G418 alone, and a 2.5-fold increase with H-24 ASO alone. Thus, antisense inhibition of NMD can augment the efficacy of a read-through compound. The ASO-induced increase in expression of truncated protein could be deleterious in some cases, but this would depend on the properties of the particular mutant protein, and on the efficacy of the RTC.
The antisense approach we present here, which we dubbed GAIN (for Gene-specific Antisense Inhibition of NMD) allows gene-specific inhibition of NMD without relying on skipping of a PTC-containing exon. This gene-specific feature, along with the clinically demonstrated efficacy and safety profiles of various ASO chemistries, such as MOE phosphorothioate, makes this a promising approach to inhibit NMD2.
Methods
ASOs
All ASOs used were uniformly modified with 2′O-MOE sugars, phosphorothioate backbone, and 5′-methyl cytosine. Synthesis, purification, and quantitation were done as described22. All ASOs were dissolved in water and stored at −20°C. The sequences of all ASOs used in this study are provided in Supplementary Table 1. ASOs were named by taking the first letter of their target gene, followed by a number representing the distance of the ASO 5′ end from the exon-exon junction.
Preparation of NMD reporter cells
All the cell lines used in this study tested negative for mycoplasma contamination. The full HBB gene and MECP2 exons 2 to 4 were cloned into the pcDNA FRT/TO plasmid (Life Technologies), with GFP sequence at the 5′ end of the first exon for monitoring protein expression. Only 200 bases at each end of MECP2 intron 3 (59,626 nt) were included in the construct. Chimeric constructs were made as follows: for Construct 1, MECP2 exon 3, along with the last 173 nt of intron 2 and the first 145 nt of intron 3, was inserted into HBB intron 2 at position IVS2+473. Likewise, for Construct 2, HBB exon 2, along with the last 60 nt of intron 1 and the first 79 nt of intron 2, was inserted into MECP2 intron 3 at position IVS3+335. Cloning was carried out by the sequence- and ligation-independent cloning strategy (SLIC)23, and PTCs were introduced by site-directed mutagenesis. HBB-T39+24 was made from HBB-T39 by replacing 6 nt at the 3′ end of HBB exon 2 with 30 nt from the 3′ end of HBB exon 1. The pcDNA FRT/TO constructs were co-transfected along with pOG44 helper vector into FRT-U2OS-TRex cells with a single integration site (Life Technologies). Cells with successful FRT-mediated integration were obtained by hygromycin selection. The expression and correct splicing of the transgenes were confirmed by radioactive RT-PCR, following induction with 1 μg/ml tetracycline.
Tissue culture and ASO transfection
U2OS cells were cultured in DMEM with 10% FBS. ASO transfections were carried out using Lipofectamine 2000 (Life Technologies) according to the manufacturer’s protocol for transfecting short oligonucleotides. Transgenes were induced with 1 μg/ml tetracycline, 6 hr after transfection. For the G418-treatment group, G418 (Sigma) was added at a final concentration of 1 mg/ml at the time of transgene induction, and again during a change of medium 24 hr after transfection.
RNA extraction and RT-PCR
Total RNA was extracted with TRIzol (Life Technologies) 48 hr post-transfection, according to the manufacturer’s protocol. Oligo dT(18)-primed reverse transcription was carried out with ImpromII Reverse Transcriptase (Roche). Semi-quantitative RT-PCR was carried out in the presence of 32P-dCTP and primer pairs corresponding to the first and last exons of each construct. MECP2 cDNA was amplified using a forward primer specific to GFP sequence, to avoid detecting the endogenous wild-type MECP2 mRNA. Primer sequences are provided in Supplementary Table 2. PCR products were detected with a Typhoon FLA7000 phosphorimager, and quantitated using MultiGauge v2.3 software (Fujifilm). Statistical significance was calculated using Student’s t-test.
Protein extraction and Western blotting
Cells were harvested 48 hr after transfection and lysed in STEN lysis buffer (50 mM Tris, pH 7.6, 150 mM NaCl, 0.1% SDS, 1% NP-40, and 2 mM EDTA + protease inhibitor cocktail (Roche)) by sonicating for 5 min at medium power using a Bioruptor (Diagenode), followed by 15-min incubation on ice. Protein concentration was measured using the Bradford assay (Bio-Rad) with BSA as a standard. Rabbit anti-GFP (Sigma G1544) and mouse anti-α-tubulin (Sigma T9026) antibodies were used with IRDye 800CW secondary antibodies (LI-COR) for Western blotting, and the blots were imaged and quantified using the Odyssey Infrared Imaging System (LI-COR).
In vitro transcription of HBB minigene
Pre-mRNA for in vitro splicing was transcribed using T7 RNA polymerase (Roche) in the presence of α-32P-UTP, using a PCR product as the template. The template contains portions of exons 2 and 3 and the intact intron 2 of HBB, and was amplified from human genomic DNA using the primer pair T7HBB-Ex2-F and HBB-Ex3-R (Supplementary Table 2). The radiolabeled transcript was purified by denaturing PAGE, and its concentration measured by liquid scintillation counting.
In vitro splicing and GST pull-down
Expression and purification of GST-eIF4A3, HeLa nuclear extract preparation, and in vitro splicing were carried out essentially as described18, 24, 25. 50-μl splicing reactions were carried out for 90 min at 30 °C with 4 μg of GST alone or GST-eIF4A3, in the presence of varying concentrations of ASOs. A 5-μl aliquot was kept as input, and 400 μl of wash buffer (20 mM Hepes, pH 8.0, 150 mM KCl, 0.05 % NP-40) was added to the remainder. mRNP was bound to 10 μl of glutathione-Sepharose beads (GE Healthcare) by nutating at 4 °C for 2 hr. The beads were washed three times with wash buffer, and resuspended in 300 μl of splice-stop solution (0.3 mM NaOAc and 0.1% SDS). RNA was then recovered by phenol extraction and ethanol precipitation, and analyzed by denaturing PAGE, followed by detection and quantitation on a Typhoon phosphorimager.
mRNA decay rate assay
U2OS cells with stably integrated HBB-T39 constructs were pre-treated with transfection reagent (control) or 50 nM H-24 ASO. Expression of the transgene was induced 24 hr post-transfection by addition of 1 μg/ml tetracycline. Two hours post-induction, transcription was halted by washing the cells once with PBS and replacing the medium with medium containing 5 μg/ml actinomycin D and no tetracycline. Starting 30 min after actinomycin D addition (defined as 0 hr), cells were harvested in TRIzol at the indicated time points to assess the decay rate of HBB-T39 mRNA. The amount of HBB mRNA was normalized to ACTB mRNA. Half-lives were calculated using the two-component mixed exponential decay model y = a × exp(b × t) + c × exp(d × t); data from our experiment were fitted to this model using the Solver function of Microsoft Excel to solve for the decay constants b and d19,26.
Supplementary Material
Acknowledgments
We thank Frank Bennett for helpful discussions. This work was supported by NIH grants R21-NS081448 and R37-GM042699, and by a research grant from the University of Pennsylvania’s Center for Orphan Disease Research and Therapy to A.R.K. We acknowledge assistance from CSHL Shared Resources, which are funded in part by Cancer Center Support Grant 5P30CA045508.
Footnotes
Author contributions
T.T.N., I.A. and A.R.K. designed the experiments. I.A. and F.R. provided critical reagents. T.T.N. performed the research. T.T.N. and A.R.K. wrote the manuscript. All authors approved the manuscript.
Competing Financial Interests
F.R. is an employee of, and A.R.K. is a consultant for, Isis Pharmaceuticals. A.R.K., I.A., and T.T.N. are inventors in a related patent application, PCT/US2014/054151.
References
- 1.Goldmann T, et al. EMBO Mol Med. 2012;4:1186–1199. doi: 10.1002/emmm.201201438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rigo F, Hua Y, Krainer AR, Bennett CF. J Cell Biol. 2012;199:21–25. doi: 10.1083/jcb.201207087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Popp MWL, Maquat LE. Annu Rev Genet. 2013;47:139–165. doi: 10.1146/annurev-genet-111212-133424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keeling KM, et al. PLoS ONE. 2013;8:e60478. doi: 10.1371/journal.pone.0060478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keeling KM, Bedwell DM. Wiley Interdiscip Rev RNA. 2011;2:837–852. doi: 10.1002/wrna.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhuvanagiri M, et al. EMBO Mol Med. 2014;6:1593–1609. doi: 10.15252/emmm.201404461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martin L, et al. Cancer Res. 2014;74:3104–3113. doi: 10.1158/0008-5472.CAN-13-2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ni JZ, et al. Genes Dev. 2007;21:708–718. doi: 10.1101/gad.1525507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karam R, Wilkinson M. RNA Biol. 2012;9:22–26. doi: 10.4161/rna.9.1.18010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jolly LA, Homan CC, Jacob R, Barry S, Gecz J. Hum Mol Genet. 2013 doi: 10.1093/hmg/ddt315. [DOI] [PubMed] [Google Scholar]
- 11.Shibuya T, Tange TO, Sonenberg N, Moore MJ. Nat Struct Mol Biol. 2004;11:346–351. doi: 10.1038/nsmb750. [DOI] [PubMed] [Google Scholar]
- 12.Kole R, Krainer AR, Altman S. Nat Rev Drug Discov. 2012;11:125–140. doi: 10.1038/nrd3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giorgi C, Moore MJ. Semin Cell Dev Biol. 2007;18:186–193. doi: 10.1016/j.semcdb.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 14.Trecartin RF, et al. J Clin Investig. 1981;68:1012–1017. doi: 10.1172/JCI110323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pan Q, et al. Genes Dev. 2006;20:153–158. doi: 10.1101/gad.1382806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gardner LB. Mol Cancer Res. 2010;8:295–308. doi: 10.1158/1541-7786.MCR-09-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Winkler J, Stessl M, Amartey J, Noe CR. ChemMedChem. 2010;5:1344–1352. doi: 10.1002/cmdc.201000156. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Z, Krainer AR. Proc Natl Acad Sci USA. 2007;104:11574–11579. doi: 10.1073/pnas.0704946104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trcek T, Sato H, Singer RH, Maquat LE. Genes & Development. 2013;27:541–551. doi: 10.1101/gad.209635.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keeling KM, Xue X, Gunn G, Bedwell DM. Annu Rev Genomics Hum Genet. 2014;15:371–394. doi: 10.1146/annurev-genom-091212-153527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loughran G, et al. Nucleic Acids Res. 2014;42:8928–8938. doi: 10.1093/nar/gku608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baker BF, et al. J Biol Chem. 1997;272:11994–12000. doi: 10.1074/jbc.272.18.11994. [DOI] [PubMed] [Google Scholar]
- 23.Jeong JY, et al. Appl Environ Microbiol. 2012;78:5440–5443. doi: 10.1128/AEM.00844-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mayeda A, Krainer AR. Methods Mol Biol. 1999;118:315–321. doi: 10.1385/1-59259-676-2:315. [DOI] [PubMed] [Google Scholar]
- 25.Mayeda A, Krainer AR. Methods Mol Biol. 1999;118:309–314. doi: 10.1385/1-59259-676-2:309. [DOI] [PubMed] [Google Scholar]
- 26.Zheng W, Finkel JS, Landers SM, Long RM, Culbertson MR. Genetics. 2008;180:1391–1405. doi: 10.1534/genetics.108.095737. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.