

Abstract
Mast cell- and basophil-associated inflammatory diseases are a considerable burden to society. A significant portion of patients have symptoms despite standard-of-care therapy. Statins, used to lower serum cholesterol, have immune modulating activities. We tested the in vitro and in vivo effects of statins on IgE-mediated mast cell and basophil activation. Fluvastatin showed the most significant inhibitory effects of the six statins tested, suppressing IgE-induced cytokine secretion among mouse mast cells and basophils. Fluvastatin's effects were reversed by mevalonic acid or geranylgeranyl pyrophosphatase, and mimicked by geranylgeranyl transferase inhibition. Fluvastatin selectively suppressed key FcεRI signaling pathways, including Syk, Akt, and ERK. While mast cells and basophils from the C57BL/6J mouse strain were responsive to fluvastatin, those from 129/SvImJ mice were completely resistant. Resistance correlated with fluvastatin-induced upregulation of the statin target HMG-CoA reductase. Human mast cell cultures from eight donors showed a wide range of fluvastatin responsiveness. These data demonstrate that fluvastatin is a potent suppressor of IgE-mediated mast cell activation, acting at least partly via blockade of geranyl lipid production downstream of HMG-CoA reductase. Importantly, consideration of statin use for treating mast cell-associated disease needs to incorporate genetic background effects, which can yield drug resistance.
Keywords: mast cell, basophil, immunoglobulin E, cholesterol, allergy, signal transduction, anaphylaxis, statin
Introduction
Statins are widely used in the treatment of hypercholesterolemia and cardiovascular disease(1). These drugs act by competitively inhibiting the 3-hydroxy-3-methyglutaryl coenzyme A reductase (HMGCR) enzyme, resulting in reduced cholesterol(1). Statin structures influence their ability to be absorbed, metabolized, distributed, and excreted (2). Statins also exhibit anti-oxidant, anti-atherosclerotic, anti-thrombotic and immunomodulatory functions (3, 4). In the rabbit atherosclerosis model, atorvastatin significantly reduced neointimal inflammation and macrophage infiltration (5). Similarly, lovastatin has been shown to decrease CD11b surface expression on monocytes and CD11b-dependent adhesiveness to fixed endothelium (6).
The anti-inflammatory effects of statins are thought to be critical to protection from cardiovascular disease (CVD). In fact, several clinical trials have demonstrated many non-lipid-lowering statin benefits on CVD (Reviewed in (7)). These effects have been attributed to reducing the production of isoprenoids that are part of the cholesterol synthesis pathway, and are involved in modifying cell signaling proteins. In particular, the geranylated and farnesylated proteins, which include the small GTPase family such as Ras, Rac and Rho, are responsible for controlling multiple cell signaling pathways. It is therefore not surprising that statins exert pleiotropic effects.
It has been shown that statins can suppress TNF and IL-1β production from macrophages (8). Fluvastatin, a synthetic lipophilic family member, inhibits mast cell degranulation in rat cell lines (2), but a mechanism has yet to be elucidated. Moreover, human and in vivo models have not been thoroughly assessed. Our data show that statins suppress a full range of FcεRI-mediated mast cell signaling in vitro and in vivo, with consistent effects on mouse basophils and primary human mast cells. Suppression appears to be exerted at the level of geranylgeranyl lipid generation. Interestingly, statins are strikingly susceptible to genetic background, likely owing to drug-induced control of HMGCR expression. Our data support consideration of statins as a means of suppressing inflammatory disease, with the caveat that genetic background may determine drug efficacy.
Materials and Methods
Cytokines and reagents
Cytokines were purchased from Biolegend. Mouse IgE for in vivo studies was generously provided by Dr. Daniel Conrad (VCU). Purified mouse IgE (clone C38-2, κ isotype) was purchased from BD Biosciences (San Diego, CA). Antibodies recognizing mouse CD49b, CD63, TNF, IL-4 and IL-6 were from Biolegend. Antibodies against c-Kit, FcεRI, Syk, IL-13, MIP-1α (CCL-3), MCP-1 (CCL-2), phosphoserine-473-AKT, and phosphothreonine 202/phosphotyrosine 204-ERK-1/2 were from Cell Signaling Technology (Danvers, MA). Propidium Iodide and dinitophenyl-coupled human serum albumin (DNP-HSA) were from Sigma-Aldrich (St. Louis, MO). Zaragozic acid and all statin drugs except Pitavastatin were from Sigma-Aldrich. Pitavastatin (ab141958) was from Abcam (Cambridge, MA). Farnesylation transferase inhibitor III (FPTIII) and geranylgeranyl transferase inhibitor-286 (GGTI-286) were from Calbiochem (Darmstadt, Germany). Farnesyl diphosphate and geranyl geranyl diphosphate were from Echelon (Salt Lake city, UT).
Animals
C57BL/6J and 129/SvImJ (henceforth 129/SvImJ) mice were from The Jackson Laboratory (Bar Harbor, ME) and used at a minimum of 10 weeks old, with approval from the VCU institutional Animal Care and Use Committee.
Mouse mast cell and basophil culture
Mouse bone marrow-derived mast cells (BMMCs) were cultured as described (9). Mouse bone marrow-derived basophils were cultured in cRPMI supplemented with recombinant IL-3 at 20ng/ml (Biolegend, San Diego, CA), for 7-10 days, then sorted by flow cytometry selecting for CD49b-positive cells (Biolegend). Peritoneal lavage cells were cultured in cRPMI containing IL-3 (5ng/ml) and SCF (10ng/ml) for 5 days. Mast cells were positively selected using the EasySep Magnet from StemCell Technologies (Vancouver, BC), with c-Kit as a positive marker. Flow cytometry confirmed purity, which was essentially 100%.
Human skin mast cell culture
All protocols involving human tissues were approved by the human studies Internal Review Board at the University of South Carolina. Surgical skin samples were obtained from the Cooperative Human Tissue Network of the National Cancer Institute. Skin MCs were prepared and cultured as described previously (10) and were used after 8-16 weeks, at which time purity was essentially 100% mast cells, as determined by staining with toluidine blue.
IgE-mediated activation
Human MC or BMMC were sensitized overnight with DNP-specific mouse IgE (1.0 μg/ml for human MC; 0.5 μg/ml for BMMC), then washed and resuspended 1×106 cells/ml in complete media with cytokines. Cells were stimulated with DNP-HSA (Ag; 30 or 20 ng/ml for human MC or mouse BMMC, respectively) for the indicated times. Cytokines were measured by standard ELISA kits from BioLegend (San Diego, CA) or BD Biosciences (San Diego, CA).
Flow cytometric analysis
Surface c-Kit and FcεRI expression was measured by standard flow cytometry on a BD FACScalibur. To detect intracellular cytokines, cells were stained as described previously(9). Basophils were also stained with FITC-anti-CD49b prior to fixation.
Degranulation assays
Cells were plated at 1×106/ml in cRPMI as described above, and treated with fluvastatin or DMSO (vehicle, diluted 1:5,000) for 24 hours ± 0.5 μg/ml of IgE, were washed twice in RPMI and activated ± DNP-HSA for 1 hour before staining with CD63, followed by flow cytometry analysis.
Migration assay
Migration was assayed as described previously(9), using 8μm polycarbonate 24-well transwell inserts from Corning.
HMG-CoA reductase qPCR
BMMCs were cultured +/− 40 μM fluvastatin for 5 hours. RNA was extracted with TRIzol reagent (Life Technologies, Grand Island, NY). cDNA was synthesized using the SuperScript ™ III Reverse Transcriptase (Invitrogen ™ by Life Technologies, Grand Island, NY) using oligo dT primers. qPCR analysis was performed with BioRad CFX96 Touch™ Real-Time PCR Detection System (Hercules, CA) and SYBR® Green detection using a relative Livak Method. Primers included: HMGCR primers q.HMGCR Forward: 5'CCTGTAACTCAGAGGGTCAAGATGAT3' and q.HMGCR Reverse: 5'CCAGCGACTGTGAGCATGAA3' or β -actin Mouse Beta-Actin Forward: 5'GATGACGATATCGCTGCGC3', Mouse Beta-Actin Reverse: 5'CTCGTCACCCACATAGGAGTC3' (housekeeping gene) primers (Invitrogen ™ by Life Technologies, Grand Island, NY). Melting curve analysis was performed between 50°C and 95°C.
Quantitative Measurement of Cholesterol
Cholesterol levels were analyzed via LC tandem mass spectrometry using a Shimadzu Nexera UPLC coupled to a QTRAP 6500 mass analyzer (AB SCIEX, Framingham, MA). Lipids were extracted from the cell pellet using a modified Bligh and Dyer method as previously described (11, 12).
Western blot analysis
Western blotting was performed as described previously (9). Blots were visualized and quanitifed using a LiCor Odyssey CLx Infrared imaging system (Lincoln, NE).
Passive Systemic Anaphylaxis
Mice were administered 200μl PBS containing 1mg fluvastatin or equivalent dilution of DMSO via intraperitoneal injection, followed by 200μl PBS containing 50μg mouse IgE anti-DNP. The following day, mice were again administered 200μl PBS containing 1mg fluvastatin or DMSO one hour before DNP-HSA (50μg) was administered via intraperitoneal injection. In some experiments, 8mg histamine was injected in place of antigen. The core body temperature of each mouse was measured using a rectal microprobe (Physitemp Instruments). Mice were euthanized and blood was collected by cardiac puncture to analyze plasma.
Statistical Analysis
P values were calculated using GraphPad Prism software, by paired or unpaired two-tailed Student's t test as appropriate. P values of <0.05 were considered statistically significant. Unless otherwise stated, results are expressed as the mean ± SEM of at least 3 independent experiments conducted in triplicate. In all figures *=p<.05; **=p<.01; ***=p<.001; ****=p<.0001.
Results
Statins inhibit IgE-mediated cytokine production by mouse mast cells
Statins alter isoprenoid generation and degranulation in RBL-2H3 cells (13). We tested several statin family members to determine their effects on IgE-activated (XL) primary mast cells. C57BL/6J BMMC treated for 24 hours prior to antigen-induced activation generally showed decreased IL-6, TNF and IL-13 production, with the exception of atorvastatin and pravastatin treatment (Figure 1A). Fluvastatin responses tended to be of the greatest magnitude, hence we focused our studies on this drug.
FIGURE 1.
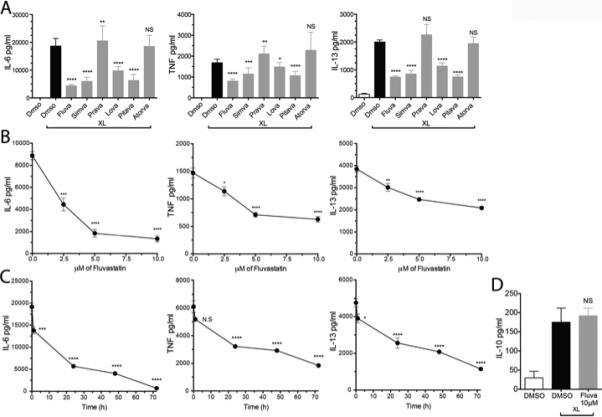
Statins suppress IgE-mediated cytokine production in mouse mast cells. (A) C57BL/6J BMMC were cultured with DMSO or 10 μM of the indicated statin for 24 hours, then activated as described. IL-6, TNF, and IL-13 levels in culture supernatants were determined by ELISA. (B) BMMC were cultured with the indicated concentrations of fluvastatin prior to activation as in (A). (C) BMMC were treated with 10μM fluvastatin as in (A) for the indicated times prior to antigen-mediated activation for 16 hours). (D) BMMC were cultured with 10μM fluvastatin as in (A), and culture supernatants were tested for the presence of IL-10 by ELISA. Data are means ± SEM of five (A) and three (B, C and D) independent experiments done in triplicate and analyzed by unpaired t-Test, comparing fluvastatin- and DMSO (control)-treated groups. For each independent experiment n=3.
Time- and dose-dependence for fluvastatin-mediated suppression were established by assessing IgE-induced IL-6, TNF and IL-13 secretion. 10μM fluvastatin yielded maximal inhibition, suppressing cytokine production 50% or greater. The IC50 for this effect was approximately 2.5-5μM (Figure 1B). A 24-hour incubation yielded significant suppression with no change in cell viability (Figure 1C). By contrast, 72-hour treatment provided the greatest suppression but decreased cell viability (not shown); hence we employed a 24-hour time point for further study. Finally, it was interesting to note that fluvastatin had no effect on production of IL-10, a cytokine that is generally anti-inflammatory (Figure 1D). This divergence from the effects on pro-inflammatory cytokines suggested some specificity in how FcεRI signaling was altered.
Fluvastatin effects are mediated via blockade of isoprenoid lipids
Fluvastatin acts by blocking HMGCR, inhibiting the production of mevalonic acid (MVA) (3, 14-16). To assess target specificity, we attempted to reverse the effects of fluvastatin with MVA. As shown in Figure 2A, MVA restored IgE-mediated cytokine production in the presence of fluvastatin, indicating that drug effects were in fact due to HMGCR blockade.
FIGURE 2.
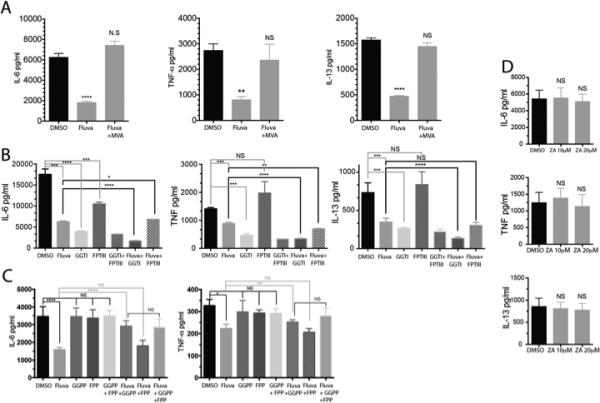
Fluvastatin-mediated suppression is linked to isoprenoid biosynthesis. (A) IgE-primed C57BL/6J BMMC were cultured for 24 hours in the presence of either DMSO or 10μM fluvastatin +/− 1000mM mevalonic acid (MVA) as indicated. Cells were then activated with antigen for 16 hours and culture supernatants were analyzed by ELISA. MVA alone did not alter cytokine production (not shown). (B) BMMC were cultured for 24 hours with IgE plus the indicated inhibitors (20μM) or fluvastatin (10μM) then activated for 16 hours with antigen. Culture supernatants were analyzed by ELISA.(C) BMMC were cultured for 24 hours with IgE in the presence of DMSO, fluvastatin (10μM) +/− the indicated farnesyl or geranyl pyrophosphate (20μM) prior to antigen activation for 16 hours. Culture supernatants were assayed by ELISA. (D) BMMC were cultured for 24 hours with IgE plus DMSO or the indicated concentration of zaragozic acid A (ZA), prior to antigen activation for 16 hours. Culture supernatants were analyzed by ELISA. Data are means ± SEM of three (A, B and D), four (C) independent experiments done in triplicate and analyzed by unpaired t-Test comparing fluvastatin- and DMSO (control)-treated groups. For each independent experiment n=3.
MVA metabolism leads to cholesterol production, but also yields farnesyl pyrophosphate and geranylgeranyl pyrophosphate, precursors of isoprenoid lipids. These are critical for protein prenylation, including modifications to small GTPases such as Ras, Rac and Rho. To determine the importance of prenylation, we employed the compounds GGTI-286 and FPTIII, selective inhibitors of geranylgeranylation and farnesylation, respectively. GGTI-286 treatment for 24 hours mimicked the effects of fluvastatin (Figure 2B). Treatment with fluvastatin plus GGTI-286 suppressed cytokine production more than either compound alone. In contrast, FPTIII treatment provided little or no inhibition (Figures 2B).
To confirm our findings with these inhibitors, we attempted to reverse fluvastatin-mediated inhibition by restoring isoprenoid lipids. BMMC were treated with fluvastatin in the presence or absence of either geranylgeranyl pyrophosphate (GGPP) or farnesyl pyrophosphate (FPP). As shown in Figure 2C, GGPP partially rescued IgE-mediated cytokine production when compared to fluvastatin-treated cells. By contrast, FPP had little or no effect.
Although isoprenoid blockade appeared to be important for fluvastatin effects, cholesterol depletion could also be critical. To test this, we used the squalene synthase inhibitor zaragozic acid A (ZA), which blocks cholesterol synthesis downstream of isoprenoid generation. ZA treatment reduced BMMC cholesterol levels 59%, similar to the effects of fluvastatin (not shown), but had no impact on IgE-mediated cytokine production (Figure 2D). Collectively, these data suggest that fluvastatin effects on antigen-induced cytokine production are predominantly due to reduced geranylgeranylation events involved in FcεRI signaling.
Fluvastatin effects can be altered by genetic background
To determine if genetic background alters fluvastatin responsiveness, we compared BMMC from C57BL/6J and 129/SvImJ mice, which possess many polymorphic variations. As shown in Figure 3A, 129/SvImJ BMMC were strikingly resistant to fluvastatin at concentrations up to 40 μM. In fact, 129/SvImJ BMMC were resistant to simvastatin, pravastatin and partially to atorvastatin (Figure S1). To establish that these differences were not due to their in vitro differentiation, we compared fluvastatin responses using peritoneal mast cells, which yielded a similar outcome (Figure 3B). We also tested BMMC cultured from BALB/c and A/J mice, both of which were also resistant to fluvastatin effects (Figure S2). These data support the hypothesis that genetic background can yield drug resistance.
FIGURE 3.
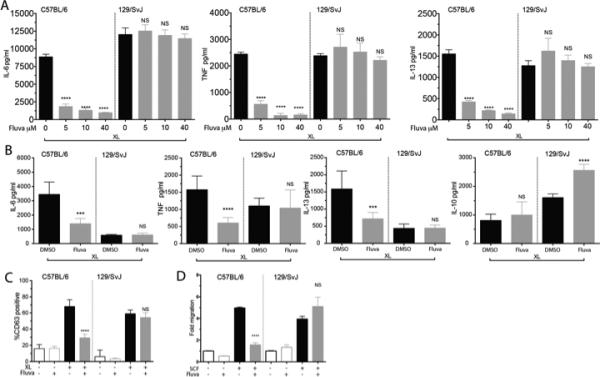
129/SvImJ mouse mast cells are resistant to fluvastatin. (A) C57BL/6J or 129/SvImJ BMMC were cultured in the indicated concentrations of fluvastatin and activated as described in Figure 1A. Culture supernatants were analyzed by ELISA. (B) Purified peritoneal mast cells from the indicated mouse strains were cultured for 24 hours with DMSO or 10μM fluvastatin for 24 hours, then activated with antigen. Culture supernatants were assessed by ELISA. (C) BMMC from the indicated strains were cultured as described in Figure 1A and activated with antigen for 1 hour prior to staining to detect surface CD63 by flow cytometry. (D) BMMC from the indicated strains were cultured with DMSO (-) or fluvastatin (10μM) for 24 hours, then tested for migration towards an SCF gradient as described in Materials and Methods. All data shown are from a minimum of three experiments. Data are means ± SEM of three independent experiments done in triplicate and analyzed by unpaired t-Test comparing fluvastatin- and DMSO (control)-treated groups. For each independent experiment n=3, (B) is representative of one of three independent experiments done in triplicate.
We further showed that fluvastatin inhibits other mast cell functions, effects that are also absent among 129/SvImJ mast cells. FcεRI-induced degranulation, as judged by membrane expression of the intra-granular protein CD63, was selectively inhibited by fluvastatin in C57BL/6J BMMC (Figure 3C). Similarly, fluvastatin diminished SCF-mediated migration of C57BL/6J but not 129/SvImJ BMMC (Figure 3D).
Fluvastatin suppresses antigen-stimulated basophils
We also investigated the effects of fluvastatin on C57BL/6J and 129/SvImJ basophils. Culture for 24 hours with fluvastatin prior to IgE+antigen-mediated activation reduced IL-4, IL-6, IL-13, TNF, and MCP-1 production, and inhibited degranulation among C57BL/6J, but had no significant effect on 129/SvImJ basophils (Figure 4).
FIGURE 4.
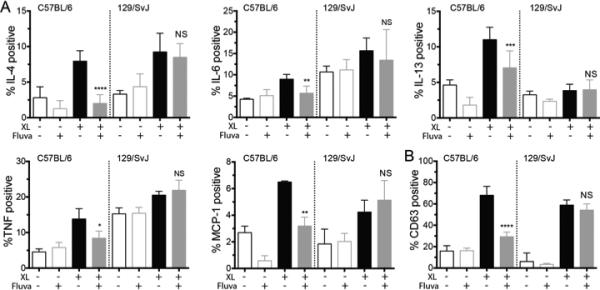
Fluvastatin suppresses IgE responses in C57BL/6J but not 129/SvImJ basophils. Basophils from the indicated mouse strains were cultured in fluvastatin as described in Figure 1, then activated with antigen prior to detecting (A) intracellular cytokines or (B) surface CD63 as described. Data are means ± SEM of three independent experiments done in triplicate and analyzed by unpaired t-Test comparing fluvastatin- and DMSO (control)-treated groups. For each independent experiment n=3.
Fluvastatin selectively alters FcεRI signaling pathways
The suppressive effects of fluvastatin could be related to reduced FcεRI or c-Kit expression. However, fluvastatin treatment with concentrations reaching 40μM for up to 4 days had no significant effect FcεRI or c-Kit surface expression as measured by flow cytometry (data not shown). Hence loss of receptor expression does not appear to explain fluvastatin effects. As such, we investigated how fluvastatin impacts FcεRI signaling.
Following antigen crosslinking, the Src family tyrosine kinases Fyn and Lyn, as well as the tyrosine kinase Syk are recruited to FcεRI (17). Fluvastatin had no effect on Fyn, Lyn or Syk expression in C57BL/6J BMMC (Figure 5A).
FIGURE 5.
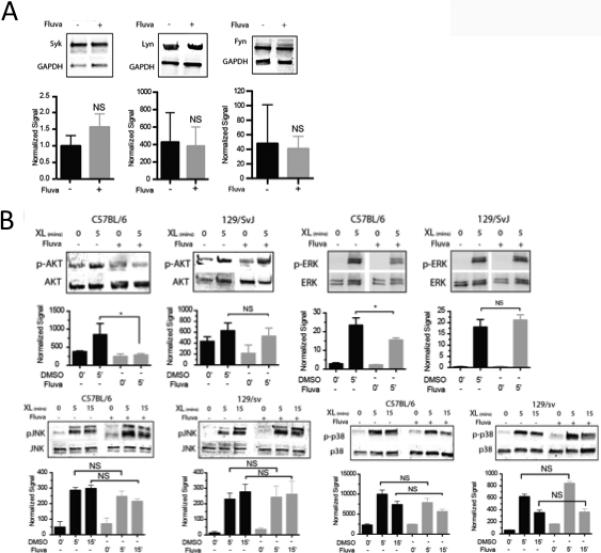
Fluvastatin suppresses FcεRI signaling. (A) C57BL/6 BMMC were cultured in DMSO or fluvastatin as described in Figure 1, then subjected to western blotting to detect Syk, Lyn or Fyn. (B) BMMC from the indicated strains were cultured for 24 hours with IgE plus DMSO or fluvastatin (10μM) prior to antigen-mediated activation for 5 minutes, then analyzed by western blotting to detect the indicated total and phosphorylated proteins. Data are means ± SEM analyzed by unpaired t-Test of (A) 9 samples and (B) three independent experiments.
Downstream of FcεRI apical tyrosine kinases, a number of signaling pathways are activated. Fluvastatin inhibited Akt, and ERK phosphorylation (Figure 5B) in C57BL/6J but not 129/SvImJ BMMC. In contrast, JNK or p38 phosphorylation was unaffected (Figure 5B). Taken together, these data demonstrate selective inhibitory effects of fluvastatin treatment, acting both apically and distally from FcεRI.
129/SvImJ resistance correlates with fluvastatin-mediated induction of HMGCR
While the C57BL/6J and 129/SvImJ strains have significant genetic variability, the fluvastatin target HMGCR is reported on the Jackson Lab Phenome site (phenome.jax.org) to lack coding polymorphisms between these strains. We confirmed this by sequencing mast cell HMGCR mRNA from these strains (not shown). However, coding variability is only one means of achieving drug resistance. Here, we demonstrate that 129/SvImJ BMMC increased HMGCR mRNA expression 2.5-fold following fluvastatin treatment, while C57BL/6J BMMC showed no change in expression (Figure 6A). Furthermore, this increased expression appeared to be functionally significant. As shown in Figure 6B, fluvastatin treatment reduced cellular cholesterol levels approximately 60% in C57BL/6J BMMC, while 129/SvImJ BMMC were unaffected. Finally, we found that FcεRI-mediated IL-6 production by 129/SvImJ BMMC could be suppressed by blocking isoprenylation events downstream of HMGCR with GGTI-286 +/− FPTIII (Figure 6C). Thus the 129/SvImJ background is responsive to isoprenoid blockade, indicating that drug resistance lies above this step. Collectively these data support the hypothesis that HMGCR induction yields drug resistance in 129/SvImJ mast cells.
FIGURE 6.
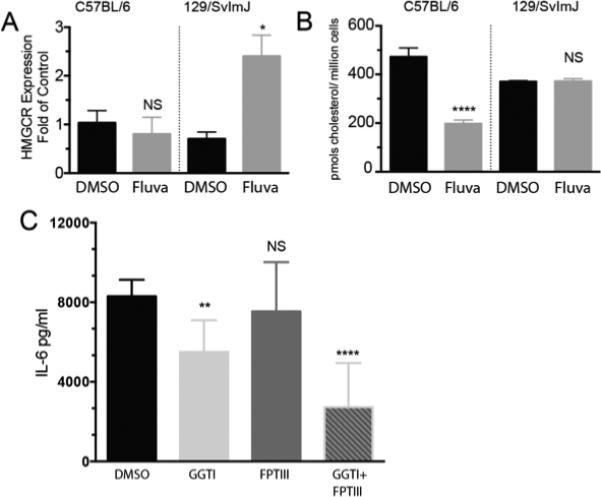
129/SvImJ mast cells exhibit fluvastatin-induced HMGCR upregulation correlating with drug resistance. (A) BMMC were cultured for 5 hours with DMSO or fluvastatin (40μM). Total RNA was analyzed for HMGCR expression by quantitative real time PCR. Data shown are from one of two independent experiments using 3 populations each analyzed by unpaired t-Test comparing fluvastatin- and DMSO (control)-treated groups. (B) BMMC were cultured with DMSO or fluvastatin (10μM) for 24 hours. Lysates were analyzed for the presence of cholesterol by mass spectroscopy. Data shown are means and SEM of three samples analyzed by paired t-Test, comparing the samples with vehicle to fluvastatin. (C) 129/SvImJ BMMC were cultured for 24 hours with IgE plus the indicated inhibitors (20μM) or fluvastatin (10μM), then activated for 16 hours with antigen prior to ELISA analysis of supernatants. Data are from three independent experiments done in triplicate and analyzed by unpaired t-Test comparing fluvastatin- and DMSO (control)-treated groups.
Fluvastatin suppresses passive systemic anaphylaxis in C57BL/6J but not 129/SvImJ mice
We next used a mast cell-dependent passive systemic anaphylaxis (PSA) model to assess the functional relevance of fluvastatin sensitivity on C57BL/6J and 129/SvImJ mice. Here, we show that fluvastatin pre-treatment significantly mitigated the loss of core body temperature in C57BL/6J mice, but had no effect on the 129/SvImJ background (Figure 7A). Plasma MIP-1α levels from these mice showed the same trend, with fluvastatin-mediated suppression observed in C57BL/6J but not 129/SvImJ mice (Figure 7B). These data show that fluvastatin dampens the early and late phases of mast cell-dependent anaphylaxis, and that genetic background can greatly alter the drug response.
FIGURE 7.
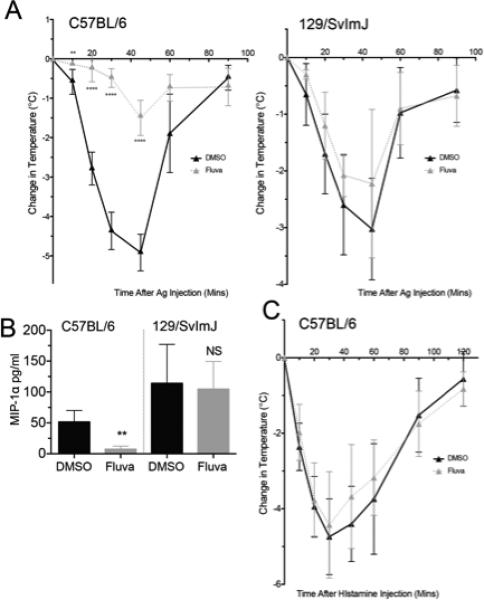
Fluvastatin suppresses passive systemic anaphylaxis in C57BL/6J but not 129/SvImJ mice. (A and B) Mice from the indicated strains were subjected to PSA as described. Plasma was analyzed for MIP-1α levels by ELISA. (C) Mice were injected with DMSO or fluvastatin as described in part (A), then challenged with 8mg histamine. Change in core body temperature was recorded. (A, C) n=5 from two independent experiments, (B) Data are means ± SEM from two independent experiments. All data were analyzed by unpaired t-Test comparing fluvastatin- and DMSO (control)-treated groups.
In addition to inhibiting mast cell degranulation, fluvastatin could be suppressing the vascular response to histamine. However, we show that fluvastatin pretreatment before histamine administration did not suppress anaphylaxis (Figure 7C). We therefore conclude that fluvastatin effects are largely due to mast cell suppression, not effects on the vasculature.
Variable responsiveness to fluvastatin is consistent among primary human mast cells
Given the stark variation in fluvastatin responses among mouse strains, we assessed primary human skin mast cells from eight healthy subjects. Interestingly, we found a large variation in fluvastatin-responses (Figure 8). When investigating TNF and MCP-1 production, we noted relative decreases in cytokine production from less than 20% to more than 80%. These data suggest that fluvastatin can significantly blunt IgE-mediated activation in human mast cells, but that genetic variability may greatly impact the extent of drug responsiveness.
FIGURE 8.
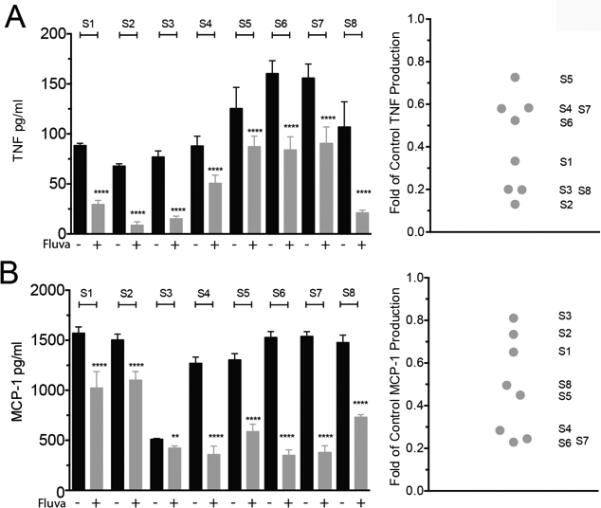
Human mast cells show varied responses to fluvastatin. Human skin mast cells from eight subjects were cultured with DMSO or fluvastatin as described in Figure 1A. Sixteen hours after antigen stimulation, culture supernatants were analyzed for the presence of TNF (A) or MCP-1 (B) by ELISA. Scatter plot shows the normalized fold of DMSO control for each subject. Each supernatant was analyzed in sextuplicate for each subject. Data are means ± SEM, analyzed by paired t-Test comparing vehicle to fluvastatin treatment.
Discussion
Statins are best known for their lipid-lowering effects, and widely-prescribed to prevent CVD. However, several clinical trials have suggested that statin anti-inflammatory benefits are important, separate from their effects on lipids (reviewed in(7)). These drugs have pleiotropic effects on mast cells. Lovastatin and fluvastatin have been reported to inhibit IgE-mediated degranulation in the rat mast cell line RBL-2H3 (2). Furthermore, cerivastatin and atorvastatin have been shown to suppress growth and IgE-mediated histamine release in human basophils (18). Our work demonstrates that a range of statins inhibit IgE-induced cytokine production from mast cells and basophils with varying degrees of efficacy. There were some exceptions: pravastatin slightly enhanced cytokine production, and atorvastatin had no significant effect. Pravastatin is more hydrophilic than the “lipophilic” simvastatin, lovastatin and fluvastatin (19). However, the most lipophilic statins, lovastatin and simvastatin (20), were not the most suppressive in our assays. Interestingly, fluvastatin's suppressive capabilities did not extend to IL-10, widely regarded as an anti-inflammatory cytokine (21, 22).
Fluvastatin effects were reversed with MVA pretreatment, confirming targeted specificity to the mevalonate pathway. Our data showed that the geranylgeranyl transfer step is critical. These data were supported by studies using the squalene synthase inhibitor zaragozic acid A(23), which did not alter IgE-induced cytokine production, despite suppressing cholesterol synthesis. These latter results suggest that fluvastatin effects are not due to large-scale changes in cholesterol-containing lipid rafts. Taken together, these data argue that geranylgeranylation has the largest effect on IgE-mediated cytokine production, and suggest this pathway as a potential target for controlling the mast cell response.
Mast cells are best known for their role in IgE-mediated inflammation. Asthma and allergy studies support broad effects of statins in mast cell-associated disease. Peripheral blood mononuclear cell (PBMC) proliferation and inflammatory responses were suppressed by fluvastatin in patients with allergic asthma (24). Furthermore, atorvastatin in conjunction with inhaled corticosteroids improved lung function and airway inflammation in atopic asthmatics (25). These and other studies have led to the conclusion that statins are beneficial for asthma management (26). Cellular and molecular mechanisms are suggested from human and animal studies. Statins may suppress T cell activation by decreasing IFNγ-induced MHC II expression on monocytes (27). T cell activation, proliferation, and migration are also suppressed by statin treatment (24). In addition to their effects on immune cells, statins have been shown to suppress bronchial wall remodeling (28).
Mouse models also support the rationale for statin therapy. Simvastatin inhibits airway hyperresponsiveness in a murine model (29). This is partly due to suppression of T cell-produced IL-4 and IL-5 (30). More recently, Kim et al showed that simvastatin reduced ovalbumin-specific IgE levels, and the number of macrophages, neutrophils and eosinophils in bronchoalveolar lavage fluid in a mast cell-independent airway hyperresponsiveness model (31). Moreover, Simvastatin also reduced thioglycolate-induced peritoneal inflammation (32) in which the predominate infiltrate is neutrophils.
The passive anaphylaxis model does not require antigen processing/presentation or a role for T and B cells. While focusing on the mast cell and vasculature, this assay demonstrated that fluvastatin treatment prior to antigen challenge dramatically reduced the severity of anaphylaxis in C57BL/6J mice. To rule out fluvastatin primarily suppressing the vascular response to histamine, we pretreated mice with fluvastatin and bypassed the mast cell response by injecting histamine, conditions under which fluvastatin had no effect. These data support the theory that fluvastatin directly suppresses mast cells in vivo and support the use of statins as a therapy for mast cell-associated disease.
Downstream of the IgE receptor, Syk, Fyn and Lyn are recruited and activated. Lyn is predominantly a negative regulator, as noted in Lyn-deficient mice (33). Along with Syk downregulation, we further show fluvastatin suppressed Akt and Erk phosphorylation in C57BL/6J but not 129/SvImJ BMMC. These are critical regulators of mast cell responses to IgE, hence their collective suppression could logically explain reduced cytokine production and anaphylaxis.
Variable responses to statins among C57BL/6J and 129/SvImJ mast cells are consistent with previous studies from our group. We found that mast cell precursors from the BALB/cJ background are resistant to IL-10 (34), and that 129/SvImJ BMMC are resistant to TGFβI-mediated suppression (9). So it was intriguing to note that 129/SvImJ, Balb/cJ, and A/J were all fluvastatin-resistant. Since C57BL/6J is Th1-prone and 129/SvImJ, BALB/c, and A/J are Th2-prone mice, a simple conclusion would be that genetic variations predisposing to strong Th1 or Th2 development have related polymorphisms yielding drug resistance. However, it is important to note that these mice have many genetic variations unrelated to T cell or inflammatory phenotypes. These observations support a deeper investigation into the mechanisms explaining this pharmacogenomic effect.
Since statins target HMGCR, we initially postulated that the genetic variation between the C57BL/6J and 129/SvImJ strains was a polymorphism or steady-state alteration in HMGCR expression between strains. Instead, we found that 129/SvImJ mast cells increase HMGCR expression after fluvastatin treatment, which correlated with resistance to fluvastatin-mediated decrease in cellular cholesterol. Importantly, statin-induced HMGCR upregulation has been noted by others. For example, Ness and co-workers found that HMGCR levels are significantly increased in rats fed atorvastatin (35). Lastly, GGTI suppressed FcεRI-mediated IL-6 production in 129/SvImJ mast cells, demonstrating that these cells can be suppressed by targeting enzymatic steps subsequent to HMGCR. We therefore propose that increased HMGCR is at least partly responsible for fluvastatin resistance in 129/SvImJ mice.
It is important to state that other explanations for fluvastatin resistance are possible and warrant further study. There is wide variation in statin responsiveness among patients, possibly due to several genetic differences (36). Clinical studies have largely focused on cytochrome P450 enzyme polymorphisms, the lipid metabolism genes apolipoprotein E and B (APOE, APOB), cholesteryl ester transfer protein (CETP), and the LDL receptor (LDLR) (37). For example, the P450 subunit Cyp2c9 is the primary pathway for fluvastatin metabolism (38). It might be that variants of the Cyp2c9 gene also contribute to fluvastatin resistance. However, the mouse Cyp2c9 orthologs Cyp2c65/66 have no coding region polymorphisms between C57BL/6J and 129/SvImJ strains on Jackson Lab's Phenome database. While Cyp2c9 polymorphisms may not explain our murine data, variations among human donors are not known, not mutually exclusive with altered HMGCR induction, and should be further examined.
The variability in fluvastatin responsiveness seen in murine mast cells was corroborated by 18% to 87% suppression among human mast cell cultures, when measuring FcεRI-mediated cytokine secretion. While larger numbers of donors are needed to corroborate these findings, these data demonstrate the possible utility of statins for treating mast cell-associated disease, but also the inherent variability that should be anticipated. It is possible that statin-induced HMGCR expression could be used to screen for statin responsiveness. Understanding how statins alter IgE-mediated signaling, and the significance of genetic background, could prove to be clinically useful. At present, a simplistic interpretation of our data would be that statins may be less efficacious among Th2-prone individuals. Our data also suggest that geranylgeranyl transfer inhibitors might be therapeutically useful, especially among statin-resistant populations. However, given the complexity of the cholesterol synthesis cascade, further study of correlations between statin-mediated mast cell suppression and polymorphisms is warranted and may provide insight for novel therapies.
Supplementary Material
Acknowledgements
The authors thank Dr. Daniel Conrad for many helpful conversations concerning this manuscript.
This work was supported by NIH grants 1R01AI101153 and 2R01AI059638 to JJR, 1R01 AI095494 to CAO, by funding from the Veteran's Administration (VA Merit Review I BX001792 to CEC and a Research Career Scientist Award 1 3F-RCS-002 to CEC), from the National Institutes of Health (HL125353 to CEC, CA154314 to CEC and NH1C06-RR17393 to Virginia Commonwealth University for renovation). Services (VCU Lipidomics/Metabolomics Core) and products in support of the research project were generated, in part, by the VCU Massey Cancer Center (shared and supported) with funding from NIH-NCI Cancer Center Support Grant, P30 CA016059.
Abbreviations
- BMMC
bone marrow-derived mast cell
- DNP-HSA
dinitophenyl-coupled human serum albumin
- MVA
mevalonic acid
- HMGCR
3-hydroxy -3-methyglutaryl coenzyme A reductase
- ZA
Zaragozic acid
- PSA
passive systemic anaphylaxis
References
- 1.Liao JK. Isoprenoids as mediators of the biological effects of statins. J. Clin. Invest. 2002;110:285–288. doi: 10.1172/JCI16421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fujimoto M, Oka T, Murata T, Hori M, Ozaki H. Fluvastatin inhibits mast cell degranulation without changing the cytoplasmic Ca2+ level. European Journal of Pharmacology. 2009;602:432–438. doi: 10.1016/j.ejphar.2008.11.040. [DOI] [PubMed] [Google Scholar]
- 3.Liao JK, Laufs U. Pleiotropic effects of statins. Annu. Rev. Pharmacol. Toxicol. 2005;45:89–118. doi: 10.1146/annurev.pharmtox.45.120403.095748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vaughan CJ, Murphy MB, Buckley BM. Statins do more than just lower cholesterol. Lancet. 1996;348:1079–1082. doi: 10.1016/S0140-6736(96)05190-2. [DOI] [PubMed] [Google Scholar]
- 5.Bustos C, Ndez-Presa MAH, Ortego MN, Tunon J, Ortega L, Perez F, Diaz C, Ndez GH, Egido JS. HMG-CoA Reductase Inhibition by Atorvastatin Reduces Neointimal Inflammation in a Rabbit Model of Atherosclerosis. Journal of the American College of Cardiology. 1998;32:2057–2064. doi: 10.1016/s0735-1097(98)00487-2. [DOI] [PubMed] [Google Scholar]
- 6.Weber C, Erl W, Weber KSC, Weber PC. HMG-CoA Reductase Inhibitors Decrease CD11b Expression and CD11b-Dependent Adhesion of Monocytes to Endothelium and Reduce Increased Adhesiveness of Monocytes Isolated From PatientsWith Hypercholesterolemia. Journal of the American College of Cardiology. 1997;30:1212–1217. doi: 10.1016/s0735-1097(97)00324-0. [DOI] [PubMed] [Google Scholar]
- 7.Tousoulis D, Psarros C, Demosthenous M, Patel R, Antoniades C, Stefanadis C. Innate and Adaptive Inflammation as a Therapeutic Target in Vascular Disease. Journal of the American College of Cardiology. 2014;63:2491–2502. doi: 10.1016/j.jacc.2014.01.054. [DOI] [PubMed] [Google Scholar]
- 8.Pahan K, Sheikh FG, Namboodiri AM, Singh I. Lovastatin and phenylacetate inhibit the induction of nitric oxide synthase and cytokines in rat primary astrocytes, microglia, and macrophages. J. Clin. Invest. 1997;100:2671–2679. doi: 10.1172/JCI119812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fernando J, Faber TW, Pullen NA, Falanga YT, Kolawole EM, Oskeritzian CA, Barnstein BO, Bandara G, Li G, Schwartz LB, Spiegel S, Straus DB, Conrad DH, Bunting KD, Ryan JJ. Genotype-Dependent Effects of TGF-1 on Mast Cell Function: Targeting the Stat5 Pathway. The Journal of Immunology. 2013;191:4505–4513. doi: 10.4049/jimmunol.1202723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kambe N, Kambe M, Kochan JP, Schwartz LB. Human skin–derived mast cells can proliferate while retaining their characteristic functional and protease phenotypes. Blood. 2001;97:2045–2052. doi: 10.1182/blood.v97.7.2045. [DOI] [PubMed] [Google Scholar]
- 11.Wijesinghe DS, Allegood JC, Gentile LB, Fox TE, Kester M, Chalfant CE. Lipidomics and Bioactive Lipids: Mass-Spectrometry–Based Lipid Analysis. The Journal of Lipid Research. 2010;11:641–651. doi: 10.1194/jlr.D000430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McDonald JG, Thompson BM, McCrum EC, Russell DW. Lipidomics and Bioactive Lipids: Mass - Spectrometry–Based Lipid Analysis. Vol. 432. Elsevier; 2007. Extraction and Analysis of Sterols in Biological Matrices by High Performance Liquid Chromatography Electrospray Ionization Mass Spectrometry. pp. 145–170. [DOI] [PubMed] [Google Scholar]
- 13.Deanin GG, Cutts JL, Pfeiffer JR, Oliver JM. Role of isopreniod metabolism in IgE receptor mediated signal transduction. The Journal of Immunology. 1991;146:3528–3535. [PubMed] [Google Scholar]
- 14.Endo A, Kuroda M, Tsuija Y. ML-236A, ML-236B, and ML-236C, new inhibitors of cholesterogenesis produced by Penicillium citrinium. The Journal of Antibiotics. 1976:1346–1349. doi: 10.7164/antibiotics.29.1346. [DOI] [PubMed] [Google Scholar]
- 15.Endo A, Tsujita Y, Kuroda M, Tanzawa K. Inhibition of Cholesterol Synthesis in vitro and in vivo by ML-236A and ML-236B, Competitive Inhibitors of 3-Hydroxy-3-methylglutaryl-Coenzyme A Reductase. European Journal of Biochemistry. 1977:1–6. doi: 10.1111/j.1432-1033.1977.tb11637.x. [DOI] [PubMed] [Google Scholar]
- 16.Tobert JA. Case history: Lovastatin and beyond: the history of the HMG-CoA reductase inhibitors. Nat Rev Drug Discov. 2003;2:517–526. doi: 10.1038/nrd1112. [DOI] [PubMed] [Google Scholar]
- 17.Kambayashi T, Koretzky GA. Proximal signaling events in FcεRI-mediated mast cell activation. Journal of Allergy and Clinical Immunology. 2007;119:544–552. doi: 10.1016/j.jaci.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 18.Majlesi Y, Samorapoompichit P, Hauswirth AW, Schernthaner G-H, Ghannadan M, Baghestanian M, Rezaie-Majd A, Valenta R, Sperr WR, Bühring H-J, Valent P. Cerivastatin and atorvastatin inhibit IL-3-dependent differentiation and IgE-mediated histamine release in human basophils and downmodulate expression of the basophil-activation antigen CD203c/E-NPP3. Journal of Leukocyte Biology. 2003;73:107–117. doi: 10.1189/jlb.0202075. [DOI] [PubMed] [Google Scholar]
- 19.Hamelin BA, Turgeon J. Hydrophilicity/ lipophilicity: relevance for the pharmacology and clinical effects of HMG-CoA reductase inhibitors. Tips. 1998;19:26–37. doi: 10.1016/s0165-6147(97)01147-4. [DOI] [PubMed] [Google Scholar]
- 20.Serajuddin ATM, Ranadive SA, Mahoney EM. Relative lipophilicities, solubilities, and structure-pharmacological considerations of 3-hydroxy-3-methylglutaryl coenzyme a (HMG-COA) reductase inhibitors pravastatin, lovastatin, mevastatin, and simvastatin. Journal of Pharmaceutical Sciences. 1990;80:830–835. doi: 10.1002/jps.2600800905. [DOI] [PubMed] [Google Scholar]
- 21.Ryan JJ, Kashyap M, Bailey D, Kennedy S, Speiran K, Brenzovich J, Barnstein B, Oskeritzian CA, Gomez G. Mast Cell Homeostasis: A Fundamental Aspect of Allergic Disease. Critical Reviews in Immunology. 2007;27:15–32. doi: 10.1615/critrevimmunol.v27.i1.20. [DOI] [PubMed] [Google Scholar]
- 22.Couper KN, Blount DG, Riley EM. IL-10: the master regulator of immunity to infection. The Journal of Immunology. 2008;180:5771–5777. doi: 10.4049/jimmunol.180.9.5771. [DOI] [PubMed] [Google Scholar]
- 23.Ness GC, Zhao Z, Keller RK. Effect of squalene synthase inhibition on the expression of hepatic cholesterol biosynthetic enzymes, LDL receptor, and cholesterol 7 alpha hydroxylase. Archives of Biochemistry and Biophysics. 1994;311:277–285. doi: 10.1006/abbi.1994.1238. [DOI] [PubMed] [Google Scholar]
- 24.Samson KTR, K Minoguchi AT, Oda N, Yokoe T, Yamamoto Y, Yamamoto M, Ohta S, Adachi M. Inhibitory effects of fluvastatin on cytokine and chemokine production by peripheral blood mononuclear cells in patients with allergic asthma. Clin Exp Allergy. 2006;36:475–482. doi: 10.1111/j.1365-2222.2006.02470.x. [DOI] [PubMed] [Google Scholar]
- 25.Hothersall EJ, Chaudhuri R, McSharry C, Donnelly I, Lafferty J, McMahon AD, Weir CJ, Meiklejohn J, Sattar N, McInnes I, Wood S, Thomson NC. Effects of atorvastatin added to inhaled corticosteroids on lung function and sputum cell counts in atopic asthma. Thorax. 2008;63:1070–1075. doi: 10.1136/thx.2008.100198. [DOI] [PubMed] [Google Scholar]
- 26.Lokhandwala T, West-Strum D, Banahan BF, Bentley JP, Yang Y. Do statins improve outcomes in patients with asthma on inhaled corticosteroid therapy? A retrospective cohort analysis. BMJ Open. 2012;2:e001279–e001279. doi: 10.1136/bmjopen-2012-001279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kwak B, Mulhaupt F, Myit S, Mach F. Statins as a newly recognized type of immunomodulator. Nature Medicine. 2000;6:1399–1402. doi: 10.1038/82219. [DOI] [PubMed] [Google Scholar]
- 28.Michalik M, Soczek E, Kosińska M, Rak M, Wójcik KA, Lasota S, Pierzchalska M, Czyź J, Madeja Z. Lovastatin-induced decrease of intracellular cholesterol level attenuates fibroblast-to-myofibroblast transition in bronchial fibroblasts derived from asthmatic patients. European Journal of Pharmacology. 2013;704:23–32. doi: 10.1016/j.ejphar.2013.02.023. [DOI] [PubMed] [Google Scholar]
- 29.Xu L, Dong X-W, Shen L-L, Li F-F, Jiang J-X, Cao R, Yao H-Y, Shen H-J, Sun Y, Xie Q-M. Simvastatin delivery via inhalation attenuates airway inflammation in a murine model of asthma. International Immunopharmacology. 2012;12:556–564. doi: 10.1016/j.intimp.2012.01.012. [DOI] [PubMed] [Google Scholar]
- 30.McKay A, Leung BP, McInnes IB, Thomson NC, Liew FY. A novel anti-inflammatory role of simvastatin in a murine model of allergic asthma. The Journal of Immunology. 2004;172:2903–2908. doi: 10.4049/jimmunol.172.5.2903. [DOI] [PubMed] [Google Scholar]
- 31.Kim DY, Ryu SY, Lim JE, Lee YS, Ro JY. Anti-inflammatory mechanism of simvastatin in mouse allergic asthma model. European Journal of Pharmacology. 2007;557:76–86. doi: 10.1016/j.ejphar.2006.11.027. [DOI] [PubMed] [Google Scholar]
- 32.Weitz-Schmidt G, Welzenbach K, Brinkmann V, Kamata T, Kallen J, Bruns C, Cottens S, TAKADA Y, Hommel U. Statins selectively inhibit leukocyte function antigen-1 by binding to a novel regulatory integrin site. Nature Medicine. 2001;7:687–692. doi: 10.1038/89058. [DOI] [PubMed] [Google Scholar]
- 33.Falanga YT, Chaimowitz NS, Charles N, Finkelman FD, Pullen NA, Barbour S, Dholaria K, Faber TW, Kolawole EM, Huang B, Odom S, Rivera J, Carlyon J, Conrad DH, Spiegel S, Oskeritzian CA, Ryan JJ. Lyn but Not Fyn Kinase Controls IgG-Mediated Systemic Anaphylaxis. The Journal of Immunology. 2012;188:4360–4368. doi: 10.4049/jimmunol.1003223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Speiran K, Bailey DP, Fernando J, Macey M, Barnstein B, Kolawole EM, Curley D, Watowich SS, Murray PJ, Oskeritzian CA, Ryan JJ. Endogenous suppression of mast cell development and survival by IL-4 and IL-10. Journal of Leukocyte Biology. 2009;85:826–836. doi: 10.1189/jlb.0708448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ness GC, Chambers CM, Lopez D. Atorvastatin action involves diminished recovery of hepatic HMG-CoA reductase activity. The Journal of Lipid Research. 1998;39:75–85. [PubMed] [Google Scholar]
- 36.Chasman DI, Posada D, Subrahmanyan L, Cook NR, Stanton VP, Ridker PM. Pharmacogenetic Study of Statin Therapy and Cholesterol Reduction. The Journal of the American medical Association. 2004;291:2821–2827. doi: 10.1001/jama.291.23.2821. [DOI] [PubMed] [Google Scholar]
- 37.Schmitz G, Drobnik W. Pharmacogenomics and pharmacogenetics of cholesterol-lowering therapy. Clinical Chemistry and Laboratory Medicine. 2003;41:581–589. doi: 10.1515/CCLM.2003.088. [DOI] [PubMed] [Google Scholar]
- 38.Kajinami K, Brousseau ME, Ordovas JM, Schaefer EJ. CYP3A4 Genotypes and Plasma Lipoprotein Levels Before and After Treatment With Atorvastatin in Primary Hypercholesterolemia. The American Journal of Cardiology. 2004;93:104–107. doi: 10.1016/j.amjcard.2003.08.078. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.