

Abstract
Protein folding is in its early stages largely determined by the protein sequence and complex local interactions between amino acids, resulting in lower energy conformations that provide the context for further folding into the native state. We compiled a comprehensive data set of early folding residues based on pulsed labeling hydrogen deuterium exchange experiments. These early folding residues have corresponding higher backbone rigidity as predicted by DynaMine from sequence, an effect also present when accounting for the secondary structures in the folded protein. We then show that the amino acids involved in early folding events are not more conserved than others, but rather, early folding fragments and the secondary structure elements they are part of show a clear trend toward conserving a rigid backbone. We therefore propose that backbone rigidity is a fundamental physical feature conserved by proteins that can provide important insights into their folding mechanisms and stability.
Introduction
Many proteins have to fold to fulfill their biological function, and it became clear early on in structural biology that the secondary and tertiary structure (fold) of a protein are more conserved in evolution than its primary structure (sequence) (1). Amino acids that are fully conserved during evolution are typically the ones directly involved in protein function (e.g., the catalytic site of an enzyme), whereas the overall fold of the protein can be maintained despite considerable sequence variation (2), although dynamic and allosteric properties put additional restraints on this variation (3, 4). At the beginning of this century a debate surfaced on whether the folding nucleus of a protein might exhibit evolutionary conservation on the sequence level (5), which ended with a rebuttal of the idea (6, 7). Evolutionary conservation of sequence in relation to protein folding is indeed unexpected considering that the protein fold, which allows considerable sequence variation, is intimately connected to the folding process (8, 9). Protein folding is, however, complex; does a protein fold at all, where does folding begin, what folding pathway(s) are followed, and how fast does the protein fold (8, 10, 11, 12, 13, 14)? Although folding in its biologically relevant context occurs in the crowded and complex environment of the cell, it is reasonable to assume that in its initial state the protein behaves as a statistical chain, where a wide range of transient conformations are adopted (15, 16), with preferential local interactions that can quickly lead to short folded fragments with lower free energy (foldons) and finally a stable fold (9). In this framework, the initial foldon(s) form locally and result in an obligate nucleus for folding; they provide the context to initiate or help the formation of a further critical nucleus that turns the free energy profile downhill to fully fold the protein (13). Proteins also tend to fold faster when an increasing fraction of the contacts in the native fold are between residues close to each other in the sequence, indicating that such local interactions steer initial folding events (17). Furthermore, because folded proteins are dynamic and can (partially) unfold and refold, local interactions continue to influence the behavior of the protein chain in terms of preferential conformations, an observation that was already alluded to in early studies of the relationship between sequence and structure (18).
Given the complexity of the folding process, the importance of local interactions in early folding, and the variability in folding pathways even for proteins of the same fold family (13), the question remains which, if any, general folding principles can be identified (19). The evolution of physical molecular characteristics related to folding could provide such insights, especially within a protein family. An obvious candidate is protein dynamics, with indications of conservation (20) and adaptation (21) of dynamics, but with no clear relationship observed between dynamics and folding. Studying this relationship is difficult because residue-level information on the dynamics of proteins is not straightforward to obtain, requiring either involved experiments (22) or computationally expensive and technically challenging molecular dynamics simulations (23). Neither of these is applicable at the moment on large sets of related sequences, although efforts within specific protein families have proven very useful (24). The recently developed DynaMine approach (25, 26), on the other hand, can rapidly predict the local dynamic properties of the backbone from the protein sequence. These predictions are based on per-residue backbone dynamics directly estimated from experimental data for proteins in solution, which incorporate the whole range of protein behavior from disordered to fully folded, including events on a local level (such as flexible loops or helix fraying). Rather than estimating the local dynamic properties of the backbone in a particular context of the protein (i.e., a specific fold), which is possible from experimental NMR data, the linear regression model underlying DynaMine extracts statistical trends, and predicts what an individual protein is capable of in terms of local backbone dynamics from its sequence only.
We focus here on early folding events: the fragments of the protein that fold first are crucial in determining what happens next in the folding pathway, and their identification from sequence can help to understand protein behavior in general. We assembled a comprehensive set of data from literature on early folding residues based on pulsed labeling (and similar) hydrogen-deuterium exchange (HDX) experiments, which identify residues that become protected from solvent early on in the folding process. In these experiments the protein is unfolded by denaturation and then diluted into a solution where it can start to fold. After very short folding times that are defined based on the folding speed of the investigated protein in each individual experiment a very short exchange pulse (usually <20 ms) is used to ensure a very fast equilibrium proton exchange process, but avoid equilibrium conformational exchange processes such as back unfolding (9), and therefore represent a kinetic rather than an equilibrium conformational state for the studied protein. These early folding residues therefore indicate which fragments of the protein adopt particular lower free energy conformations when only local interactions are in play, so resulting in a more defined and rigid backbone and providing the context for further folding (Fig. 1). We show that these early folding residues are indeed much more likely to reside in protein regions with higher predicted local backbone rigidity as computed by DynaMine, whose predictions thus accurately reflect where sequence fragments favor specific conformations due to purely local interactions. An analysis of multiple sequence alignments from HHblits (27) and Jackhmmer (28) then indicates that the amino acids involved in early folding events tend to conserve this tendency toward a rigid backbone in evolution. Finally, the overall predicted backbone rigidity of native secondary structure elements that contain early folding residues tends to be higher than their nonearly folding counterparts, a characteristic that is also conserved in evolution. We therefore propose that early folding fragments of proteins tend to maintain a sequence context in evolution that enables local interactions favoring specific conformations, and show that a resulting increase in the local rigidity of the protein backbone is predicted from sequence by our method.
Figure 1.

Conceptual overview of how the approaches used in this work relate to protein folding.
Materials and Methods
Creation of early folding data sets
The early folding data from NMR- or mass spectroscopy-coupled HDX pulsed labeling or quenched flow experiments are taken from the Start2Fold database (29). The EARLY sets of Start2Fold comprise residues whose amide protons become protected from solvent at a very early stage during the folding process, as determined by methods that address folding starting from the completely unfolded state. Due to the heterogeneity of the relevant methods (pulsed labeling, quenched flow, dead time labeling, and competition-based HDX coupled with NMR or mass spectroscopy), the investigated proteins (folding rates, fold types and sizes) and the measures used to describe folding rates (protection factors, folding rate constants,), generic thresholds for protection or folding rate could not be applied. The residue classifications for the proteins were adopted either from Li and Woodward (30), or from the corresponding original publications. In those cases where good quality measures of residue folding rates were available without a classification, we determined a threshold that selected the first foldon unit(s) for three-state folders (possibly not exceeding 10–20% of their residues) and all residues simultaneously folding for two-state folders. We tried to avoid unnatural thresholds that would place residues with very similar protection rates into different groups.
When assembling the data set for this study, we only selected those EARLY sets of the database where the protection of amides was defined at residue resolution. If multiple measurements were available for the same protein (e.g., horse cytochrome c), the experiment with the best time resolution was selected. We also excluded two proteins due to different reasons: for apoflavodoxin the folding was monitored in a different type of experiment in the presence of very high denaturant concentration, whereas for BLA the measurement was not following the course of folding from the completely unfolded state. In two cases (Escherichia coli RNase HI and horse ferricytochrome c) we also added the INTERMEDIATE residue set as early folding because only a very low fraction of the residues was part of the EARLY set in the database despite a high number of probes applied in the corresponding experiments (E. coli RNase HI and horse ferricytochrome c). At this point the earlyFold data set contained 32 proteins with <90% sequence identity.
We then further filtered this data set for exchange pulse length and excluded two proteins (ubiquitin (31) and β-lactoglobulin (32)) with unusually long exchange pulses (>30 ms) without any justification from the authors. For each of the 30 remaining proteins (Table S1 in the Supporting Material), totaling 3393 residues, the 482 residues that were experimentally determined as early folding were classified as belonging to class F, the remaining 2911 residues as belonging to class N. An additional earlyFoldFragment data set was created based on earlyFold where all residues from class N that are within three sequence positions from F residues in the protein sequence were reclassified as F. An analysis of these data is available in Supporting Materials and Methods and Figs. S1–S5.
Secondary structure data was only used if the sequence of the protein subjected to the HDX experiment exactly matched a sequence in the Protein Data Bank (PDB), resulting in a smaller earlyFoldSs set for the secondary structure analyses, comprising 26 proteins and 2797 residues (Table S1). For each of these proteins the secondary structure elements were determined from the corresponding structure in the PDB using the POLYVIEW server (33), which is based on DSSP (34). Secondary structure elements (SSE) containing at least one early folding residue were separated from the ones that did not and are termed early folding SSEs.
Generation of multiple sequence alignment
Multiple sequence alignments (MSAs) were generated for each sequence in the earlyFold data set using HHblits (27) and Jackhmmer (28) with three iterations and E value threshold of 10−4. All the retrieved homologs have minimum 90% coverage with the query sequence. By using HHfilter, a postprocessing tool provided in the HHblits package, we built two different sets of MSAs by varying the maximum pairwise sequence identity threshold between the collected homologs in each MSA. Sequences in the MSAs HHBLITS_lowSeqId and HMMER_lowSeqId have at most 60% sequence identity, whereas the ones in HHBLITS_highSeqId and HMMER_highSeqId have up to 90% sequence identity.
Sequence and MSA-derived data
The DynaMine predictions were run locally with a recently modified version of the software to correct for bias in the prediction for N- and C-terminal residues (see Supporting Material), which is important in the context of early folding as residues close to the termini might be involved. These original predictions were then normalized by shifting them in a way that the maximum prediction value for each protein is always 1.0. By doing this, the relative differences between the prediction values within each protein remain unchanged, but the residue(s) with the highest tendency toward rigidity have more similar values among the different proteins. The backbone dynamics of the sequences in the earlyFold data set were predicted this way. The (ungapped) sequences in the HHblits and HMMER MSAs were predicted without normalization to preserve the differences within a protein family, and mapped back to the full (gapped) MSA. To avoid bias in the analysis because of limited data, MSAs with <10 aligned proteins, and residues with <10 prediction values for the corresponding column in the MSA were discarded. For the most restrictive HHBLITS_lowSeqId set, this resulted in six proteins being removed (Fadd-DD, GB1, hFGF-1, hisactophilin-1, Protein A B domain, and onconase). Within each MSA, the sequence entropy was calculated according to the classical definition (35), and as reduced sequence entropy, which is defined in (5) by grouping the amino acids in the following classes: (Ala, Val, Leu, Ile, Met, Cys), (Phe, Tyr, Trp, His), (Ser, Thr, Asn, Gln), (Lys, Arg), (Asp, Glu), and (Gly, Pro).
Distribution comparisons and plot generation
Plot generation was done by R (36) through custom Python scripts. In the notched box plots, the colored box shows the interquartile range, the whiskers indicate the maximum value, or the respective quartile value times 1.5 the interquartile range, whichever is less. The notch displays a confidence interval based on the median plus/minus 1.57 times the interquartile range divided by the square root of the number of points. If the notches of two boxes do not overlap, this is strong evidence that their medians differ significantly (37). A solid circle shows the mean of each distribution. The number of data points for each distribution is, for the per-amino acid plots, indicated above the boxes. Distributions of F and N values were also compared using the Wilcoxon rank-sum test in R (38), and for the per-amino acid comparisons only p-values that remained significant after applying the Benjamini-Hochberg multiple hypotheses testing correction were retained (39). Throughout the work we indicate the retained p-values with ∗∗∗ for a highly significant one <0.001, ∗∗ a very significant one <0.01, and ∗ a significant one <0.05. The comparisons of distributions over all amino acids are biased because certain amino acids are more likely to be early folding than others. This bias is strong enough to create significant differences, so we corrected it by subtracting first, for each amino acid type, the median value over the F and N classes from the actual value, and then renormalizing to the expected value range by adding the median value over all amino acids to the actual value. These are indicated by NoB in the plots.
Results
Relation of predicted backbone dynamics to early folding residues
DynaMine predicts per-residue local dynamic properties of a protein backbone from sequence only, and accounts for local interactions by using a 51-residue sequence window where amino acid contributions are position-specific; higher values (toward 1.0) indicate increased rigidity for that residue, lower values more flexibility. Residues involved in early folding events, as determined by HDX experiments, are precisely influenced by local interactions only, and the DynaMine predictions, if accurate, should pick up these residues. We also asserted that the pulsed labeling HDX data is distinct from native exchange HDX data through analysis of the Start2Fold database (Fig. S6).
We therefore compiled the earlyFold data set, which contains information on early folding residues for 30 proteins (see Material and Methods), and predicted their expected local backbone rigidity with DynaMine. We observed that the predictions for early folding residues are higher than those for the remaining residues (Fig. 2, All column), and mostly indicate very high rigidity (values above 0.80) (26). The difference between the distributions is highly significant (see Materials and Methods for more information on the distribution comparison and the information displayed in these figures). Although the DynaMine predictions take the sequence context into account, typical order-inducing amino acids such as Val tend to have higher predicted values (Fig. 2) and are overrepresented as an early folding residue (see Fig. S4), therefore introducing a bias into the overall distribution. When subdividing the set by individual amino acid type, significant differences in distribution remain present for 13 amino acids despite the limited data sample sizes: highly significant for Lys, Met, Ser, and Tyr, very significant for Ala, Asp, Glu, Phe, His, and significant for Asn, Gln, Thr, and Val (indicated at the bottom of each boxplot in Fig. 2; Table 1). Given the nature of HDX experiments, no data is available for Pro, and for Cys, Gly, Ile, Leu, and Trp the median and average of the distribution is higher for the early folding residues, but not significantly so. A bias-corrected overall distribution, where the median value for each amino acid type is taken into account (see Materials and Methods), also confirms that early folding residues have a significantly higher predicted rigidity (Fig. 2, NoB; Table 1). We performed the same analyses on the earlyFoldSs data set, where the data were subdivided by secondary structure elements as observed in the native fold (see Materials and Methods). About 21% of the residues in α-helices are early folding ones, with a similar amount in β-sheets (23%), and the bias-corrected distributions confirm that the trend for higher predicted backbone rigidity is also present within each secondary structure element class (Fig. S7), showing that the higher predictions for early folding residues are not attributable to secondary structure bias. On a per-amino acid level, the number of data points is generally too low to observe significant differences, although the distributions for early folding residues have higher or equal medians compared to other residues. If we assume that it is the whole fragment around the early folding residues that is relevant for folding, and we include the three residues preceding and succeeding the experimentally detected early folding one, the results are very or highly significant for all amino acids except Cys and Trp (Table 1; Fig. S8).
Figure 2.

Boxplots showing the distribution per amino acid residue of the original predicted backbone rigidity divided into normal and early folding classes. The number of amino acids in each distribution is indicated at the top of each graph, whereas the significance of the difference between the distributions is reported under the amino acid three-letter code. To see this figure in color, go online.
Table 1.
Significance of the Difference between the Distributions of DynaMine Predictions for Early Folding and Other Residues
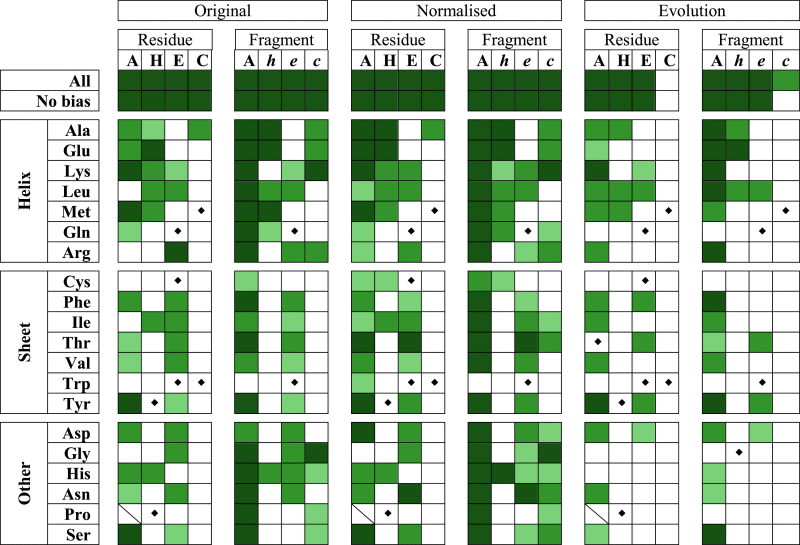 |
The p-values are indicated by dark (<0.001), medium (<0.01), and light green (<0.05). The original and normalized predictions for the individual early folding residues (Residue), the early folding fragment comprising ± 3 residues around early folding ones (Fragment), and the median over the MSA (Evolution) are included. At the column level the early folding residues are assessed as all residues (A), residues within the early folding secondary structure element for respectively helices (H), sheets (E), and coil/other (C), and residues in those secondary structure elements when part of an early folding fragment (respectively h, e, and c). At the row level distributions for all residues (All), bias-corrected values (No bias), and individual amino acids were assessed. The amino acids are grouped by typical helix-forming (top), β-sheet (middle), and other (bottom) residues. Significance was not assessed for five or less data points (♦), and if no data are available (/). To see this table in color, go online.
These findings attest that DynaMine accurately predicts from sequence where purely local amino acid interactions limit the backbone movement when the protein is not yet additionally restrained by long-range native interactions, or in other words the predictions tend to pick up the regions of the protein where conformations with significantly lower free energies exist before folding. However, proteins fold differently and at different speeds, and the maximum value of the DynaMine prediction within each protein in the earlyFold set varies considerably (from 0.88 to 1.03, a 17% change). We tried to compensate for these innate differences in overall flexibility between proteins by equalizing residue(s) with the highest tendency toward rigidity: the original predictions were shifted so that the maximum predicted value for each protein is always 1.0, which leaves the relative differences between the prediction values within each protein unchanged. These normalized predictions result in distributions on a per-amino acid basis that are at least significant for 16 amino acids, with Asp, Ile, and Trp not significant (but with higher medians for early folding residues) (Table 1; Fig. S9). For the fragment-based analysis, the differences are highly significant except for Cys (very significant) and Trp (no significance) (Table 1; Fig. S10). Early folding events have been related to hydrophobic collapse, surface accessibility, and the tendency of the protein toward order; we confirmed that these are not as accurate as DynaMine in detecting early folding residues by testing 22 different hydrophobicity scales, NetSurfP (40) solvent accessibility predictions, s2D secondary structure population prediction (41), and two flavors of the ESpritz order/disorder predictor (42) (see Supporting Material). The protein structure-based DSSP relative solvent accessibility (34) also does not perform on par, whereas the contact S2 parameter (43) gives good results, indicating that early folding residues tend to become the residues with the most and closest backbone interactions in the folded protein (see Supporting Material).
Relation between early folding, predicted backbone dynamics, and secondary structure elements in the folded protein
In early folding fragments purely local interactions permit conformations with lower free energy that have an initiating role in the rest of the folding process. We examined how this relates to the SSE as observed in the experimentally determined native fold: we delineated the α-helices, β-sheets, and other/coil elements in the earlyFoldSs data set (see Materials and Methods), separated these SSEs on the basis of whether they contained early folding residues or not, and analyzed them in relation to the DynaMine predictions. Residues that are part of early folding SSEs have significantly higher predicted values than the ones in the remaining SSEs, especially for sheets and helices, but also for the coil/other class (Table 1; see Fig. S16). More than half of the residues are in early folding SSEs for helices (62%) and sheets (59%), and far fewer in coil/other (22%). Helices and sheets that contain early folding residues also tend to have a higher average rigidity over the SSE (Fig. 3 A), with a more significant difference for sheets, although there are, percentage-wise, slightly more helices that contain early folding residues (46/94 or 49%) than sheets (72/155 or 46%) in our data set. The same trend is found in the coil/other class (Fig. 3 A). SSEs that contain early folding residues also tend to be significantly longer (Fig. 3 B). This trend is especially relevant for helices and sheets, where in general longer SSE length is slightly correlated with higher overall flexibility of the SSE (Fig. S17). On the individual amino acid level (Table 1), there are intriguing differences in the distributions of especially the normalized predicted backbone rigidity (Table 1). In helices, the significantly different distributions include 5 out of 7 residues with high helix propensity (Table 1) (44) along with the helix-indifferent His and Ile residues and the helix-breaking Cys. In sheets these include the 5 out of 7 sheet-favoring residues, with not enough data available for the other two (Table 1) along with the indifferent Leu and Arg and the sheet-breaking Asp, Ser, Gly, Asn, and Lys, whereas no coil favoring residues are covered in the coil/other class. In a similar analysis, residues in the previously defined early folding fragments were considered and assessed per SSE subclass; the conclusions here are similar to those based on early folding SSE, with no significant differences in the distributions for the coil/other classes focused on coil and helix-favoring residues (Normalized/Fragment/h,e,c). Overall, these results are in line with the observation that some SSEs, as experimentally observed in the native fold, are strongly determined by local interactions (18): our analysis shows that especially for early folding SSEs the local sequence context supports specific lower free energy conformations. The DynaMine predictions that pick up this trend are particularly elevated for amino acids that favor the conformation of the SSE formed in the native fold.
Figure 3.

Boxplots showing the distribution of the average predicted backbone rigidity per secondary structure element as observed in the native fold, divided by whether they do or do not contain early folding residues (A) and the distributions of the length of these secondary structure elements (B). The significance of the difference between the distributions is indicated under the secondary structure element class. To see this figure in color, go online.
Evolution of backbone dynamics for early folding residues
The sequence context in a protein determines where local interactions are possible that favor particular conformations, therefore reducing backbone dynamics and initiating folding. When studying early folding in evolutionary terms, it should therefore be more relevant to study where related proteins tend to adopt specific conformations based on local interactions, instead of focusing on the evolution of individual amino acids. To investigate this, we generated MSAs for all sequences in the earlyFold data set (see Materials and Methods), and analyzed the distributions of the original DynaMine predictions per MSA column; no normalization was performed because the comparisons are within a protein family. This approach is possible because the DynaMine predictions require a single sequence and do not depend on evolutionary information. The median value of the DynaMine prediction per MSA column then indicates how rigid the residue in this sequence position remains in evolution, whereas the root mean-square deviation (RMSD) of the values per column with respect to the target sequence value indicates the variation of the rigidity in evolution. We also calculated the sequence entropy and reduced entropy for each MSA column. In the following discussion we focus on the HHBLITS_lowSeqId MSA data set, where the maximum sequence identity to the original sequence is 60% and the minimum coverage is set to be 90%, because the conclusions drawn from the other alignments are similar.
The first conclusion is that the median backbone rigidity is not correlated to (reduced) entropy, with only a very weak correlation between higher entropy and increased flexibility (Fig. S18); even fully conserved residues show a large spread in median backbone rigidity. The variation of the rigidity is somewhat correlated with entropy, with higher entropy related to a slightly higher RMSD (Fig. S18). These correlations are similar when considering only residues within each secondary structure element class, and for each individual protein MSA. More conserved residues therefore tend to have more similar backbone rigidity (lower RMSD), but there is no relation between the conservation of a residue and its evolutionary tendency toward backbone rigidity or flexibility. Note that this study concerns proteins that fold, and that these results will likely differ for intrinsically disordered regions of proteins (45, 46).
The second conclusion is that early folding residues are more conserved, but a lot less so when accounting for the bias in their amino acid composition: early folding residues are more conserved than nonearly folding residues, especially when using reduced entropy (Fig. S19, All), but this effect is greatly reduced when correcting for amino acid bias (Fig. S19, NoB). The underlying cause is that residues such as Cys, Trp, and Phe, which tend to be more conserved (Fig. S19), are also overrepresented in the early folding set. In addition, very few significant differences are observed on the individual amino acid level and on the SSE level. Our analysis therefore indicates that early folding residues favor amino acid types that are better conserved, but there is very little preferential conservation within each type.
The third conclusion is that early folding residues tend to conserve their higher backbone rigidity in evolution. The median of the DynaMine predictions is significantly higher for these residues, also after correcting amino acid bias (Fig. 4; Table 1). This trend is also observed after correcting amino acid bias for the helix and sheet SSE subclasses (Fig. S20) and in all four alignment approaches, so the effect cannot be attributed to the higher occurrence of early folding residues in SSEs or to an artifact of the alignment approaches. On a per-amino acid level, the distribution difference is significant for 13 amino acids (Table 1), a trend also observed in the three other alignment approaches. The overall effect is even stronger for the residues in the early folding fragments, where 15 of the amino acids have significantly higher distributions of the median predicted backbone rigidity (Table 1).
Figure 4.

Boxplots showing the distribution of the median of the predicted backbone rigidity values per MSA column in the HHBLITS_lowSeqId set, with the data divided into normal and early folding classes, by the amino acid as observed in the target sequence for that MSA column. The number of MSA columns in each distribution is indicated at the top of each graph, whereas the significance of the difference between the distributions is reported under the amino acid three-letter code. To see this figure in color, go online.
Finally, residues in the early folding SSEs except the coil/other class maintain significantly higher predicted backbone rigidity in evolution, also after correction for amino acid bias (Table 1; Fig. S21). Again, helix-promoting residues appear to be more rigid in helices (Ala, Leu, Met), and sheet-promoting residues in sheets (Phe, Tyr, Thr). This trend is also present for helix and sheet when considering the average of the median predicted backbone rigidity for all residues in helices and sheets (Fig. S22) and within the helix and sheet SSE classes for the early folding fragments (Table 1). SSEs involved in early folding therefore tend to maintain their higher overall rigidity in evolution, and this higher rigidity seems to be especially significant for the residues with high propensity for the particular SSE.
Case studies
Two well-studied proteins illustrate the dynamics and evolutionary properties of early folding residues. For sperm whale apo-myoglobin (apo-Mb) and horse ferricytochrome c (cyt c) recent folding experiments were carried out with sophisticated techniques that have very good time resolution (47, 48). In apo-Mb (Fig. 5 A) early folding residues occur within the N-terminal helix and the two C-terminal helices. They correspond well to local maxima of the DynaMine backbone rigidity predictions (red line), both for the target sequence itself and in terms of its preservation in homologous sequences, which show high sequence entropy (darker blue shading means more conserved). In contrast, no early folding residues were reported within the N-terminal helix of the homologous horse apo-Mb, which has remarkably lower DynaMine predictions for this region than whale apo-Mb (Fig. S20). Ultrafast HDX experiments on cyt c detected that in the initial stages of folding only residues within the C-terminal half of the protein become protected from solvent (Fig. 5 B), whereas the N-terminal half remains highly unstructured (47). The only exception was His-26, which is the only potential heme ligand in the measured H33N mutant of cyt c. Its tendency to interact with the heme iron in the denatured state likely explains its well-protected nature (49). Interestingly, this His-26 is predicted as having one of the lowest rigidity values within the protein, indicating that it is not a true early folding residue. The real early folding residues in the C-terminal part correspond to helices or helix-neighboring residues of coils and have very high predicted backbone rigidity, which is again well preserved in evolution despite a general lack of sequence conservation in this region.
Figure 5.

The structural, dynamics and evolutionary properties of sperm whale apo-Mb (A) and horse ferricytochrome c (B) are shown as a function of their residue positions on the left, whereas the corresponding three-dimensional structures are on the right (PDB: 1MBC and 1HRC, respectively; structures are only available for the proteins together with their heme cofactors (black), and for cyt c the wild-type protein is depicted). In both panels, early folding residues are marked with green shading on the graphs and with green stick representations within the three-dimensional structures, with their residue positions and types indicated. A red line depicts the per-residue DynaMine-predicted backbone rigidity of the protein. The medians of predicted values in the corresponding HHBLITS_lowSeqId alignment columns are shown as a black line, whereas their first and third quartiles of the distribution are marked in dark gray, their minima and maxima with lighter gray. The blue shading between the dark gray quartile lines represents the sequence entropy for each alignment position, with darker blue indicating lower entropy (high evolutionary conservation). The secondary structure elements assigned by the POLYVIEW server are also provided, with early folding helices shown as green cylinders and others as gray cylinders. To see this figure in color, go online.
Discussion
The view of proteins as dynamic rather than static objects is steadily gaining recognition, and is especially relevant to explain processes such as folding, allostery, and aggregation, which are related to how and where the protein can change. In this context, it is not only important to understand the precise interactions between amino acid residues in the native state, but also what the protein might be capable of in terms of dynamics and other conformations. Here, we show an excellent correlation between early folding residues, which indicate where in the protein specific lower free energy conformations are possible through local interactions only, and segments of the protein chain with reduced backbone dynamics as determined by DynaMine from sequence. The importance of local interactions in determining protein conformation has been pointed out decades ago (50, 51), especially in relation to formation and prediction of α-helices (52), although secondary structure predictions from sequence evidence that there is a limit to how well conformation can be predicted from local information (53). The final native fold, after all, provides ample context to other residues, and helices or sheets can be formed even if local sequence-based interactions are not favorable (53). These local sequence-based interactions, however, are crucial for the formation of early folding fragments, and likely remain relevant in the dynamic protein: the interactions between side chains of residues that are closely connected by covalent bonds are always relevant. This is corroborated by our observation that SSEs that contain early folding residues have a higher overall tendency toward rigidity. We also show that these local interactions are more complex than dictated by hydrophobicity, which does not give a signal for early folding. Although sequence-based accessible surface area predictors and sophisticated disorder predictors like ESpritz do give good results (42), DynaMine outperforms them. The reason why it is better in detecting early folding residues is, in our view, because it uses a simple linear regression model that was not trained on structural information, but rather on estimations of per-residue behavior from experimental data (NMR chemical shifts) directly measured for the protein in solution (42, 54, 55, 56). The predictions therefore cover movements from fast (ps) to slower (high μs) timescales, between which they do not distinguish, include folded to intrinsically disordered proteins, and portray the statistical trends in the data instead of attempting to predict the highly context-dependent native fold. Interestingly, the contact S2 parameter, which estimates the backbone dynamics from structure, is also a very good indicator for early folding residues, implying that the backbone of these residues tends to be the most closely surrounded by heavy atoms in the folded protein.
In an evolutionary context the DynaMine predictions, which require a single sequence without any additional information, enable a different view of evolution: one that is based on conservation of a physical characteristic of the overall sequence, not on individual amino acids. Based on alignments from two state-of-the art methods, we observe that there is no clear correlation, in our data set, between the sequence-determined rigidity of residues and their sequence conservation. This again points out the complexity of the local interactions in the sequence, which cannot be captured by only the amino acid, but instead requires the local sequence context. We do observe that early folding residues are slightly more conserved overall, but that the main reason for this is an amino acid bias: the amino acid types that are more commonly detected as early folding also tend to be the ones that are more conserved in evolution. This observation in part contradicts earlier conclusions that residues important for folding are not preferentially conserved in evolution (6), which disputed theoretical predictions (57, 58) and a statistical study (5). However, there are fundamental differences between our approach and the one adopted at the time. First, we are using early folding data from pulsed labeling HDX experiments, which are directly related to early folding events in the wild-type protein, and not Φ values, which give information about the folding transition state based on the effect of amino acid mutations in the wild-type protein. These Φ values are widely used and are especially useful when values close to 0 and values close to 1 are obtained, which indicate that the mutated residue and surrounding region are respectively unstructured or native-like in the folding transition state. The experimental design and choice of mutation is however crucial, and if not performed correctly folding pathways might be affected, and overall protein stability can bias the results (59). It was shown for the chymotrypsin inhibitor 2 (CI2) protein that folding pathways as determined by Φ values do not relate well to native exchange HDX data (60). Because no pulsed labeling HDX experiments are available for CI2, we could not establish whether early folding residues relate better to these Φ values. We do show however that the pulsed labeling HDX experiments generally result in distinctly different residue sets compared to native exchange HDX data. A statistical comparison between Φ values and early folding residues proved to be very difficult because a certain level of interpretation is required for Φ values, and not many reliable data points are available. There are only a few proteins for which both early folding HDX data and Φ values are available (ACBP, protein A B domain, LB1, GB1, ubiquitin), but these are all two-state folders. For these, folding HDX data are mostly uniform for all residues within the secondary structure elements of the protein chain and hence do not provide a good basis for comparison. A second important advantage in our study is the much reduced sampling bias, as in HDX experiments most protein residues are observed, whereas for Φ values the residue sampling is highly dependent on which mutations were scanned (6). Third, we were able to look at the signal on a per-amino acid type level, which shows that increased conservation of folding residues is mainly an effect of their composition bias. The main confounding factors for the pulsed labeling HDX experiments themselves are first the length of the labeling pulse, which has to be very short to avoid equilibrium processes such as back unfolding, and which we could address by retaining only experiments where this is the case. More difficult to address is the heterogeneity in the experimental conditions, which could change folding by, for example, (de-)protonation of a residue compared to physiological conditions. In our data set, experiments were performed in the pH3.0–8.0 range.
We show here that early folding residues do tend to preserve their tendency toward forming specific conformations in evolution, as encompassed by higher rigidity predictions. Even with our relatively limited data set this effect is visible on the individual residue level for 13 out of the 20 amino acids, and includes 6 out of 7 helix-favoring residues (except for Gln), four sheet-favoring residues, and the coil-favoring Asp, Ser, and Asn (Fig. 4). An outlier in all our results is Trp, which barely shows differences in distribution at the individual sequence or at the evolution level from any approach, even though it is one of the more common early folding residues. This indicates that Trp is not much affected by sequence context, even if it strongly affects other residues itself and might trigger folding. Also striking is that the increased predicted backbone rigidity is especially significant for the residues that strongly prefer the SSE as observed in the native fold (see Table 1). In other words, the typical SSE favoring residues determine the type of specific conformation formed in regions with stabilizing local interactions. This is confirmed by the subset in which we delineated SSEs from the native fold, and divided them into ones that contain early folding residues or not. If we use these early folding SSEs as a proxy for foldons, it is the overall rigidity of the foldon that is important compared to the rest of the protein. Within a foldon, it is then especially the residues with a high propensity for the required SSE that are predicted to be more rigid. Both of these features seem to be preserved in evolution, indicating that these early folding regions have to be maintained as regions that can locally preorganize (microscopically) to steer the rest of the macroscopic folding process (9). Interestingly, the early folding SSEs also tend to be longer. Foldons are expected to be around 20 residues long (9), with preorganized secondary structure an important determinant of protein folding (61), and the length of SSEs in the native fold related to folding speed (62). This was also alluded to in earlier studies, where sequence segments that strongly determine secondary structure were suggested as nuclei around which folding occurs (18).
This study is limited by the availability of high-quality protein folding data, and our results are therefore biased on the full sequence level toward monomeric proteins that fold easily, form a single stable conformation, and have been (extensively) studied with experimental approaches. However, the data we use relate to most residues in these proteins, so there is no bias on an individual residue level, and because early folding regions are determined by local interactions, protein length should not play a direct role in their formation. Interestingly, the difference between the per-amino acid distributions becomes highly to very significant for 19 amino acid types when assessing early folding fragments around the specific early folding residues. This could be related to the limited data size, or alternatively implies that neighboring residues are equally important in shielding a residue’s protein backbone in early folding events. Finally, the view we present here of early folding events has only become possible due to a rigorous meta-study of literature data in combination with novel prediction methodology. We hope that it will contribute to especially the understanding of transient events leading to the formation of foldons on fast (sub-ms) timescales; as indicated by Sosnick and Barrick (16), the early, weakly populated species going up the (initial) barrier leading to the first stably collapsed species are particularly hard to characterize.
Author Contributions
R.P. and W.F.V. designed the research, analyzed the data, and wrote the article; D.R., E.C., and W.F.V. contributed analytic tools.
Acknowledgments
We thank Shoshana Wodak and Mainak Guharoy for their useful comments on the manuscript.
W.F.V. acknowledges the Brussels Institute for Research and Innovation (Innoviris) grant BB2B 2010-1-12; E.C. is a postdoctoral researcher with the Belgian Fonds de la Recherche Scientifique (F.R.S.-FNRS).
Editor: Michele Vendruscolo.
Footnotes
Supporting Materials and Methods, Supporting Results, twenty-three figures, and one table are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(15)04812-2.
Supporting Citations
References (63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97) appear in the Supporting Material.
Supporting Material
References
- 1.Bajaj M., Blundell T. Evolution and the tertiary structure of proteins. Annu. Rev. Biophys. Bioeng. 1984;13:453–492. doi: 10.1146/annurev.bb.13.060184.002321. [DOI] [PubMed] [Google Scholar]
- 2.Luheshi L.M., Crowther D.C., Dobson C.M. Protein misfolding and disease: from the test tube to the organism. Curr. Opin. Chem. Biol. 2008;12:25–31. doi: 10.1016/j.cbpa.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 3.Law A.B., Fuentes E.J., Lee A.L. Conservation of side-chain dynamics within a protein family. J. Am. Chem. Soc. 2009;131:6322–6323. doi: 10.1021/ja809915a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Süel G.M., Lockless S.W., Ranganathan R. Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nat. Struct. Biol. 2003;10:59–69. doi: 10.1038/nsb881. [DOI] [PubMed] [Google Scholar]
- 5.Mirny L., Shakhnovich E. Evolutionary conservation of the folding nucleus. J. Mol. Biol. 2001;308:123–129. doi: 10.1006/jmbi.2001.4602. [DOI] [PubMed] [Google Scholar]
- 6.Larson S.M., Ruczinski I., Plaxco K.W. Residues participating in the protein folding nucleus do not exhibit preferential evolutionary conservation. J. Mol. Biol. 2002;316:225–233. doi: 10.1006/jmbi.2001.5344. [DOI] [PubMed] [Google Scholar]
- 7.Tseng Y.Y., Liang J. Are residues in a protein folding nucleus evolutionarily conserved? J. Mol. Biol. 2004;335:869–880. doi: 10.1016/j.jmb.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 8.Dill K.A., MacCallum J.L. The protein-folding problem, 50 years on. Science. 2012;338:1042–1046. doi: 10.1126/science.1219021. [DOI] [PubMed] [Google Scholar]
- 9.Englander S.W., Mayne L. The nature of protein folding pathways. Proc. Natl. Acad. Sci. USA. 2014;111:15873–15880. doi: 10.1073/pnas.1411798111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Sancho D., Doshi U., Muñoz V. Protein folding rates and stability: how much is there beyond size? J. Am. Chem. Soc. 2009;131:2074–2075. doi: 10.1021/ja808843h. [DOI] [PubMed] [Google Scholar]
- 11.Kubelka J., Hofrichter J., Eaton W.A. The protein folding ‘speed limit’. Curr. Opin. Struct. Biol. 2004;14:76–88. doi: 10.1016/j.sbi.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 12.Lindberg M.O., Oliveberg M. Malleability of protein folding pathways: a simple reason for complex behaviour. Curr. Opin. Struct. Biol. 2007;17:21–29. doi: 10.1016/j.sbi.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 13.Nickson A.A., Wensley B.G., Clarke J. Take home lessons from studies of related proteins. Curr. Opin. Struct. Biol. 2013;23:66–74. doi: 10.1016/j.sbi.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rollins G.C., Dill K.A. General mechanism of two-state protein folding kinetics. J. Am. Chem. Soc. 2014;136:11420–11427. doi: 10.1021/ja5049434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schuler B., Hofmann H. Single-molecule spectroscopy of protein folding dynamics--expanding scope and timescales. Curr. Opin. Struct. Biol. 2013;23:36–47. doi: 10.1016/j.sbi.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 16.Sosnick T.R., Barrick D. The folding of single domain proteins--have we reached a consensus? Curr. Opin. Struct. Biol. 2011;21:12–24. doi: 10.1016/j.sbi.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Plaxco K.W., Simons K.T., Baker D. Contact order, transition state placement and the refolding rates of single domain proteins. J. Mol. Biol. 1998;277:985–994. doi: 10.1006/jmbi.1998.1645. [DOI] [PubMed] [Google Scholar]
- 18.Rooman M.J., Wodak S.J. Extracting information on folding from the amino acid sequence: consensus regions with preferred conformation in homologous proteins. Biochemistry. 1992;31:10239–10249. doi: 10.1021/bi00157a010. [DOI] [PubMed] [Google Scholar]
- 19.Daggett V., Fersht A.R. Is there a unifying mechanism for protein folding? Trends Biochem. Sci. 2003;28:18–25. doi: 10.1016/s0968-0004(02)00012-9. [DOI] [PubMed] [Google Scholar]
- 20.Gagné D., Charest L.-A., Doucet N. Conservation of flexible residue clusters among structural and functional enzyme homologues. J. Biol. Chem. 2012;287:44289–44300. doi: 10.1074/jbc.M112.394866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stafford K.A., Robustelli P., Palmer A.G., 3rd Thermal adaptation of conformational dynamics in ribonuclease H. PLOS Comput. Biol. 2013;9:e1003218. doi: 10.1371/journal.pcbi.1003218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mittermaier A.K., Kay L.E. Observing biological dynamics at atomic resolution using NMR. Trends Biochem. Sci. 2009;34:601–611. doi: 10.1016/j.tibs.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 23.Shaw D.E., Maragakis P., Wriggers W. Atomic-level characterization of the structural dynamics of proteins. Science. 2010;330:341–346. doi: 10.1126/science.1187409. [DOI] [PubMed] [Google Scholar]
- 24.Benson N.C., Daggett V. Dynameomics: large-scale assessment of native protein flexibility. Protein Sci. 2008;17:2038–2050. doi: 10.1110/ps.037473.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cilia E., Pancsa R., Vranken W.F. From protein sequence to dynamics and disorder with DynaMine. Nat. Commun. 2013;4:2741. doi: 10.1038/ncomms3741. [DOI] [PubMed] [Google Scholar]
- 26.Cilia E., Pancsa R., Vranken W.F. The DynaMine webserver: predicting protein dynamics from sequence. Nucleic Acids Res. 2014;42:W264–W270. doi: 10.1093/nar/gku270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Remmert M., Biegert A., Söding J. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods. 2012;9:173–175. doi: 10.1038/nmeth.1818. [DOI] [PubMed] [Google Scholar]
- 28.Finn R.D., Clements J., Eddy S.R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pancsa R., Varadi M., Vranken W.F. Start2Fold: a database of hydrogen/deuterium exchange data on protein folding and stability. Nucleic Acids Res. 2015 doi: 10.1093/nar/gkv1185. Published online November 17, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li R., Woodward C. The hydrogen exchange core and protein folding. Protein Sci. 1999;8:1571–1590. doi: 10.1110/ps.8.8.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Briggs M.S., Roder H. Early hydrogen-bonding events in the folding reaction of ubiquitin. Proc. Natl. Acad. Sci. USA. 1992;89:2017–2021. doi: 10.1073/pnas.89.6.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Forge V., Hoshino M., Goto Y. Is folding of beta-lactoglobulin non-hierarchic? Intermediate with native-like beta-sheet and non-native alpha-helix. J. Mol. Biol. 2000;296:1039–1051. doi: 10.1006/jmbi.1999.3515. [DOI] [PubMed] [Google Scholar]
- 33.Porollo A.A., Adamczak R., Meller J. POLYVIEW: a flexible visualization tool for structural and functional annotations of proteins. Bioinformatics. 2004;20:2460–2462. doi: 10.1093/bioinformatics/bth248. [DOI] [PubMed] [Google Scholar]
- 34.Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 35.Shenkin P.S., Erman B., Mastrandrea L.D. Information-theoretical entropy as a measure of sequence variability. Proteins. 1991;11:297–313. doi: 10.1002/prot.340110408. [DOI] [PubMed] [Google Scholar]
- 36.Team R.D.C. R Foundation for Statistical Computing; Vienna, Austria: 2008. R: A Language and Environment for Statistical Computing. [Google Scholar]
- 37.Chambers J.M. Chapman and Hall/CRC; 1983. Graphical Methods for Data Analysis. [Google Scholar]
- 38.Mann H.B., Whitney D.R. On a test of whether one of two random variables is stochastically larger than the other. Ann. Math. Stat. 1947;18:50–60. [Google Scholar]
- 39.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statist. Soc. Ser. B. 1995;57:289–300. [Google Scholar]
- 40.Petersen B., Petersen T.N., Lundegaard C. A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct. Biol. 2009;9:51. doi: 10.1186/1472-6807-9-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sormanni P., Camilloni C., Vendruscolo M. The s2D method: simultaneous sequence-based prediction of the statistical populations of ordered and disordered regions in proteins. J. Mol. Biol. 2015;427:982–996. doi: 10.1016/j.jmb.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 42.Walsh I., Martin A.J.M., Tosatto S.C.E. ESpritz: accurate and fast prediction of protein disorder. Bioinformatics. 2012;28:503–509. doi: 10.1093/bioinformatics/btr682. [DOI] [PubMed] [Google Scholar]
- 43.Zhang F., Brüschweiler R. Contact model for the prediction of NMR N-H order parameters in globular proteins. J. Am. Chem. Soc. 2002;124:12654–12655. doi: 10.1021/ja027847a. [DOI] [PubMed] [Google Scholar]
- 44.Costantini S., Colonna G., Facchiano A.M. Amino acid propensities for secondary structures are influenced by the protein structural class. Biochem. Biophys. Res. Commun. 2006;342:441–451. doi: 10.1016/j.bbrc.2006.01.159. [DOI] [PubMed] [Google Scholar]
- 45.Brown C.J., Takayama S., Dunker A.K. Evolutionary rate heterogeneity in proteins with long disordered regions. J. Mol. Evol. 2002;55:104–110. doi: 10.1007/s00239-001-2309-6. [DOI] [PubMed] [Google Scholar]
- 46.Radivojac P., Obradović Z., Dunker A.K. Improving sequence alignments for intrinsically disordered proteins. Pac. Symp. Biocomput. 2002:589–600. [PubMed] [Google Scholar]
- 47.Fazelinia H., Xu M., Roder H. Ultrafast hydrogen exchange reveals specific structural events during the initial stages of folding of cytochrome c. J. Am. Chem. Soc. 2014;136:733–740. doi: 10.1021/ja410437d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Uzawa T., Nishimura C., Wright P.E. Hierarchical folding mechanism of apomyoglobin revealed by ultra-fast H/D exchange coupled with 2D NMR. Proc. Natl. Acad. Sci. USA. 2008;105:13859–13864. doi: 10.1073/pnas.0804033105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Colón W., Wakem L.P., Roder H. Identification of the predominant non-native histidine ligand in unfolded cytochrome c. Biochemistry. 1997;36:12535–12541. doi: 10.1021/bi971697c. [DOI] [PubMed] [Google Scholar]
- 50.Rooman M.J., Rodriguez J., Wodak S.J. Relations between protein sequence and structure and their significance. J. Mol. Biol. 1990;213:337–350. doi: 10.1016/S0022-2836(05)80195-0. [DOI] [PubMed] [Google Scholar]
- 51.Rooman M.J., Wodak S.J. Identification of predictive sequence motifs limited by protein structure data base size. Nature. 1988;335:45–49. doi: 10.1038/335045a0. [DOI] [PubMed] [Google Scholar]
- 52.Compiani M., Fariselli P., Casadio R. An entropy criterion to detect minimally frustrated intermediates in native proteins. Proc. Natl. Acad. Sci. USA. 1998;95:9290–9294. doi: 10.1073/pnas.95.16.9290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rost B. Review: protein secondary structure prediction continues to rise. J. Struct. Biol. 2001;134:204–218. doi: 10.1006/jsbi.2001.4336. [DOI] [PubMed] [Google Scholar]
- 54.Berjanskii M.V., Wishart D.S. A simple method to predict protein flexibility using secondary chemical shifts. J. Am. Chem. Soc. 2005;127:14970–14971. doi: 10.1021/ja054842f. [DOI] [PubMed] [Google Scholar]
- 55.Berjanskii M.V., Wishart D.S. The RCI server: rapid and accurate calculation of protein flexibility using chemical shifts. Nucleic Acids Res. 2007;35:W531–W537. doi: 10.1093/nar/gkm328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Berjanskii M.V., Wishart D.S. Application of the random coil index to studying protein flexibility. J. Biomol. NMR. 2008;40:31–48. doi: 10.1007/s10858-007-9208-0. [DOI] [PubMed] [Google Scholar]
- 57.Mirny L.A., Abkevich V.I., Shakhnovich E.I. How evolution makes proteins fold quickly. Proc. Natl. Acad. Sci. USA. 1998;95:4976–4981. doi: 10.1073/pnas.95.9.4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shakhnovich E., Abkevich V., Ptitsyn O. Conserved residues and the mechanism of protein folding. Nature. 1996;379:96–98. doi: 10.1038/379096a0. [DOI] [PubMed] [Google Scholar]
- 59.Naganathan A.N., Muñoz V. Insights into protein folding mechanisms from large scale analysis of mutational effects. Proc. Natl. Acad. Sci. USA. 2010;107:8611–8616. doi: 10.1073/pnas.1000988107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Best R.B., Vendruscolo M. Structural interpretation of hydrogen exchange protection factors in proteins: characterization of the native state fluctuations of CI2. Structure. 2006;14:97–106. doi: 10.1016/j.str.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 61.Myers J.K., Oas T.G. Preorganized secondary structure as an important determinant of fast protein folding. Nat. Struct. Biol. 2001;8:552–558. doi: 10.1038/88626. [DOI] [PubMed] [Google Scholar]
- 62.Huang J.-T., Cheng J.-P., Chen H. Secondary structure length as a determinant of folding rate of proteins with two- and three-state kinetics. Proteins. 2007;67:12–17. doi: 10.1002/prot.21282. [DOI] [PubMed] [Google Scholar]
- 63.Agashe V.R., Shastry M.C., Udgaonkar J.B. Initial hydrophobic collapse in the folding of barstar. Nature. 1995;377:754–757. doi: 10.1038/377754a0. [DOI] [PubMed] [Google Scholar]
- 64.Arrington C.B., Robertson A.D. Microsecond protein folding kinetics from native-state hydrogen exchange. Biochemistry. 1997;36:8686–8691. doi: 10.1021/bi970872m. [DOI] [PubMed] [Google Scholar]
- 65.Bai Y., Karimi A., Wright P.E. Absence of a stable intermediate on the folding pathway of protein A. Protein Sci. 1997;6:1449–1457. doi: 10.1002/pro.5560060709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bull H.B., Breese K. Surface tension of amino acid solutions: a hydrophobicity scale of the amino acid residues. Arch. Biochem. Biophys. 1974;161:665–670. doi: 10.1016/0003-9861(74)90352-x. [DOI] [PubMed] [Google Scholar]
- 67.Choe S.E., Matsudaira P.T., Shakhnovich E.I. Folding kinetics of villin 14T, a protein domain with a central beta-sheet and two hydrophobic cores. Biochemistry. 1998;37:14508–14518. doi: 10.1021/bi980889k. [DOI] [PubMed] [Google Scholar]
- 68.Di Paolo A., Balbeur D., Matagne A. Rapid collapse into a molten globule is followed by simple two-state kinetics in the folding of lysozyme from bacteriophage λ. Biochemistry. 2010;49:8646–8657. doi: 10.1021/bi101126f. [DOI] [PubMed] [Google Scholar]
- 69.Greene L.H., Li H., Wilson K. Folding of an all-helical Greek-key protein monitored by quenched-flow hydrogen-deuterium exchange and NMR spectroscopy. Eur. Biophys. J. 2012;41:41–51. doi: 10.1007/s00249-011-0756-6. [DOI] [PubMed] [Google Scholar]
- 70.Hooke S.D., Radford S.E., Dobson C.M. The refolding of human lysozyme: a comparison with the structurally homologous hen lysozyme. Biochemistry. 1994;33:5867–5876. doi: 10.1021/bi00185a026. [DOI] [PubMed] [Google Scholar]
- 71.Hsieh H.C., Kumar T.K., Yu C. Refolding of a small all beta-sheet protein proceeds with accumulation of kinetic intermediates. Arch. Biochem. Biophys. 2006;447:147–154. doi: 10.1016/j.abb.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 72.Hu W., Walters B.T., Englander S.W. Stepwise protein folding at near amino acid resolution by hydrogen exchange and mass spectrometry. Proc. Natl. Acad. Sci. USA. 2013;110:7684–7689. doi: 10.1073/pnas.1305887110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jones B.E., Matthews C.R. Early intermediates in the folding of dihydrofolate reductase from Escherichia coli detected by hydrogen exchange and NMR. Protein Sci. 1995;4:167–177. doi: 10.1002/pro.5560040204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kato H., Vu N.-D., Bai Y. The folding pathway of T4 lysozyme: an on-pathway hidden folding intermediate. J. Mol. Biol. 2007;365:881–891. doi: 10.1016/j.jmb.2006.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kern G., Handel T., Marqusee S. Characterization of a folding intermediate from HIV-1 ribonuclease H. Protein Sci. 1998;7:2164–2174. doi: 10.1002/pro.5560071014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Koide S., Dyson H.J., Wright P.E. Characterization of a folding intermediate of apoplastocyanin trapped by proline isomerization. Biochemistry. 1993;32:12299–12310. doi: 10.1021/bi00097a005. [DOI] [PubMed] [Google Scholar]
- 77.Kuszewski J., Clore G.M., Gronenborn A.M. Fast folding of a prototypic polypeptide: the immunoglobulin binding domain of streptococcal protein G. Protein Sci. 1994;3:1945–1952. doi: 10.1002/pro.5560031106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu C., Gaspar J.A., Wong H.J., Meiering E.M. Conserved and nonconserved features of the folding pathway of hisactophilin, a beta-trefoil protein. Protein Sci. 2002;11:669–679. doi: 10.1110/ps.31702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Miranker A., Radford S.E., Dobson C.M. Demonstration by NMR of folding domains in lysozyme. Nature. 1991;349:633–636. doi: 10.1038/349633a0. [DOI] [PubMed] [Google Scholar]
- 80.Morozova-Roche L.A., Jones J.A., Dobson C.M. Independent nucleation and heterogeneous assembly of structure during folding of equine lysozyme. J. Mol. Biol. 1999;289:1055–1073. doi: 10.1006/jmbi.1999.2741. [DOI] [PubMed] [Google Scholar]
- 81.Mullins L.S., Pace C.N., Raushel F.M. Investigation of ribonuclease T1 folding intermediates by hydrogen-deuterium amide exchange-two-dimensional NMR spectroscopy. Biochemistry. 1993;32:6152–6156. doi: 10.1021/bi00075a006. [DOI] [PubMed] [Google Scholar]
- 82.Pan J., Han J., Konermann L. Characterizing short-lived protein folding intermediates by top-down hydrogen exchange mass spectrometry. Anal. Chem. 2010;82:8591–8597. doi: 10.1021/ac101679j. [DOI] [PubMed] [Google Scholar]
- 83.Parker M.J., Dempsey C.E., Clarke A.R. Acquisition of native beta-strand topology during the rapid collapse phase of protein folding. Biochemistry. 1997;36:13396–13405. doi: 10.1021/bi971294c. [DOI] [PubMed] [Google Scholar]
- 84.Radford S.E., Dobson C.M., Evans P.A. The folding of hen lysozyme involves partially structured intermediates and multiple pathways. Nature. 1992;358:302–307. doi: 10.1038/358302a0. [DOI] [PubMed] [Google Scholar]
- 85.Roder H., Wüthrich K. Protein folding kinetics by combined use of rapid mixing techniques and NMR observation of individual amide protons. Proteins. 1986;1:34–42. doi: 10.1002/prot.340010107. [DOI] [PubMed] [Google Scholar]
- 86.Samuel D., Kumar T.K., Yu C. Structural events during the refolding of an all beta-sheet protein. J. Biol. Chem. 2001;276:4134–4141. doi: 10.1074/jbc.M005921200. [DOI] [PubMed] [Google Scholar]
- 87.Schulenburg C., Löw C., Arnold U. The folding pathway of onconase is directed by a conserved intermediate. Biochemistry. 2009;48:8449–8457. doi: 10.1021/bi900596j. [DOI] [PubMed] [Google Scholar]
- 88.Silow M., Oliveberg M. Transient aggregates in protein folding are easily mistaken for folding intermediates. Proc. Natl. Acad. Sci. USA. 1997;94:6084–6086. doi: 10.1073/pnas.94.12.6084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sivaraman T., Kumar T.K., Yu C. Events in the kinetic folding pathway of a small, all beta-sheet protein. J. Biol. Chem. 1998;273:10181–10189. doi: 10.1074/jbc.273.17.10181. [DOI] [PubMed] [Google Scholar]
- 90.Teilum K., Kragelund B.B., Poulsen F.M. Formation of hydrogen bonds precedes the rate-limiting formation of persistent structure in the folding of ACBP. J. Mol. Biol. 2000;301:1307–1314. doi: 10.1006/jmbi.2000.4003. [DOI] [PubMed] [Google Scholar]
- 91.Udgaonkar J.B. Polypeptide chain collapse and protein folding. Arch. Biochem. Biophys. 2013;531:24–33. doi: 10.1016/j.abb.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 92.Udgaonkar J.B., Baldwin R.L. Early folding intermediate of ribonuclease A. Proc. Natl. Acad. Sci. USA. 1990;87:8197–8201. doi: 10.1073/pnas.87.21.8197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Varley P., Gronenborn A.M., Clore G.M. Kinetics of folding of the all-beta sheet protein interleukin-1 beta. Science. 1993;260:1110–1113. doi: 10.1126/science.8493553. [DOI] [PubMed] [Google Scholar]
- 94.Walkenhorst W.F., Edwards J.A., Markley J.L., Roder H. Early formation of a beta hairpin during folding of staphylococcal nuclease H124L as detected by pulsed hydrogen exchange. Protein Sci. 2002;11:82–91. doi: 10.1110/ps.28202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Walsh I., Giollo M., Tosatto S.C.E. Comprehensive large-scale assessment of intrinsic protein disorder. Bioinformatics. 2015;31:201–208. doi: 10.1093/bioinformatics/btu625. [DOI] [PubMed] [Google Scholar]
- 96.Wilkins M.R., Gasteiger E., Hochstrasser D.F. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 1999;112:531–552. doi: 10.1385/1-59259-584-7:531. [DOI] [PubMed] [Google Scholar]
- 97.Yi Q., Scalley M.L., Baker D. Characterization of the free energy spectrum of peptostreptococcal protein L. Fold. Des. 1997;2:271–280. doi: 10.1016/S1359-0278(97)00038-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.