

Abstract
Long-term memory (LTM) of fear stores activity dependent modifications that include changes in amygdala signaling. Previously, we identified an enhanced probability of release of glutamate mediated signaling to be important in rat fear potentiated startle (FPS), a well-established translational behavioral measure of fear. Here, we investigated short- and long-term synaptic plasticity in FPS involving metabotropic glutamate receptors (mGluRs) and associated downstream proteomic changes in the thalamic-lateral amygdala pathway (Th-LA). Aldolase A, an inhibitor of phospholipase D (PLD), expression was reduced, concurrent with significantly elevated PLD protein expression. Blocking the PLD-mGluR signaling significantly reduced PLD activity. While transmitter release probability increased in FPS, PLD-mGluR agonist and antagonist actions were occluded. In the unpaired group (UNP), blocking the PLD-mGluR increased while activating the receptor decreased transmitter release probability, consistent with decreased synaptic potentials during tetanic stimulation. FPS Post-tetanic potentiation (PTP) immediately following long-term potentiation (LTP) induction was significantly increased. Blocking PLD-mGluR signaling prevented PTP and reduced cumulative PTP probability but not LTP maintenance in both groups. These effects are similar to those mediated through mGluR7, which is co-immunoprecipitated with PLD in FPS. Lastly, blocking mGluR-PLD in the rat amygdala was sufficient to prevent behavioral expression of fear memory. Thus, our study in the Th-LA pathway provides the first evidence for PLD as an important target of mGluR signaling in amygdala fear-associated memory. Importantly, the PLD-mGluR provides a novel therapeutic target for treating maladaptive fear memories in posttraumatic stress and anxiety disorders.
1. Introduction
1.1
Storage of long-term memory (LTM) plays an important role in pathological states of fear and anxiety. However, the mechanisms underlying these conditions are yet to be understood. Fear potentiated startle (FPS) is an ideal translational paradigm for studying memory mechanisms of anxiety and fear in humans (Grillon et al., 1991; Grillon and Davis, 1997; Grillon et al., 2011; Schmitz et al., 2014) and animals (Campeau and Davis, 1995a; b; Kazama et al., 2013; Parsons and Davis, 2011; Sananes and Davis, 1992). In FPS (a classical conditioning paradigm), a neutral conditioned stimulus (CS, tone) paired with an aversive unconditioned stimulus (UCS, a footshock) results in fear associated learning, where subsequent CS alone elicits fear as heightened startle responses (Davis, 1986; Davis et al., 1993). Hence, FPS is an appropriate model to investigate the physiological and neurochemical mechanisms expressed during long-term memory of specific cue-induced fear conditioning.
1.2
The amygdala is an essential brain area that is involved in anxiety, fear, and other forms of emotional learning and memory (Johansen et al., 2011; Rogan et al., 1997). In brain imaging studies, the degree of activation in the amygdala during fear recall correlates with the extent of conditioning (LaBar et al., 1998). In addition, lesions in the amygdala result in selective deficits of facial and auditory recognition of fear in humans (Feinstein et al., 2011). The lateral nucleus of the amygdala (LA) receives auditory sensory inputs directly from the thalamus as well as indirectly from the cortex, and serves as a sensory interface (LeDoux et al., 1990a; LeDoux et al., 1990b; LeDoux and Farb, 1991; McDonald, 1998; Pitkanen et al., 1997). Functional inactivation (Muller et al., 1997; Wilensky et al., 1999) or lesions (Amorapanth et al., 2000; LeDoux et al., 1990a; Nader et al., 2001) in the LA result in inability to acquire/recall fear memory. We previously demonstrated that synaptic plasticity recorded after FPS involves alterations in N-methyl-D-aspartate receptor (NMDARs) and non-NMDAR neurotransmission in the thalamic-LA (Th-LA) neural circuit (McKernan and Shinnick-Gallagher, 1997; Zinebi et al., 2002; Zinebi et al., 2001; Zinebi et al., 2003).
1.3
Long-term synaptic potentiation (LTP) is one type of synaptic plasticity that underlies the cellular mechanism of learning and memory (Bliss and Collingridge, 2013; Malenka and Bear, 2004; Tsvetkov et al., 2002). Depending on the induction protocol, LTP elicited in the Th-LA neural circuit can be prevented by inhibition of NMDARs, group I metabotropic glutamate receptors (mGluR5, mGluR1) or voltage-gated calcium channels (Bauer et al., 2002; Shin et al., 2010; Weisskopf et al., 1999). These results raise the possibility of additional excitatory signaling in FPS LTM in the LA, particularly via mGluRs, well known for their role in synaptic plasticity.
1.4
Metabotropic GluRs make attractive therapeutic targets in a variety of pathological states through modulation of synaptic plasticity (Nicoletti et al., 2011; Nicoletti et al., 2015; Ribeiro et al., 2010). The mGluRs can be divided into three groups (I, II and III) on the basis of sequence similarity, pharmacology, and the preferred signal transduction mechanisms. Group I mGluR (consisting of mGluR1 and mGluR5) antagonists block contextual conditioning (Nielsen et al., 1997), leading to a transient up-regulation of mGluR5 in the hippocampus (Riedel et al., 2000) while activation of mGluR5 is necessary for fear memory and LTP in the amygdala (Fendt and Schmid, 2002; Lee et al., 2002; Rodrigues et al., 2002). Additionally, within group III mGluRs, (consisting of mGluR4, mGluR6, mGluR7 and mGluR8), mGluR7, which is the most widely distributed of mGluRs (Flor et al., 1997) and closely associated with vesicle release sites (Shigemoto et al., 1996), has a role in acquisition and extinction of fear memory (Fendt et al., 2013; Masugi et al., 1999). Though phospholipase C (PLC) is the canonical pathway associated with group I mGluR activation, group III mGluRs, which are classically linked to inhibition of adenylyl cylase, are similarly known to associate with PLC (Perroy et al., 2000). However, mGluRs are also known to signal via other phospholipases. For example, cysteine sulfinic acid (CSA), an amino acid released in the hippocampus during specific conditions of high frequency stimulation (Klancnik et al., 1992), is an agonist at the mGluR isoform linked to another phospholipase, phospholipase D (PLD) (Boss et al., 1994). This unique receptor subtype is defined by its exclusive response to a specific antagonist, PCCG-13 (Albani-Torregrossa et al., 1999). Consequently, PLD activation downstream from selective mGluR activation can mediate aspects of glutamate signaling (Frohman, 2015; Klein, 2005) but the mechanisms of PLD-mGluR actions on synaptic transmission are largely unknown (Cuellar et al., 2005; Fuortes et al., 2010; Rico and Merlin, 2004).
1.5
In the present study, we investigated neuronal plasticity in the Th-LA pathway during FPS LTM (Duvarci and Nader, 2004). We examined the downstream elements underlying mGluR signaling during FPS LTM, as well as neurochemical changes in the expression of rat amygdala proteins. We identified a decreased expression of aldolase A, the aldolase isoform most abundantly expressed in human brain (Buono et al., 2001) which directly inhibits phospholipase D (PLD) (Kim et al., 2002), a downstream signaling target of mGluRs (Boss and Conn, 1992; Klein et al., 1997; Pellegrini-Giampietro et al., 1996; Shinomura et al., 2000). We showed that anti-PLD antibodies immunoprecipitate aldolase A in the amygdala and that PLD activity and proteins are increased in FPS LTM. Subsequently, we analyzed changes in synaptic plasticity produced by the PLD-mGluR agonist and antagonist. Finally, we tested whether PLD-mGluR in the LA plays a role in the expression of long-term fear memory.
2. Methods
2.1 Animals
All animal procedures were carried out in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health (NIH) and approved (Approval ID: 8907176) by the Institutional Animal Care and Use Committee (IACUC) at the University of Texas Medical Branch at Galveston (UTMB). Male Sprague-Dawley albino rats (Harlan, Houston, TX, USA) aged three-four weeks and weighing approximately 45 gm at arrival, were used as subjects. After three days acclimation, animals were randomly divided into unpaired (UNP) and paired (PA) groups and housed in a temperature-controlled room at 22–24°C with a 12 h light/dark cycle and fed a standard laboratory chow diet and water ad libitum. Animals were housed two per cage and except for routine handling were kept undisturbed in the isolated stress-free animal facility throughout the experimental schedule.
2.2 Apparatus
Acoustic startle is augmented in the presence of a conditioned stimulus (CS) (Brown et al., 1951; Davis and Astrachan, 1978) and this response is termed fear-potentiated startle (FPS) (Davis, 1986). Animals were trained and tested using enclosures equipped with a foot-shock grid fixed atop a stabilimeter/accelerometer (San Diego Instruments, CA, USA). Startle was measured by the accelerometer, converted to voltage, and stored on computer. Startle was defined as the maximum displacement during 200 ms following the onset of the startle stimulus. FPS testing sessions were performed in separate chambers to control for effects of contextual conditioning.
2.3 Fear Potentiated Startle
Training and testing were performed as described earlier (McKernan and Shinnick-Gallagher, 1997; Scott and Shinnick-Gallagher, 2005). Briefly, the animals were acclimated to the training and testing chambers for 10 min each on the first day (day one). On day two, the animals were habituated to the startle stimulus by exposing them to 20 white noise bursts [95 decibels (dB)]. Following habituation, the animals received either a paired (PA) or unpaired (UNP) protocol. The CS, a 72 dB filtered, white noise tone of 3.7 s duration, was paired 10 times a day for two days and co-terminated with the unconditioned stimulus (UCS), a 0.5 mA foot shock of 0.5 s duration in the PA group using a pseudorandom inter-trial interval. UNP controls received the same number of CS and UCS stimuli presented, independently without pairing, in a pseudorandom fashion. On day three (24 h after the last FPS training), the rats were tested using 10 startle stimuli to habituate the animals. This was followed by 10 startle stimuli paired to CS and randomly interspersed with 10 startle stimuli alone. FPS was measured as a change in startle magnitude elicited by CS-startle stimulus pairing compared to that elicited by the startle stimulus alone. A startle greater than 30% of baseline startle was defined as FPS (Walker and Davis, 2002). Due to variability caused by non-responding, animals were tested prior to pharmacological and electrophysiological studies as described below. Since consolidation of fear memory are rapid in the amygdala (Nader and Hardt, 2009; Rodrigues et al., 2004; Schafe et al., 2005; Schafe and LeDoux, 2000) slices were obtained 24 hours after the FPS test (Duvarci and Nader, 2004; Monfils et al., 2009).
2.4 Brain Slice Preparation
Twenty-four hours after testing, animals were sacrificed by decapitation and the brains quickly removed. Typically, only one slice/animal was used in each experimental paradigm where “n” equals the number of animals/slices. No anesthetics were used prior to decapitation to avoid their influence on neuronal plasticity. The brains were sliced (500 μm thick) on a Vibroslicer while being superfused with ice-cold artificial cerebrospinal fluid (ACSF) consisting of the following: 117 mM NaCl; 3.0 mM KCl; 2.5 mM CaCl2; 1.2 mM MgCl2; 25 mM NaHCO3; 1.2 mM NaH2PO4; and 11.5 mM glucose. ACSF was continually bubbled with 95% O2 and 5% CO2 (Carbogen) to maintain a pH of 7.4 ± 0.1. Slices were allowed to gradually rise to room temperature (RT) for approximately 2 h prior to electrophysiological recording.
2.5 Two Dimensional Gel Electrophoresis
Pooled protein samples (13 animals/group) as described in later sections contained approximately 150 μg of protein and were brought up to a volume of 350 μl with 2-D lysis buffer (containing 1.8 μl IPG buffer, Amersham Pharmacia Biotech). Isoelectric focusing strips (pH range 3–10, 18 cm) were passively hydrated with sample for 12 hr. Isoelectric focusing was carried out using a total of 58.5 kV. Prior to SDS-PAGE, the sample loaded strips were incubated for 10 min in a reducing equilibration buffer consisting of: 50 mM Tris HCl pH 6.8, 6 M urea, 30% (v/v) glycerol, 2% (w/v) sodium dodecyl sulfate (lauryl sulfate, SDS), and 10 mg/ml dithiothreitol (DTT). Following reduction, the strips were incubated for 10 min in alkylating equilibration buffer composed of: 50 mM Tris HCl pH 8.8, 6 M urea, 30% (v/v) glycerol, 2% (w/v) SDS, and 40 mg/ml iodoacetamide. For the second dimension separation, the strips were loaded onto 10% SDS-polyacrylamide gels and separated for 1 h at 75 V and 9 hr 45 min at 140 V. The spots were visualized on the acrylamide gel for imaging and subsequent quantification using silver iodide treatment as specified in the Silver Stain Plus kit (Bio-Rad, Hercules, CA). The intensity of each protein spot was determined, normalized to the sum of intensities of all spots on a gel, and quantified as a percentage volume in each gel using Progenesis 2D software. Individual protein spots were then matched with the identical protein spot in gels obtained from each animal within a treatment group. The matched spots were averaged from the separate gels within each animal group. The average normalized volume of each spot in PA group was compared to that of the matched spot in the UNP group. Differences greater than two-fold in expression were considered significant. The spots were identified with matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS). These studies were repeated seven times. Changes in protein expression were verified by Western blotting. Image analyses and 2-D gel running conditions of the blots were performed in the UTMB Proteomic Laboratory essentially as reported earlier (Straub et al., 2009).
2.6 PLD Activity Assay
Twenty-four hours after testing, animals were decapitated without anesthesia to avoid drug effects on plasticity induced in brain tissue. Coronal brain slices (350 μm) were placed in ice-cold (0–6°C) aerated ACSF. Amygdalae were dissected from each brain slice of the two treatment groups and placed in test tubes containing about 2 ml of Kreb’s bicarbonate buffer consisting of 22 mM NaCl; 3.1 mM KCl; 1.2 mM MgSO4; 0.4 mM KH2PO4; and 1.3 mM CaCl2. The pH of the buffer containing the slices was maintained at 7.4 throughout the day by continuous aeration with carbogen. The amygdala slices remained in the test tubes at RT for 30 min, and were then placed in a water bath (37°C) for 30 additional minutes. Kreb’s buffer was removed from the test tubes and replaced by 1 ml ACSF containing 30 μCi of tritiated glycerol per sample. The slices were exposed to the ACSF containing the radioactive material for 2 hr. Amygdala slices were then washed with 2 ml ACSF and replaced with 500 μl ACSF, containing ethanol (5 μl per sample), ethanol and a drug, or an additional ACSF equivalent for a negative control. The slices remained exposed to the drug and ethanol for 1 h while remaining in the water bath. The reaction was stopped by addition of 2 ml of ice-cold chloroform/methanol/HCl (100:200:2) to each test tube. The samples were sonicated for 30 min and centrifuged at 3000 rpm for 2 min. The lower organic layer was then dried by N2 gas and then dissolved in 70 μl of chloroform. The solubilized organic layers were spotted on silica gel coated TLC plates in about 3 μl increments till all the 70 μl slurry was spotted in the same region (adding in increments reduced diffusion to greater extent than spotting the entire amount at one time). The plate was placed in a large rectangular glass chamber containing a minimum of 100 ml of a solvent system consisting of ethyl acetate: 2,2,4–trimethyl pentane (also called iso-octane): acetic acid: methanol: water in 60:80:20:20:10 ratio. After 30 min, the plate was removed from the glass chamber and placed in another chamber containing a bottom layer of iodine crystals for 15 min. The phosphatidyl ethanol (PEtOH) sample was determined by collecting 1 mm of sample from the plate, which was run between standards. The phospholipids remaining in the lanes of each sample were scraped from the plate and collected in individual vials of scintillation fluid to determine the ratio of PEtOH/Total phospholipids.
2.7 Western Blot Analysis
Twenty four hours after testing, the dissected brain tissue was removed from the rat and was frozen rapidly on dry ice to remove the lateral amygdala as described elsewhere (Lamprecht et al., 2006). Briefly, the removal of the specialized small area of the lateral amygdala nucleus, which is difficult to recover using conventional surgical instruments, was punched from this frozen tissue using a special corer, Deluxe Aluminum Harris Micro-Punch (Ted Pella Inc., Redding, CA), that allows isolation of 0.5 mm, under the dissecting microscope and the punched tissue was stored at −80°C until homogenization. Frozen amygdala tissues were homogenized in cold (4°C) modified radioimmunoprecipitation (RIPA) buffer which contained 50 mM Tris, 150 mM NaCl, 1% NP-40, 0.5% Na-deoxycholate, 0.1% SDS, in the presence of protease inhibitor cocktail consisting of 1 mM AEBSF, 15 μM pepstatin A, 14 μM E-64, 40 μM bestatin, 22 μM leupeptin, and 0.8 μM aprotinin or Complete™ Mini EDTA-free Protease Inhibitor Cocktail Tablets (Roche Biochemica, Indianapolis, IN). Homogenates were centrifuged twice at 10,000 g for 10 min at 4°C. The supernatant was collected and stored at −80°C until use. Protein concentrations of the fractions were determined using the BioRad DC protein assay kit (BioRad Laboratories, Hercules, CA). Equal amounts of protein (whole cell lysate) were separated by SDS-PAGE, and transferred onto a polyvinylidene difluoride (PVDF) membrane. After blocking with 5% non-fat milk in TBS-Tween buffer, the transferred membrane was then incubated for 1.5 h at RT with the primary antibodies. The blots were washed four times for 10 min with TBS-Tween buffer and then incubated in horseradish peroxidase–conjugated IgG for 1.5 h at RT. The bands were visualized by enhanced chemiluminescence (ECL Plus; Amersham, Arlington Heights, IL) and quantified by densitometry using Lynx V software. To control for variability in sample loading and protein concentrations between samples, the ratio of densities of actin was used to normalize values between samples. For co-immunoprecipitation studies, antibodies (45 μg per reaction) were covalently crosslinked onto protein A/G resin according to the manufacturer’s instructions (Pierce© Crosslink Immunoprecipitation Kit, Pierce Biotechnology, Rockford, IL). Prior to the immunoprecipitation, 400–500 μg of synaptosomal protein was incubated for 1 hr at RT with Control Agarose Resin© to reduce non-specific protein binding. The pre-cleared synaptosomal lysate was incubated with the antibody-crosslinked resin overnight at 4°C with constant shaking. The suspension was centrifuged at 1,000 g for 30 sec and the flow-through was collected (unbound antigen). The bound antigen-antibody-crosslinked resin was washed twice with IP Lysis/Wash buffer, then once with 1X Conditioning Buffer©. The complex was incubated for 5 min at RT in Elution Buffer©. Neutralization buffer (1 M Tris pH 9.5; 1/3 of the Elution buffer volume) was added to the collection tube to adjust the eluate to physiological pH. Samples were centrifuged and the flow-through was collected. The eluted antigen was subjected to cold acetone precipitation (four volumes of acetone for 3 hr at −20°C). The sample was centrifuged at 15,000 g for 10 min at 4°C, the acetone was decanted and the precipitated protein was resuspended in 2X loading buffer and subjected to SDS-PAGE. Immunoblotting with appropriate antibodies were performed as described above. Antibodies were obtained from different sources rabbit polyclonal antibodies for PLD1 (sc-25512), PLD2 (sc-25513) aldolase A (sc-30082) at 1:200 dilution, and goat polyclonal actin (sc-1616) at 1:1000 dilution from Santa Cruz Biotechnologies (San Diego, CA); rabbit polyclonal antibody for mGluR4 (AB15097) at 1:5000; mGluR7 (07-239) at 1:400 from Millipore (Temecula, CA).
2.8 Field Potential Recordings
Extracellular field potentials were recorded using a tungsten electrode (2–5 MΩ) and amplified using a microelectrode AC amplifier (A-M Systems, Carlsborg, WA). Bipolar concentric stimulating electrodes (50–75 KΩ) were placed within fiber pathways arising from the thalamus (McKernan and Shinnick-Gallagher, 1997), and orthodromic synaptic potentials were elicited with an isolated pulse stimulator (A-M Systems, Carlsborg, WA). Biphasic pulses (100 μsec), typically ranging from 4 V to 15 V, evoked field excitatory postsynaptic potentials (fEPSPs) in the LA and fEPSPs were included for study if the maximum amplitude was above 0.8 mV (without spiking). To ensure monosynaptic fEPSPs, only fEPSPs that had a short synaptic latency (time from stimulus to onset of fEPSP) and an initial slope that was graded smoothly with increases in stimulus intensity were considered acceptable for recording. For analysis of synaptic plasticity, the stimulus intensity was adjusted to 20–50% of maximum fEPSP amplitude. Experimenters were blinded to the animal groups and slices from experimental and control animals were recorded simultaneously in the same chamber to control for day-to-day variation. Stimulation protocols were generated and off-line analyses of fEPSP recordings performed using pClamp 9.0 (Axon Instruments, Foster City, CA, USA). Averaged traces of six successive sweeps were used for analysis of fEPSPs. Slope (10–90% amplitude) was used as the measure of fEPSP amplitude as it is directly proportional to peak conductance and sums in a linear fashion. Slices were maintained at 32 ± 1°C and drugs applied by bath application. High frequency stimulation (HFS) consisting of five trains of stimuli (100 Hz for 1 sec, 3 min intervals) applied to the thalamic pathway was used to evoke long-term potentiation (LTP) in some experiments. Drug application preceded the HFS by 5 min and was continued for the duration of the stimulation.
2.9 Cannula placement and infusion experiments
After acclimating to a low noise and stress environment after arrival, rats were anesthetized with i.p. injections of 90 mg/kg ketamine plus 10 mg/kg xylazine (Vedco, St. Joseph, MO). The rat remained on a isothermal heating pad set to a low temperature from inception of the surgical procedure until recovery to a fully awake state. Based on animal size, age adjustments were made to the co-ordinates to guide the cannulae into the lateral amygdala (LA) for infusions. Specifically, bilateral cannulae (Plastics One, Roanoke, VA) were stereotaxically placed in the LA [anterioposterior −2.8 mm from bregma, lateral +/−4.9 mm and dorsoventral −5.0 mm (Paxinos and Watson, 1998)], according to previously described procedures (Gardiner and Toth, 1999). Except during injections, solid dummy needles were placed in the cannulae to prevent clogging. The area surrounding the cannulae implant was cleaned carefully and topical analgesic Xylocaine® 2% jelly (lidocaine HCl) and triple antibiotic were applied at the incision sites to ameliorate any localized pain during recovery from anesthesia and prevent infection respectively. Following recovery, rats were returned to their home cages, weighed, and monitored daily for recovery. Animals were allowed to recuperate from surgery for five days and then FPS training and testing was performed using procedures described above. Twenty-four hours following the FPS testing session, animals were bilaterally infused with 0.5 μl of either PCCG-13 (8.5 μM) in vehicle (0.0134% beta cyclo-dextrin, BCD, in 0.9% saline) or vehicle alone over a period of 20 min and the injection needle was kept in place for 5 min of post-infusion time. The animals were then allowed to remain in their home cage for 15 min prior to testing FPS. This second testing occurred after the animals had acquired and reconsolidated FPS. Twenty four hours after the test, animals were infused with 0.5 μl of methylene blue dye in dimethyl sulfoxide (DMSO), sacrificed and brain slices prepared to verify cannulae placement.
2.10 Statistics
Slices were cut from both hemispheres and one slice per hemisphere was used in each set of experimental treatments. Analyses were performed using Clampfit 9.0 software (Molecular Devices, Sunnyvale, CA). Individual traces were filtered and six consecutive traces were averaged. Averaged responses were measured 50–60 min after wash-out and normalized to baseline responses, for subsequent statistical analyses. For immunoblotting analysis, the means and standard error were calculated from a minimum of three samples. To account for non-normal distribution of data, non-parametric tests were used for statistical analysis. Behavioral data was analyzed using either one-way ANOVA (Kruskal-Wallis test) or repeated measures two-way ANOVA followed by a paired or unpaired t-test when significance was achieved. Western blot and electrophysiological data were analyzed using a Kruskal-Wallis test followed by a Mann-Whitney U or Wilcoxon matched pair test as appropriate for pair-wise comparison. Statistical significance was defined at p<0.05.
3. Results
3.1 Amygdala aldolase A expression levels are decreased in FPS LTM
3.1.1
The amygdala is a crucial brain region involved in fear memory mechanisms (Adolphs et al., 1995; Blair et al., 2001; Cahill et al., 1999; Charney, 2003; Davis, 1992; 1997; Davis et al., 1993; Ehrlich et al., 2009; LeDoux, 2000; Maren, 1996; 2003; Shinnick-Gallagher et al., 2003) and therefore plays an important role in FPS LTM state. However, the molecular signaling events exhibited in FPS LTM in the amygdala, particularly downstream from mGluRs, are not well characterized. By using two-dimensional gel electrophoresis, we investigated potential signaling elements in the amygdala contributing to FPS LTM. Rats were subjected to FPS training and amygdala proteins obtained from the crude synaptosomal preparations (6–7 animals/group) were separated using the 2-D gel electrophoresis. The total number of averaged spots on the 2-D gel from the paired (PA) group was 900 compared to 1259 in the unpaired (UNP) group, at 24 hr after the last conditioning session. However, there was greater than two-fold difference using normalized volume as a parameter, where 37 proteins showed increased expression, 44 were decreased, 141 proteins were essentially not measurable, and 2 new spots appeared (Figure 1A) in the PA amygdala compared to the UNP group. Because of the relative low abundance of amygdala protein, we pooled individual spots from naïve, UNP, and PA animals for identification using MALDI-TOF spectral analyses. Despite the pooling, there was still insufficient protein available for analysis. As an alternative approach, we selected the largest protein spot on the raw gel that showed a visually distinct difference between the UNP and PA animals, followed by the MALDI-TOF analyses and identified aldolase A. Analysis of 2-D gels showed a significant decrease in the normalized volume of aldolase A protein in amygdala from PA compared to UNP animals (p<0.05; Figure 1B). Western blot analysis of crude synaptosomal preparations confirmed the decreased expression of aldolase A in amygdala from PA relative to UNP animals when naïve levels were taken as 100% and aldolase A expression was normalized to actin expression (PA: 71.1 ± 9.7%, n=11; UNP: 111.1 ± 16.2%, n=11; p<0.05). Because of the low protein abundance, no other spots were identified by MALDI-TOF. Since aldolase A is widely known for its glycolytic action (Funari et al., 2007; Penhoet et al., 1969) and its probes consequently are not sufficiently discriminating for analysis of neurotransmission, we examined signaling partners of aldolase A, in particular, phospholipase D (PLD) that is known to be co-immunoprecipitated with aldolase A and whose activity is inversely related to aldolase A concentration (Kim et al., 2002). Importantly, specific drugs and assays to quantify PLD activity, particularly via mGluR, in mammalian systems have been reported (Albani-Torregrossa et al., 1999; Attucci et al., 2001; Bhattacharya et al., 2004; Boss and Conn, 1992; Boss et al., 1994).
Figure 1.

FPS decreases amygdala aldolase A expression. A) Top panel shows the decrease in spot in 2-D gels of amygdala protein acquired from PA animals compared to Naïve and UNP. Protein spot (white oval) was identified as aldolase A using MALDI-TOF. B) Normalized volume of protein spot identified as aldolase A demonstrated a 29% decrease in the PA (black bar) compared to UNP (white bar). The boxes above the graph represent the volume difference between PA and UNP as observed in the gels. C) Western blot analysis also showed decreased levels of aldolase A protein in PA (black bar) compared to UNP (white bar). Protein expression relative to the loading control is plotted along the Y-axis. Representative immunoblots are shown in the panels above the graph. * denotes significant difference (p<0.05) compared to UNP group.
3.2 Amygdala PLD protein expression levels are increased in FPS LTM
3.2.1
At low concentrations, aldolase A inhibits PLD activity (Kim et al., 2002). We analyzed whether amygdala protein levels of PLD were affected in the PA group using Western blot analysis. Amygdala PLD1 and PLD2 protein expression were increased in PA compared to UNP animals (Figure 2). In whole amygdala homogenate from PA animals, PLD1 expression levels (as a ratio of actin levels) were significantly greater than those in UNP animals (UNP: 0.50 ± 0.03; PA: 0.63 ± 0.04; n=3, p<0.05, Figure 2A) whereas, PLD2 expression was increased primarily in crude synaptosomal fractions from slices of PA animals as compared to UNP animals (UNP: 0.10 ± 0.005; PA: 0.12 ± 0.009; n=12, p<0.05), suggesting that PLD as a signaling partner in amygdala mediated fear memory mechanisms may be correlated with reduced aldolase A levels. Additionally, we tested whether PLD and aldolase A were coupled in the amygdala using co-immunoprecipitation assays. Naïve amygdala incubated with PLD1/2 antibodies immunoprecipitated a band around 40 kDa that corresponded to aldolase A (Figure 2C). Reciprocal assays with aldolase A antibodies showed only immunoglobulin bands (data not shown). One possible explanation could be an inaccessibility to the antibody epitope when protein is bound to other proteins (Hall, 2004). These data indicated that in FPS LTM the increased expression of PLD might be directly linked to decreased aldolase A in the amygdala.
Figure 2.
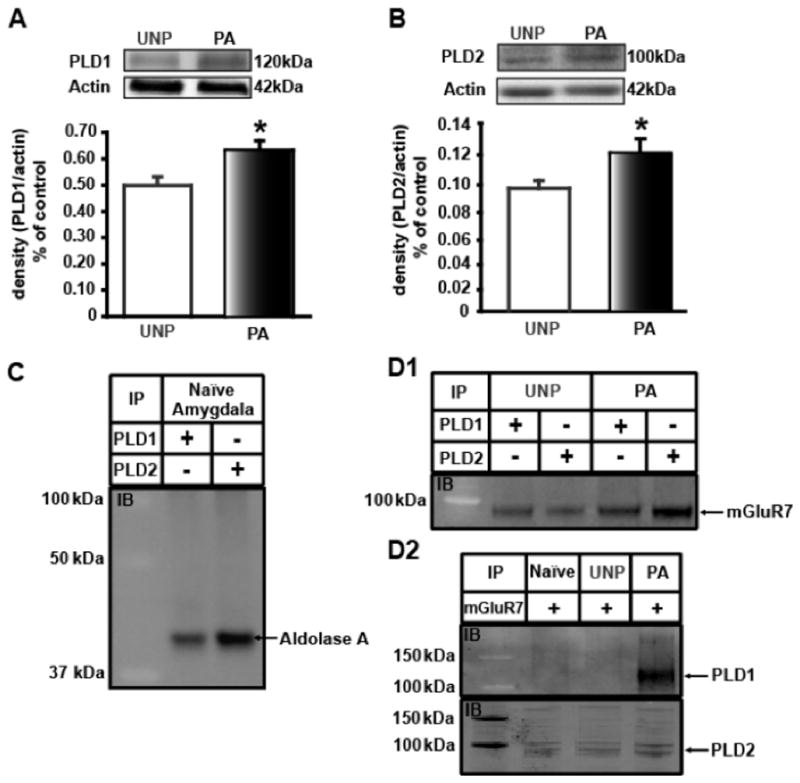
FPS increases amygdala PLD expression. Protein expression relative to the loading control is plotted along the Y-axis. Representative immunoblots are shown in the panels above graph. A) Western blot analysis of whole amygdala homogenate show a significant increase in PLD1 protein expression (by 28%) in the PA group (black bar) compared to UNP animals (white bar). B) Western blot analysis of amygdala crude synaptosomal fractions indicated a significant increase in PLD2 protein expression (by 20%) in the PA group (black bar) compared to UNP animals (white bar). C) Immunoprecipitation (IP) with either PLD1 or PLD2 antibody using naïve amygdala crude synaptosomal fractions was immunoblotted (IB) with Aldolase A antibody. D1) IP with PLD1 or PLD2 antibody using UNP or PA amygdala crude synaptosomal fractions (top panel) immunoprecipitates mGluR7 but not mGluR4 (not shown). D2) The reciprocal experiment using mGluR7 antibody immunoprecipitates PLD1 only in the PA group while PLD2 is pulled down in all three animal groups. The molecular weight markers using protein standards are depicted in kDa in the far left lane of the IP experiments (C, D). * denotes significant difference (p<0.05) compared to UNP.
3.2.2
Previously, we have demonstrated that PLD1/2 protein was co-immunoprecipitated (Krishnan et al., 2011) with mGluR1 and mGluR5 which primarily are found postsynaptically (Shigemoto et al., 1993). Here we assessed whether PLD is associated with presynaptically located group III mGluR receptors (Shigemoto et al., 1997), mGluR4 and mGluR7 (Figure 2D). The other group III presynaptic receptor, mGluR8, was not tested since in the LA it is not associated with changes in LTP or cued fear responses although it is linked to depressed basal transmission (Fendt et al., 2013). Amygdala from fear conditioned animals incubated with anti-PLD1/2 antibodies immunoprecipitated a band around 102 kDa that corresponded to mGluR7 (Figure 2D1) but not the mGluR4 expected band of 100 kDa (data not shown) whereas anti-mGluR7, but not mGluR4 antibody, immunoprecipitated a band around 120 kDa for PLD1 and 100 kDa for PLD2 (Figure 2D2). Moreover, western blot analysis of amygdala crude synaptosomal fractions from UNP and PA groups showed no difference in expression of either mGluR7 or mGluR4 (data not shown). In an earlier study (Krishnan et al., 2011), we also found an increased association of PLD with mGluR1 and mGluR5 despite no increase in the mGluR1 or mGluR5 protein expression. These data suggest that mGluR7 is associated with PLD1/2 in fear learning.
3.3 Amygdala PLD activity is increased in FPS LTM
3.3.1
Since we detected an increase in amygdala PLD expression, we next examined whether the PLD activity was also changed to ascertain if function of this protein was affected in FPS LTM. We observed that basal PLD activity was increased four-fold in amygdala slices from PA animals compared to UNP animals (UNP VEH: 13 ± 2, n=11; PA VEH: 55 ± 8, n=15; p<0.05, Figure 3).
Figure 3.
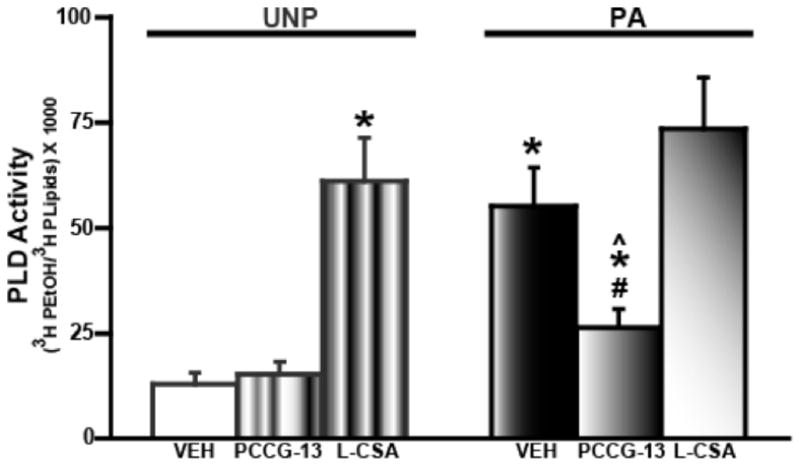
FPS increases and PLD-mGluR antagonist (PCCG-13) reduces amygdala PLD activity. PLD activity is expressed as a ratio of the amount of tritiated phosphatidyl ethanol (3H PEtOH) to total phospholipids (3H PLipids). PLD activity is significantly increased in the PA VEH (black bar) group compared to UNP VEH (white bar). The PLD-mGluR antagonist, PCCG-13, (2 μM) has no effect in the UNP group but decreases the PLD activity in amygdala from PA animals. The PLD-mGluR agonist, L-CSA, (100 μM) increases the PLD activity in the UNP group compared to UNP VEH group but not in the PA L-CSA group compared to PA VEH and UNP L-CSA groups. * denotes p<0.05 compared to UNP VEH control; # indicates p<0.05 compared to PA VEH control and ^ represents p<0.05 compared to UNP PCCG-13.
3.3.2
As we found that both PLD activity and protein expression were increased during FPS LTM (Figures 2, 3), we next investigated whether the increase of PLD was linked to activation of mGluRs. Towards this, we analyzed effects of mGluR antagonists and agonists on PLD activity. Initially, we tested the effect of PCCG-13, a potent antagonist for the PLD-mGluR (Pellicciari et al., 1999), on PLD activity in amygdala slices from PA and UNP animals. PCCG-13 (2 μM) significantly reduced PLD activity by >50% in PA but not UNP amygdala slices (UNP PCCG-13: 15 ± 2, n=11; PA PCCG-13: 26 ± 3, n=15; p<0.05; Figure 3) suggesting that the increased PLD activity was due in part to the PLD-mGluR. We subsequently tested whether L-CSA, an agonist for the PLD-mGluR (Boss et al., 1994), affected PLD activity. It is known that the PLD-mGluR was not blocked by iGluR antagonists or activated by other excitatory amino acids (EAA) (Boss et al., 1994). Thus, L-CSA (100 μM), without addition of iGluR or other EAA antagonists, was applied to amygdala slices from PA and UNP groups. The agonist increased PLD activity in UNP but PLD activity was not significantly different than in PA group (UNP L-CSA: 61 ± 10, n=11; PA L-CSA: 73 ± 12, n=15; p<0.05) when compared to respective vehicle (ethanol) groups. The activity levels observed with application of L-CSA to slices from UNP and PA groups were similar to that observed in the PA vehicle group, suggesting that PLD activity may have been maximally activated in the PA group and that stimulation with L-CSA could not further increase PLD activity.
3.4 Metabotropic GluR signaling via PLD enhanced short-term plasticity is occluded in FPS LTM
3.4.1
Previously our group (McKernan and Shinnick-Gallagher, 1997) reported an increase in transmitter release probability, at the Th-LA synapse in amygdala slices from PA. We next addressed whether the increased PLD activity downstream from mGluR activation alters synaptic plasticity in the thalamo-LA neural circuit. In the present study, we similarly examined the effect of PLD-mGluR on transmitter release probability using mean paired pulse ratios (PPRs). Field EPSPs (fEPSPs) recorded in slices from PA group showed a decrease in PPR (50 ms interstimulus interval at 0.05 Hz) compared to that recorded in the UNP group (Figure 4A), indicating an increase in transmitter release probability. Addition of the mGluR antagonist, PCCG-13, significantly decreased PPR in slices from UNP animals but had no effect in slices from PA animals (UNP: 1.50 ± 0.16, n=6; UNP PCCG-13: 0.98 ± 0.10, n=6; p<0.05 and PA: 1.15 ± 0.06, n=8; PA PCCG-13: 1.21 ± 0.09, n=5; ns; Figure 4B). This suggests that blocking the PLD-mGluR increased presynaptic release of neurotransmitter in the UNP and that its effect was occluded in the PA group. Additionally, PCCG-13 had no significant effect on baseline magnitude of fEPSPs elicited at low stimulus frequencies (0.05 Hz) in either animal group (Figure 4C) which is similar to that reported for an mGluR5 antagonist (Fendt and Schmid, 2002) and mGluR7 agonist (Fendt et al., 2008) in naïve slices.
Figure 4.
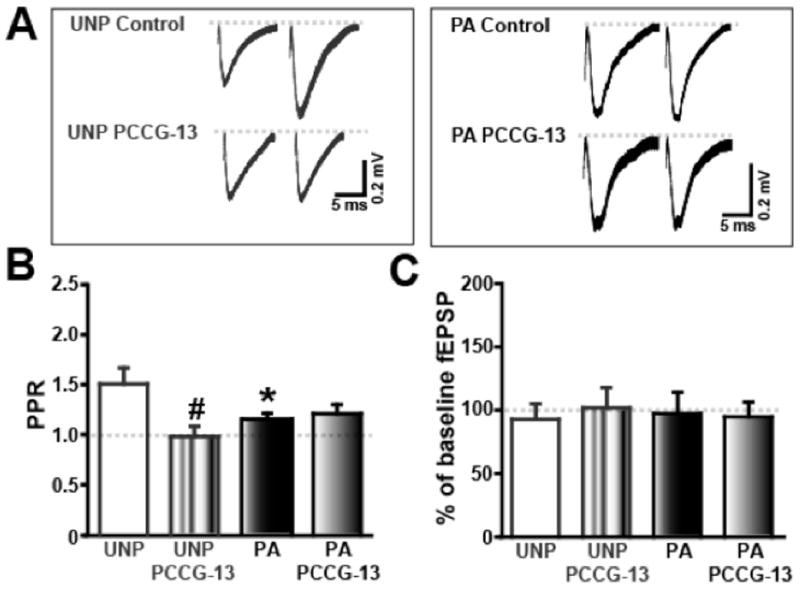
FPS occludes paired pulse facilitation. A) Raw traces of pairs of fEPSPs depict the decreases in the PPR of the PA compared to the UNP group. B) The PLD-mGluR antagonist, PCCG-13, significantly reduces the PPR in UNP group while no effect is measured in the PA group. C) No change is recorded in fEPSP magnitude expressed in either the UNP or PA group with or without PCCG-13. * denotes significant difference (p<0.05) in UNP and PA groups; # indicates p<0.05 in UNP and UNP PCCG-13 treatments. PPR = mean 2nd fEPSP/mean 1st fEPSP.
3.5 PLD-mGluR antagonist decreases post-tetanic potentiation but not LTP maintenance
3.5.1
Our lab (McKernan and Shinnick-Gallagher, 1997) first demonstrated that a lasting increase in synaptic efficacy between afferent and efferent connections recorded in vitro, is a candidate mechanism underlying cued fear responses at LA synapses. In the present study, we utilized LTP and post-tetanic potentiation (PTP) to analyze PLD-mGluR mechanisms in fear memory. We studied the effects of the PLD-mGluR antagonist, PCCG-13, on LTP evoked by high frequency stimulation (HFS, 5 trains, 100 Hz, 1 s, 3 min interval) in amygdala slices from UNP and PA animals (Figure 5A, B). PCCG-13 was applied and fEPSP magnitudes measured. During the last 10 min of LTP (80–90 min of recording) in the maintenance phase of LTP, fEPSPs were not significantly altered in either the UNP (UNP: 144.4 ± 26.4%, n=6; UNP PCCG-13: 152.0 ± 12.4%, n=6; ns; Figure 5A, inset) or the PA (PA: 179.2 ± 27.5%, n=8; PA PCCG-13: 176.6 ± 13.4%, n=5; ns; Figure 5B, inset) or between the PA and UNP groups. There are recent studies that also report an increase in synaptic transmission due to postsynaptic modifications (Rumpel et al., 2005; Yeh et al., 2006) that was not the focus of the current study. We speculate that the fear conditioning paradigm could have resulted in hyperpolarization of the postsynaptic membrane that can result in failure to observe LTP (Malinow and Miller, 1986). Our earlier study (Schroeder and Shinnick-Gallagher, 2004) as well as other groups (Tsvetkov et al., 2002) have reported an occluded LTP in the cortico-LA synapse and another group (Hong et al., 2012) demonstrated a similar occlusion in the Th-LA synapse, typically interpreted as being due to the fact that synapses in fear conditioned animals have already undergone LTP-like changes in synaptic strength and are therefore less capable of showing additional LTP (Sigurdsson et al., 2007).
Figure 5.
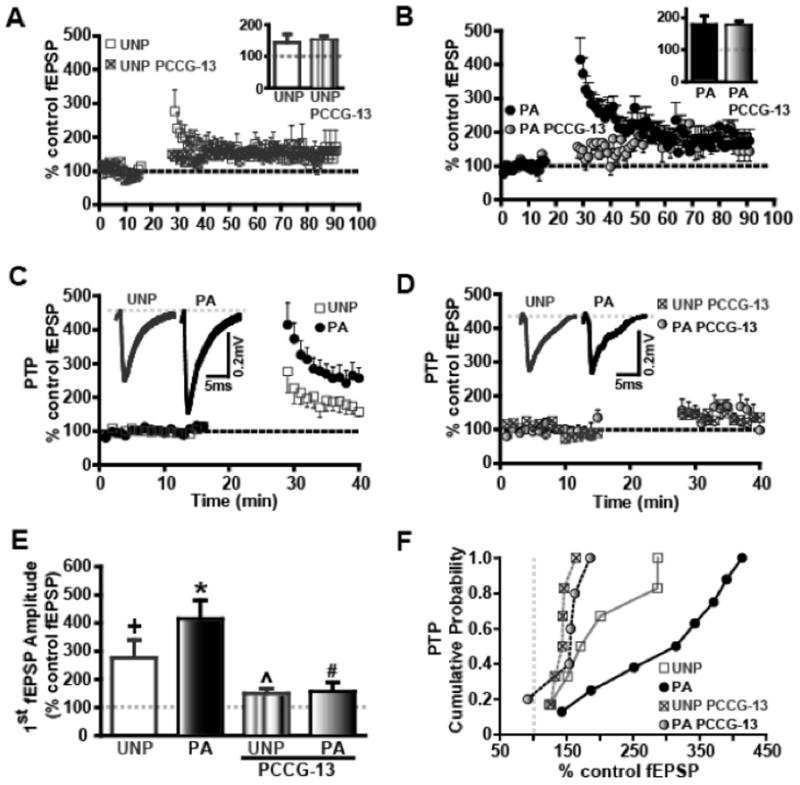
FPS increases first fEPSP post tetanus and PTP (first 5–10min post tetanus) but not maintenance phase of long-term potentiation (LTP). fEPSP measured the last 10 min, at 80–90 min (or 50–60 min following HFS) in the presence of the PLD-mGluR antagonist, PCCG-13, are not significantly different either in the A) UNP or B) PA groups. The percent of control fEPSP is plotted along the Y-axis as a function of time in min along the X-axis. Inset bar graphs depict the mean fEPSP values for last 10 min of the recording with or without PCCG-13 treatment. C) PTP is greater in PA than in UNP groups as recorded during the first 10 min following HFS. Representative traces illustrate the differences recorded between the two groups. D) Following PCCG-13 treatment, PTP in PA and UNP groups is significantly reduced compared to their respective controls. Representative traces recorded within the first 10 min following HFS+PCCG-13 treatment are in figures above graphs. E) Analysis of the first fEPSPs reveals a significant increase in the PA group (black bar) compared to the UNP group (white bar) while first fEPSP amplitudes are significantly diminished following PCCG-13 treatment in both groups. F) Cumulative release probability in PTP was significantly different in UNP and PA groups before but not after PCCG-13 treatment. + denotes p<0.05 in UNP compared to baseline levels; * denotes p<0.05 in PA compared to UNP groups; ^ indicates p<0.05 compared to UNP control group; # symbolizes p<0.05 compared to control PA group.
3.5.2
However, PTP, defined as mean fEPSP potentiation 5–10 min after the tetanus, was significantly enhanced (Figure 5C, E) compared to baseline in both UNP (baseline: 99.5 ± 1.1%; PTP: 214.9 ± 28.3%, n=6; p<0.05) and PA groups (baseline: 97.9 ± 1.9%; PTP: 331.8 ± 38.7%, n=8; p<0.05) and the mean PTP magnitude was significantly greater in the PA group compared to the UNP group (p<0.05, Figure 5C, E). Mean PTP was significantly reduced by PCCG-13 (2 μM) in slices from the UNP group (141.7 ± 10.4, n=6; p<0.05; Figure 5A and 5D), a 115% difference, and from the PA group (144.6 ± 18.1, n=5; p<0.05; Figure 5B and 5D), a 233% difference, as compared to the respective controls but final values were not significantly different between the two groups (ns; Figure 5F). Cumulative probability curves for PTP showed a significant shift to the right in the PA group compared to the UNP control group (p<0.05; Figure 5F). After PCCG-13, cumulative probability was reduced and was not significant between the two groups suggesting that the PLD-mGluR may mediate PTP in both PA and UNP groups. Additionally, when compared between PA and UNP groups, the first fEPSP after the tetanus was significantly greater in the PA compared to the UNP groups (UNP: 276.7 ± 63.4%, n=6; PA: 414.8 ± 65.8%, n=8; p<0.05; Figure 5E). The first fEPSP (Figure 5E) was significantly reduced after PCCG-13 (2 μM) application in slices from both UNP (149.5 ± 19.0%, n=6; p<0.05) and PA (157.0 ± 33.5%, n=5; p<0.05) compared to their respective untreated controls.
3.6 PLD-mGluR agonist decreases fEPSP magnitude and transmitter release probability in UNP but not PA groups
3.6.1
Subsequently, we tested the specificity of the signaling mechanism by studying the effect of activating PLD-mGluR utilizing a selective agonist, L-CSA. However, L-CSA also activates N-methyl D-aspartate (NMDA) receptors (EC50 = 59 μM) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors (EC50 = 3.3 mM) (Curras and Dingledine, 1992). To address these confounding effects, we utilized (2R)-amino-5-phosphonovaleric acid (APV, antagonist for NMDA receptors) and lower concentrations of L-CSA to reduce an effect on AMPA receptors (Boss and Conn, 1992; Boss et al., 1994). Under these conditions, LTP at Th-LA synapses is blocked. We found that repetitive 10 Hz tetani applied at 5 sec intervals, a frequency that associated with release of endogenous CSA in the hippocampus (Klancnik et al., 1992), did not cause a persistent rundown of fEPSPs. The stable amplitude of the last ten fEPSPs and unchanging magnitude of first fEPSP at low concentrations of L-CSA in UNP and UNP groups indicate a reproducible response to the tetanus (without decrement of fEPSPs with repetitive tetani). We tested the effect of different concentrations of L-CSA on fEPSPs in the presence of APV (50 μM) to block NMDA receptor activation (Figure 6A). Initial fEPSP magnitudes in APV-treated slices from PA animals at low concentrations of L-CSA were not different than those in slices from UNP animals (Figure 6A inset and 6B). However, L-CSA depressed the first fEPSP in slices from UNP animals at the two higher concentrations (UNP APV: 99.32 ± 9.22%, n=9; UNP L-CSA 1 μM: 100.15 ± 13.19%, n=6; UNP APV/L-CSA 10 μM: 94.42 ± 7.38%, n=5; PA APV: 94.71 ± 7.50%, n=9; PA APV/L-CSA 1 μM: 113.15 ± 13.40%, n=6; PA APV/L-CSA 10 μM: 114.95 ± 10.77%, n=6; Figure 6C) compared to the PA group, where L-CSA had no significant effect on the first fEPSP except at the 1 mM concentration (UNP APV/L-CSA 100 μM: 77.21 ± 11.71%, n=5; UNP APV/L-CSA 1 mM: 52.30 ± 12.27%, n=5; PA APV/L-CSA 100 μM: 117.70 ± 14.50%, n=6; PA APV/L-CSA 1 mM: 49.94 ± 12.85%, n=6; Figure 6C) which was not significantly different than APV/L-CSA (1mM) in the UNP group. The mean values of the last 10 fEPSPs of the tetanus were also not significantly different at lower concentrations of L-CSA (<1 mM) in the UNP and PA groups except in control (UNP: 38.55 ± 2.27%, n=9; PA: 55.30 ± 6.11%, n=8; p<0.05) and APV (UNP: 39.15 ± 2.20%, n=9; PA: 48.69 ± 4.63%, n=9; p<0.05) treatment groups (Figure 6B). This inhibitory effect of a high concentration (1 mM) of L-CSA (Figure 6B, C) in both groups may be due to activation and desensitization of AMPA receptors. These data suggested that activating the PLD-mGluR depressed tetanic synaptic transmission at a concentration (100 μM) less likely to activate AMPA receptors in the UNP group, but not the PA groups.
Figure 6.

The PLD-mGluR agonist inhibits fEPSPs during a 10Hz tetanus in the UNP group A) fEPSP amplitudes induced by 5 s tetani (10 Hz) are plotted at different concentrations of the PLD-mGluR agonist, L-CSA. APV (50 μM) is added to block L-CSA activation of NMDA receptors. fEPSP amplitude is % of baseline control. Inset shows expanded version of graph indicated by Ψ in A. PA is represented by black circles while UNP group is indicated by light grey squares. B) Bar graph plots of fEPSP magnitudes evoked by the last 10 stimuli within the 5 s tetanus show that L-CSA at 1 mM concentration has a pronounced inhibitory effect on the last 10 sweeps of fEPSPs in the PA group (dark bars) and UNP group (white bars). Representative traces depict the differences observed between the two groups, where the numbers correspond to the bars that they represent. C) A significant decrease in the first fEPSP is observed in PA (black circles) as well as UNP (light grey squares) groups at higher concentrations of L-CSA. * represents significant difference (p<0.05) between PA and UNP groups; # denotes significant difference (p<0.05) between L-CSA 100 μM and 1 mM in the PA group.
3.6.2
PPR measurements (Figure 7) were examined to determine whether the L-CSA-induced fEPSP decrease could be related to changes in release probability. In UNP slices treated with APV (0.97 ± 0.06, n=6), L-CSA significantly increased PPR at 100 μM (1.43 ± 0.16, n=6; p<0.05) and 1 mM (1.42 ± 0.13, n=6; p<0.05) suggesting a decrease in neurotransmitter release probability in the UNP group. Between group comparisons showed L-CSA (in the presence of APV) increases PPR in UNP slices (UNP APV/L-CSA 100 μM: 1.43 ± 0.16, n=6; PA APV/L-CSA 100 μM: 0.99 ± 0.05, n=6; UNP APV/L-CSA 1 mM: 1.42 ± 0.13, n=6; PA APV/L-CSA 100 μM: 1.28 ± 0.09, n=7; Figure 7B). In contrast, no changes in the PPR ratio were measured in the PA group at any concentration of L-CSA, suggesting that in FPS LTM, agonist effects of L-CSA may be attenuated or occluded during short-term tetanic stimulation. Note that PPR is reduced from ~1.5 in control (Figure 4B) to ~1.0 in APV in the UNP group (Figure 7B). Previously, we have shown that the NMDA component (125%) of paired pulse facilitation (PPF), a similar calculation of transmitter release probability, is greater than the AMPA component (30%) (Zinebi et al., 2001) and in the present experiment compared to control the larger NMDA component would be blocked in APV resulting in a reduced PPR. Additionally, in FPS, NMDA EPSCs are decreased due to reduced NR2A, NR2B, and phosphorylated NR1 protein in the amygdala (Zinebi et al., 2003), contributing to a relatively small change in PPR with APV.
Figure 7.
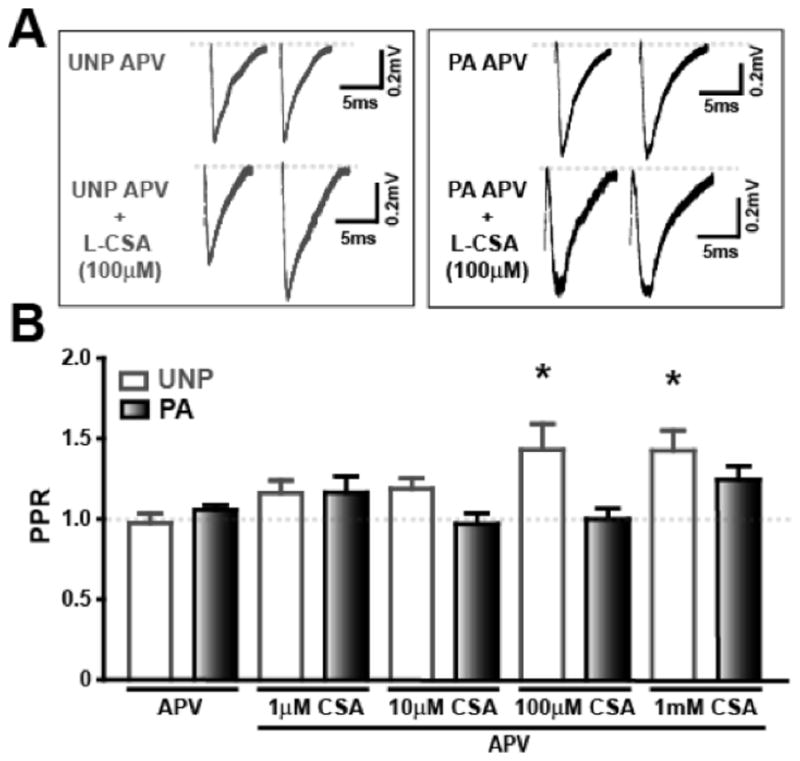
FPS occludes PLD-mGluR agonist-induced paired pulse facilitation in the Th-LA synapse. A) Traces show changes in PPR associated with administration of the PLD-mGluR agonist, L-CSA (1 mM) in the presence of the NMDA antagonist, APV (50 μM). B) PPR ratio is increased at 100 μM and 1 mM L-CSA + APV in the UNP group (white bars) while there are no significant effects in the PA group (black bars). * denotes significant difference (p<0.05) compared to UNP + APV control group.
3.7 PLD-mGluR antagonist attenuates FPS recall
3.7.1
By blocking receptors upstream from PLD signaling via PCCG-13 infusion into the LA, we studied the functional role of PLD-mGluR on recall of FPS memory. Such an approach allowed us to directly address whether the neurochemical changes were necessary and sufficient for the behavior associated with the acoustic cue-induced associative memory mechanisms in the LA. On the drug-testing day (day four), the animals were divided into two groups, and infused with vehicle (0.0134% beta cyclo-dextrin in 0.9% saline), or the drug in vehicle (PCCG-13 at 8.5 μM in vehicle). PCCG-13 at concentrations higher than 8.5 μM significantly affected startle amplitude itself and impaired the ability to discriminate FPS in those animals (data not shown) and therefore, we utilized the above concentration for the experiments described here. FPS magnitude elicited by the CS was increased in PA animals when compared to UNP animals (UNP: 5.01 ± 3.44%, n=39; PA: 78.01 ± 5.61%, n=50; p<0.05; Figure 8A). PA animals treated with PCCG-13 in vehicle did not show FPS while vehicle-treated animals demonstrated FPS (vehicle: 63.47 ± 5.81%, n=16; PCCG-13: 7.97 ± 6.73%, n=15; p< 0.05; Figure 8B). Infusions in the UNP group failed to show changes (No infusions: 5.96 ± 4.92%, n=12; vehicle: 5.61 ± 7.53%, n=13; PCCG-13: 5.95 ± 4.32%, n=12, data not shown). Thus, direct infusion of PCCG-13 into the LA attenuated the cue-induced associative enhanced response to acoustic startle.
Figure 8.
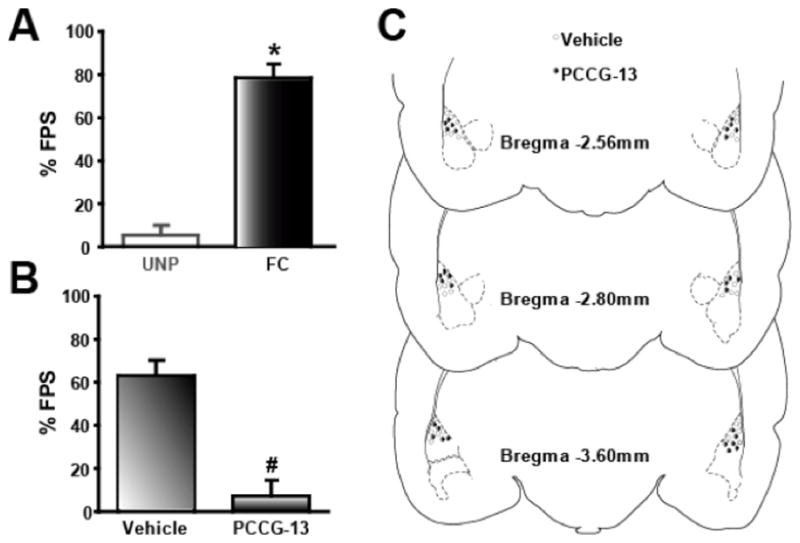
PLD-mGluR antagonist attenuates the retrieval of associative fear memory. A) Graph shows that FPS in PA animals wherein the cue (tone) is associated with the US (footshock) is significantly higher compared to UNP animals in which cue and US are presented randomly. B) PCCG-13 significantly reduces FPS in the PA group. C) Location of the injection site in the LA using figures from (Paxinos and Watson, 1998) and adapted to show vehicle infusion (white circles) and PCCG-13 (black circles). * denotes p<0.05 compared to UNP; # indicates significant difference (p<0.05) compared to PA group infused with vehicle alone.
4. Discussion
4.1
In the present study, we report on presynaptic cellular mechanisms involved in FPS LTM by examining downstream elements of mGluR signaling in the Th-LA synaptic pathway. Following FPS, proteomic analyses identified a 30% decrease in amygdala aldolase A (Figure 1), that immunoprecipitated with amygdala anti-PLD1/2 antibodies (Figure 2). Amygdala aldolase A has a reverse profile to the expression of PLD, a downstream target of mGluR signaling. PA group PLD expression (Figure 2) and activity (Figure 3) increased significantly, attesting that our hypothesis of increased PLD (in response to aldolase A reduction) is important for storage mechanisms of FPS LTM. The fEPSPs during baseline and LTP maintenance remain unaltered (Figure 4, 5), but the PPR ratios in the PA group are reduced compared to the UNP (Figure 4B) confirming an increased release probability in FPS, previously reported in our lab (McKernan and Shinnick-Gallagher, 1997). Neither inhibition nor stimulation of mGluR signaling via PLD affected the PA group PPR ratios (Figure 4, 7) suggesting that the increased probability of release coupled to the increased PLD activity may contribute to multiple vesicular release resulting in a constant PPR (Chamberland et al., 2014; Christie and Jahr, 2006; Conti and Lisman, 2003; Oertner et al., 2002). HFS recruits PLD signaling since the first fEPSP and PTP (Figure 5E, F) are affected following block of PLD-mGluR signaling. These effects are similar to those mediated through mGluR7, which is co-immunoprecipitated with PLD in the PA group (Figure 2D1, D2). Finally, blocking PLD signaling upstream at mGluR level attenuated the associative memory mechanism underlying FPS response (Figure 8), underscoring the importance of PLD-mGluR in cue-induced anxiety and fear in emotional learning.
4.2 Amygdala PLD expression is increased while aldolase A expression is reduced in FPS LTM
4.2.1
Of 240 proteins with altered expression profiles, aldolase A was significantly decreased in PA compared to the UNP group. Aldolase A, a glycolytic enzyme (Funari et al., 2007; Penhoet et al., 1969), was also reported to bind the light neurofilament mRNA, contributing to neuronal homeostasis (Canete-Soler et al., 2005) and inhibits postnatal cortical neurite outgrowth (Blackmore et al., 2010). Oxidized cortical aldolase A, found in aged human cortex (Martinez et al., 2008), is a preclinical marker for Parkinson’s disease (Ferrer et al., 2011). Reduced levels of Aldolase A, a major autoantigen (Mor et al., 2005), were reported in the hippocampus in Alzheimer’s disease (Sultana et al., 2007), establish this protein as an important biomarker in memory mechanisms in neuropathological states.
4.2.2
In our investigation, reduced aldolase A in FPS LTM is pulled down with PLD1/2 antibodies (Figure 1, 2). Since aldolase A directly inhibits PLD (Kim et al., 2002) we hypothesized and confirmed that PLD levels would be increased following FPS in the amygdala (Figure 2). Excellent in vitro studies of PLD for expression/activity (Gibbs and Meier, 2000) established that inducible PLD1/constitutive PLD2 isoforms are involved with neurotransmitter release (Humeau et al., 2001), cytoskeletal reorganization (Colley et al., 1997), exocytosis (Vitale et al., 2001) and neurite outgrowth (Watanabe et al., 2004a; Watanabe et al., 2004b; Zhang et al., 2005), all of which are important in synaptic plasticity. In the present study, the first in vivo of its kind, both the inducible PLD1 and constitutive PLD2 isoforms were increased in the amygdala of PA group (see Figure 2) suggesting both isoforms play a role in FPS induced modulation of PLD activity (Figure 3).
4.2.3
A 20–30% increase in PA PLD expression (Figure 2) raises PLD activity four-fold (Figure 3). This four-fold increase of PLD activity was also observed in the UNP group, but only following application of a PLD-mGluR agonist. The same application did not significantly increase the already elevated PA PLD activity, suggesting a ceiling effect of amygdala PLD functionality. An alternative explanation could be where PLD-mGluR agonist depolarizes synaptic membranes (Turski et al., 1987) that inhibits PLD activity (Waring et al., 1999).
4.3 PLD-mGluR signaling in FPS LTM occludes Th-LA PPRs without affecting LTP maintenance
4.3.1
During the maintenance phase of LTP, fEPSPs were not significantly altered in either the UNP or the PA (Figure 5) or between the PA and UNP groups. This suggests that there are increases in synaptic transmission due to postsynaptic modifications (Rumpel et al., 2005; Yeh et al., 2006) that were not recorded in the present paradigm. We predict that the fear conditioning paradigm hyperpolarizes the postsynaptic membrane and contributes to the failure in LTP (Malinow and Miller, 1986). Our earlier study (Schroeder and Shinnick-Gallagher, 2004) as well as other groups (Tsvetkov et al., 2002) have reported an occluded LTP in the cortico-LA synapse and the Th-LA synapse (Hong et al., 2012), typically interpreted as being due to the fact that synapses in fear conditioned animals have already undergone LTP-like changes in synaptic strength and are therefore less capable of showing additional LTP (Sigurdsson et al., 2007).
4.3.2
Catalytically inactive PLD1 reduces the number of functional neurotransmitter release sites (Humeau et al., 2001) in Aplysia neurons suggesting that active PLD1 may increase operative neurotransmitter release sites. PPR measures synaptic vesicle release probability (Debanne et al., 1996; Dobrunz and Stevens, 1997; Manabe et al., 1993). Neither PLD-mGluR agonist (Figure 7) nor antagonist (Figure 4B) had any effect on PA group Th-LA PPR. We, therefore, propose that the increased probability of release coupled with the increased PLD activity may contribute to multiple vesicular release that results in the constant PPR (Chamberland et al., 2014; Christie and Jahr, 2006; Conti and Lisman, 2003; Oertner et al., 2002) observed and establishes a new synaptic homeostasis that is unresponsive to the pharmacological treatments. Alternatively, mGluR internalization due to increased probability of release in FPS LTM may contribute to the unresponsiveness (Bhattacharya et al., 2004; Pelkey et al., 2005; Suh et al., 2013). A third possibility is that of a ligand-independent action (Lorez et al., 2003) involving mGluR7. mGluR7 (a class III mGluR), associates with PLD (Figure 2), and is located presynaptically in all amygdala nuclei (Kinoshita et al., 1998) and thalamic afferents (Dobi et al., 2013). It can operate in a ligand independent manner, where prolonged action by agonists can cause release and activation of presynaptic mGluR7 normally bound to munc-18-1 (Nakajima et al., 2009) via increased calcium binding to the Ca/CaM site on mGluR7 (Mochida, 2011; Nakajima et al., 2009; Nakajima et al., 1999) and facilitating SNARE-mediated vesicle fusion (Mochida, 2011; Nakajima et al., 2009; Rizo and Sudhof, 2002; Shen et al., 2007), thereby acting as an inhibitory modulator of vesicle secretion.
4.3.3
mGluR7 is also known to be linked to phospholipase C (PLC) (Martin et al., 2010; Perroy et al., 2000). Moreover, release of diacyglyceride (DAG) from PLC directly stimulates protein kinase C (PKC), which subsequently stimulates PLD, where the PKC-PLD interaction may be independent of phosphorylation and kinase activity (McDermott et al., 2004), supporting a role for PLD in PTP. With prolonged application of agonist, mGluR7 activation potentiates release, an effect independent of calcium channels or inhibitors of PKC, acting at ATP binding sites, but blocked by DAG antagonists acting at non-kinase DAG binding sites (Martin et al., 2010), where mGluR7 and munc 13-1 (essential for priming synaptic vesicles) along with other proteins, is recruited to the active zone of the nerve terminal (Andrews-Zwilling et al., 2006). Munc 13-1, a phorbol ester receptor, is itself activated by DAG moving it from soluble to particulate fractions in nerve terminals (Martin et al., 2010). Taken together, these data together suggest a significant role for mGluR7, PKC, and PLD in potentiating transmitter release. Thus, like the bidirectional modulation of glutamate release mediated through mGluR7 (Martin et al., 2010), the PLD-mGluR mediates inhibition and potentiation at Th-LA synapses (Figure 6C vs Figure 5). These studies suggest that PTP effects mediated by the PLD-mGluR at the Th-LA synapse may be comparable to those mediated by mGluR7.
4.4 FPS retrieval is attenuated by block of the amygdala PLD-mGluR signaling pathway
4.4.1
Our study presents evidence that the PLD-mGluR antagonist blocked recall in FPS LTM, suggesting that PLD-mGluR signaling is important in amygdala long-term fear memory. Prior to testing the PLD-mGluR antagonist, animals were exposed to 10 CS presentations during the initial testing for FPS, thereby making it possible that some extinction occurred and implicating PLD-mGluR in enhancing extinction. Indeed, mGluR7 deficient mice show less acquisition, expression, and extinction (Fendt et al., 2013; Masugi et al., 1999). On the other hand, activation of mGluR7 by an allosteric agonist also reduces acquisition of FPS (Siegl et al., 2008) and cued freezing (Fendt et al., 2013) but enhances extinction by essentially acting as an antagonist (Fendt et al., 2008), an effect thought to be due to mGluR7 internalization (Pelkey et al., 2005). In contrast, a novel antagonist of mGluR7 slightly delayed acquisition but had no effect on the expression of cued freezing (Gee et al., 2014). Thus, the novel mGluR7 antagonist effects in the fear-conditioning paradigm are incongruent with those of mGluR7 deficient animals, the mGluR7 agonist, and also the PLD-mGluR antagonist. The mGluR7 link to enhancement of extinction is a significant finding (Fendt et al., 2008), which provides an important therapeutic target in the treatment of cued anxiety pathologies. There are unanswered questions regarding the identity of the PLD-mGluR and its relationship to mGluR7. Such studies will require detailed analyses of synaptic mechanisms and the different aspects of fear learning, including acquisition, (consolidation), reconsolidation and extinction.
4.4.2
In conclusion, these neurophysiological investigations of the Th-LA pathway provide the first indication that PLD is a critical downstream target of mGluR signaling in an amygdala fear-associated memory pathway and that neuroadaptive changes in PLD activity may be important in fear memory.
Highlights.
In the present study, we report on presynaptic cellular mechanisms involved in fear potentiated startle long-term memory (FPS LTM) by examining downstream elements of metabotropic glutamate receptor (mGluR) signaling in the thalamic-lateral amygdala (Th-LA) synaptic pathway. Following fear potentiated startle behavior (FPS) in rats, proteomic analyses identified a 30% decrease in amygdala aldolase A, that immunoprecipitated with amygdala anti-phospholipase D1 and D2 isoform (PLD1/2) antibodies. Amygdala aldolase A has a reverse profile to the expression of PLD, a downstream target of mGluR signaling. Paired (PA) group PLD expression and activity increased significantly, attesting that our hypothesis of increased PLD (in response to aldolase A reduction) is important for storage mechanisms of FPS LTM. The fEPSPs during baseline and long-term potentiation (LTP) maintenance remain unaltered, but the PPR ratios in the PA group are reduced compared to the unpaired (UNP) confirming an increased release probability in FPS. Neither inhibition nor stimulation of mGluR signaling via PLD affected the PA group PPR ratios suggesting that the increased probability of release coupled to the increased PLD activity may contribute to multiple vesicular release resulting in a constant PPR. High frequency stimulation (HFS) recruits PLD signaling since the first field excitatory post synaptic potential (fEPSP) and post-tetanic potentiation (PTP) are affected following block of PLD-mGluR signaling. These effects are similar to those mediated through mGluR7, which is co-immunoprecipitated with PLD in the PA group. Finally, blocking PLD signaling upstream at mGluR level attenuated the associative memory mechanism underlying FPS response, underscoring the importance of PLD-mGluR in cue-induced anxiety and fear in emotional learning.
Acknowledgments
This work was supported by NRSA F32 Ruth L. Kirschstein postdoctoral grant DA023316, and NIDA R03 DA033428 to B.K., and an NHLBI Proteomics Center Award N01-HV-00245. The authors acknowledge Robert Fox, David V. Herin (deceased), Luis F. Orozco-Cabal for technical assistance with survival surgeries. The authors acknowledge Kelly Dineley and especially Gladys Ko, Anusha Srinivasan for their suggestions and critical reading of the manuscript.
Footnotes
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adolphs R, Tranel D, Damasio H, Damasio AR. Fear and the human amygdala. J Neurosci. 1995;15:5879–5891. doi: 10.1523/JNEUROSCI.15-09-05879.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albani-Torregrossa S, Attucci S, Marinozzi M, Pellicciari R, Moroni F, Pellegrini-Giampietro DE. Antagonist pharmacology of metabotropic glutamate receptors coupled to phospholipase D activation in adult rat hippocampus: focus on (2R,1′S,2′R,3′S)-2-(2′-carboxy-3′-phenylcyclopropyl)glycine versus 3, 5-dihydroxyphenylglycine. Mol Pharmacol. 1999;55:699–707. [PubMed] [Google Scholar]
- Amorapanth P, LeDoux JE, Nader K. Different lateral amygdala outputs mediate reactions and actions elicited by a fear-arousing stimulus. Nat Neurosci. 2000;3:74–79. doi: 10.1038/71145. [DOI] [PubMed] [Google Scholar]
- Andrews-Zwilling YS, Kawabe H, Reim K, Varoqueaux F, Brose N. Binding to Rab3A-interacting molecule RIM regulates the presynaptic recruitment of Munc13-1 and ubMunc13-2. J Biol Chem. 2006;281:19720–19731. doi: 10.1074/jbc.M601421200. [DOI] [PubMed] [Google Scholar]
- Attucci S, Albani-Torregrossa S, Moroni F, Pellegrini-Giampietro DE. Metabotropic glutamate receptors stimulate phospholipase D via different pathways in the adult and neonate rat hippocampus. Neurochem Res. 2001;26:1151–1155. doi: 10.1023/a:1012327007733. [DOI] [PubMed] [Google Scholar]
- Bauer EP, Schafe GE, LeDoux JE. NMDA receptors and L-type voltage-gated calcium channels contribute to long-term potentiation and different components of fear memory formation in the lateral amygdala. J Neurosci. 2002;22:5239–5249. doi: 10.1523/JNEUROSCI.22-12-05239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya M, Babwah AV, Godin C, Anborgh PH, Dale LB, Poulter MO, Ferguson SS. Ral and phospholipase D2-dependent pathway for constitutive metabotropic glutamate receptor endocytosis. J Neurosci. 2004;24:8752–8761. doi: 10.1523/JNEUROSCI.3155-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackmore MG, Moore DL, Smith RP, Goldberg JL, Bixby JL, Lemmon VP. High content screening of cortical neurons identifies novel regulators of axon growth. Mol Cell Neurosci. 2010;44:43–54. doi: 10.1016/j.mcn.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair HT, Schafe GE, Bauer EP, Rodrigues SM, LeDoux JE. Synaptic plasticity in the lateral amygdala: a cellular hypothesis of fear conditioning. Learn Mem. 2001;8:229–242. doi: 10.1101/lm.30901. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. Expression of NMDA receptor-dependent LTP in the hippocampus: bridging the divide. Mol Brain. 2013;6:5. doi: 10.1186/1756-6606-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boss V, Conn PJ. Metabotropic excitatory amino acid receptor activation stimulates phospholipase D in hippocampal slices. J Neurochem. 1992;59:2340–2343. doi: 10.1111/j.1471-4159.1992.tb10131.x. [DOI] [PubMed] [Google Scholar]
- Boss V, Nutt KM, Conn PJ. L-cysteine sulfinic acid as an endogenous agonist of a novel metabotropic receptor coupled to stimulation of phospholipase D activity. Mol Pharmacol. 1994;45:1177–1182. [PubMed] [Google Scholar]
- Brown JS, Kalish HI, Farber IE. Conditioned fear as revealed by magnitude of startle response to an auditory stimulus. J Exp Psychol. 1951;41:317–328. doi: 10.1037/h0060166. [DOI] [PubMed] [Google Scholar]
- Buono P, D’Armiento FP, Terzi G, Alfieri A, Salvatore F. Differential distribution of aldolase A and C in the human central nervous system. J Neurocytol. 2001;30:957–965. doi: 10.1023/a:1021828421792. [DOI] [PubMed] [Google Scholar]
- Cahill L, Weinberger NM, Roozendaal B, McGaugh JL. Is the amygdala a locus of “conditioned fear”? Some questions and caveats. Neuron. 1999;23:227–228. doi: 10.1016/s0896-6273(00)80774-6. [DOI] [PubMed] [Google Scholar]
- Campeau S, Davis M. Involvement of subcortical and cortical afferents to the lateral nucleus of the amygdala in fear conditioning measured with fear-potentiated startle in rats trained concurrently with auditory and visual conditioned stimuli. J Neurosci. 1995a;15:2312–2327. doi: 10.1523/JNEUROSCI.15-03-02312.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau S, Davis M. Involvement of the central nucleus and basolateral complex of the amygdala in fear conditioning measured with fear-potentiated startle in rats trained concurrently with auditory and visual conditioned stimuli. J Neurosci. 1995b;15:2301–2311. doi: 10.1523/JNEUROSCI.15-03-02301.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canete-Soler R, Reddy KS, Tolan DR, Zhai J. Aldolases a and C are ribonucleolytic components of a neuronal complex that regulates the stability of the light-neurofilament mRNA. J Neurosci. 2005;25:4353–4364. doi: 10.1523/JNEUROSCI.0885-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassella JV, Davis M. The design and calibration of a startle measurement system. Physiol Behav. 1986;36:377–383. doi: 10.1016/0031-9384(86)90032-6. [DOI] [PubMed] [Google Scholar]
- Chamberland S, Evstratova A, Toth K. Interplay between synchronization of multivesicular release and recruitment of additional release sites support short-term facilitation at hippocampal mossy fiber to CA3 pyramidal cells synapses. J Neurosci. 2014;34:11032–11047. doi: 10.1523/JNEUROSCI.0847-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charney DS. Neuroanatomical circuits modulating fear and anxiety behaviors. Acta Psychiatr Scand Suppl. 2003:38–50. doi: 10.1034/j.1600-0447.108.s417.3.x. [DOI] [PubMed] [Google Scholar]
- Christie JM, Jahr CE. Multivesicular release at Schaffer collateral-CA1 hippocampal synapses. J Neurosci. 2006;26:210–216. doi: 10.1523/JNEUROSCI.4307-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colley WC, Sung TC, Roll R, Jenco J, Hammond SM, Altshuller Y, Bar-Sagi D, Morris AJ, Frohman MA. Phospholipase D2, a distinct phospholipase D isoform with novel regulatory properties that provokes cytoskeletal reorganization. Curr Biol. 1997;7:191–201. doi: 10.1016/s0960-9822(97)70090-3. [DOI] [PubMed] [Google Scholar]
- Conti R, Lisman J. The high variance of AMPA receptor- and NMDA receptor-mediated responses at single hippocampal synapses: evidence for multiquantal release. Proc Natl Acad Sci U S A. 2003;100:4885–4890. doi: 10.1073/pnas.0630290100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuellar JC, Griffith EL, Merlin LR. Contrasting roles of protein kinase C in induction versus suppression of group I mGluR-mediated epileptogenesis in vitro. J Neurophysiol. 2005;94:3643–3647. doi: 10.1152/jn.00548.2005. [DOI] [PubMed] [Google Scholar]
- Curras MC, Dingledine R. Selectivity of amino acid transmitters acting at N-methyl-D-aspartate and amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptors. Mol Pharmacol. 1992;41:520–526. [PubMed] [Google Scholar]
- Davis M. Pharmacological and anatomical analysis of fear conditioning using the fear-potentiated startle paradigm. Behav Neurosci. 1986;100:814–824. doi: 10.1037//0735-7044.100.6.814. [DOI] [PubMed] [Google Scholar]
- Davis M. The role of the amygdala in fear and anxiety. Annu Rev Neurosci. 1992;15:353–375. doi: 10.1146/annurev.ne.15.030192.002033. [DOI] [PubMed] [Google Scholar]
- Davis M. Neurobiology of fear responses: the role of the amygdala. J Neuropsychiatry Clin Neurosci. 1997;9:382–402. doi: 10.1176/jnp.9.3.382. [DOI] [PubMed] [Google Scholar]
- Davis M, Astrachan DI. Conditioned fear and startle magnitude: effects of different footshock or backshock intensities used in training. J Exp Psychol Anim Behav Process. 1978;4:95–103. doi: 10.1037//0097-7403.4.2.95. [DOI] [PubMed] [Google Scholar]
- Davis M, Falls WA, Campeau S, Kim M. Fear-potentiated startle: a neural and pharmacological analysis. Behav Brain Res. 1993;58:175–198. doi: 10.1016/0166-4328(93)90102-v. [DOI] [PubMed] [Google Scholar]
- Debanne D, Guerineau NC, Gahwiler BH, Thompson SM. Paired-pulse facilitation and depression at unitary synapses in rat hippocampus: quantal fluctuation affects subsequent release. J Physiol. 1996;491(Pt 1):163–176. doi: 10.1113/jphysiol.1996.sp021204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobi A, Sartori SB, Busti D, Van der Putten H, Singewald N, Shigemoto R, Ferraguti F. Neural substrates for the distinct effects of presynaptic group III metabotropic glutamate receptors on extinction of contextual fear conditioning in mice. Neuropharmacology. 2013;66:274–289. doi: 10.1016/j.neuropharm.2012.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- Duvarci S, Nader K. Characterization of fear memory reconsolidation. J Neurosci. 2004;24:9269–9275. doi: 10.1523/JNEUROSCI.2971-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, Luthi A. Amygdala inhibitory circuits and the control of fear memory. Neuron. 2009;62:757–771. doi: 10.1016/j.neuron.2009.05.026. [DOI] [PubMed] [Google Scholar]
- Feinstein JS, Adolphs R, Damasio A, Tranel D. The human amygdala and the induction and experience of fear. Curr Biol. 2011;21:34–38. doi: 10.1016/j.cub.2010.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fendt M, Imobersteg S, Peterlik D, Chaperon F, Mattes C, Wittmann C, Olpe HR, Mosbacher J, Vranesic I, van der Putten H, McAllister KH, Flor PJ, Gee CE. Differential roles of mGlu(7) and mGlu(8) in amygdala-dependent behavior and physiology. Neuropharmacology. 2013;72:215–223. doi: 10.1016/j.neuropharm.2013.04.052. [DOI] [PubMed] [Google Scholar]
- Fendt M, Schmid S. Metabotropic glutamate receptors are involved in amygdaloid plasticity. Eur J Neurosci. 2002;15:1535–1541. doi: 10.1046/j.1460-9568.2002.01988.x. [DOI] [PubMed] [Google Scholar]
- Fendt M, Schmid S, Thakker DR, Jacobson LH, Yamamoto R, Mitsukawa K, Maier R, Natt F, Husken D, Kelly PH, McAllister KH, Hoyer D, van der Putten H, Cryan JF, Flor PJ. mGluR7 facilitates extinction of aversive memories and controls amygdala plasticity. Mol Psychiatry. 2008;13:970–979. doi: 10.1038/sj.mp.4002073. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Martinez A, Blanco R, Dalfo E, Carmona M. Neuropathology of sporadic Parkinson disease before the appearance of parkinsonism: preclinical Parkinson disease. J Neural Transm (Vienna) 2011;118:821–839. doi: 10.1007/s00702-010-0482-8. [DOI] [PubMed] [Google Scholar]
- Flor PJ, Van Der Putten H, Ruegg D, Lukic S, Leonhardt T, Bence M, Sansig G, Knopfel T, Kuhn R. A novel splice variant of a metabotropic glutamate receptor, human mGluR7b. Neuropharmacology. 1997;36:153–159. doi: 10.1016/s0028-3908(96)00176-1. [DOI] [PubMed] [Google Scholar]
- Frohman MA. The phospholipase D superfamily as therapeutic targets. Trends Pharmacol Sci. 2015;36:137–144. doi: 10.1016/j.tips.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funari VA, Crandall JE, Tolan DR. Fructose metabolism in the cerebellum. Cerebellum. 2007;6:130–140. doi: 10.1080/14734220601064759. [DOI] [PubMed] [Google Scholar]
- Fuortes MG, Rico MJ, Merlin LR. Distinctions between persistent and reversible group I mGluR-induced epileptiform burst prolongation. Epilepsia. 2010;51:1633–1637. doi: 10.1111/j.1528-1167.2010.02682.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee CE, Peterlik D, Neuhauser C, Bouhelal R, Kaupmann K, Laue G, Uschold-Schmidt N, Feuerbach D, Zimmermann K, Ofner S, Cryan JF, van der Putten H, Fendt M, Vranesic I, Glatthar R, Flor PJ. Blocking metabotropic glutamate receptor subtype 7 (mGlu7) via the Venus flytrap domain (VFTD) inhibits amygdala plasticity, stress, and anxiety-related behavior. J Biol Chem. 2014;289:10975–10987. doi: 10.1074/jbc.M113.542654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs TC, Meier KE. Expression and regulation of phospholipase D isoforms in mammalian cell lines. J Cell Physiol. 2000;182:77–87. doi: 10.1002/(SICI)1097-4652(200001)182:1<77::AID-JCP9>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Grillon C, Ameli R, Woods SW, Merikangas K, Davis M. Fear-potentiated startle in humans: effects of anticipatory anxiety on the acoustic blink reflex. Psychophysiology. 1991;28:588–595. doi: 10.1111/j.1469-8986.1991.tb01999.x. [DOI] [PubMed] [Google Scholar]
- Grillon C, Davis M. Fear-potentiated startle conditioning in humans: explicit and contextual cue conditioning following paired versus unpaired training. Psychophysiology. 1997;34:451–458. doi: 10.1111/j.1469-8986.1997.tb02389.x. [DOI] [PubMed] [Google Scholar]
- Grillon C, Heller R, Hirschhorn E, Kling MA, Pine DS, Schulkin J, Vythilingam M. Acute hydrocortisone treatment increases anxiety but not fear in healthy volunteers: a fear-potentiated startle study. Biol Psychiatry. 2011;69:549–555. doi: 10.1016/j.biopsych.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall RA. Co-immunoprecipitation as a strategy to evaluate receptor-receptor or receptor-protein interactions. In: George SR, O’Dowd BF, editors. G Proten Coupled Receptor-Protein Interactions. John Wiley and Sons, Inc; 2004. pp. 165–178. [Google Scholar]
- Hong I, Kim J, Song B, Park K, Shin K, Eom KD, Han PL, Lee S, Choi S. Fear conditioning occludes late-phase long-term potentiation at thalamic input synapses onto the lateral amygdala in rat brain slices. Neurosci Lett. 2012;506:121–125. doi: 10.1016/j.neulet.2011.10.063. [DOI] [PubMed] [Google Scholar]
- Humeau Y, Vitale N, Chasserot-Golaz S, Dupont JL, Du G, Frohman MA, Bader MF, Poulain B. A role for phospholipase D1 in neurotransmitter release. Proc Natl Acad Sci U S A. 2001;98:15300–15305. doi: 10.1073/pnas.261358698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen JP, Cain CK, Ostroff LE, LeDoux JE. Molecular mechanisms of fear learning and memory. Cell. 2011;147:509–524. doi: 10.1016/j.cell.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazama AM, Schauder KB, McKinnon M, Bachevalier J, Davis M. A novel AX+/BX- paradigm to assess fear learning and safety-signal processing with repeated-measure designs. J Neurosci Methods. 2013;214:177–183. doi: 10.1016/j.jneumeth.2013.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Lee S, Kim JH, Lee TG, Hirata M, Suh PG, Ryu SH. Phospholipase D2 directly interacts with aldolase via Its PH domain. Biochemistry. 2002;41:3414–3421. doi: 10.1021/bi015700a. [DOI] [PubMed] [Google Scholar]
- Kinoshita A, Shigemoto R, Ohishi H, van der Putten H, Mizuno N. Immunohistochemical localization of metabotropic glutamate receptors, mGluR7a and mGluR7b, in the central nervous system of the adult rat and mouse: a light and electron microscopic study. J Comp Neurol. 1998;393:332–352. [PubMed] [Google Scholar]
- Klancnik JM, Cuenod M, Gahwiler BH, Jiang ZP, Do KQ. Release of endogenous amino acids, including homocysteic acid and cysteine sulphinic acid, from rat hippocampal slices evoked by electrical stimulation of Schaffer collateral-commissural fibres. Neuroscience. 1992;49:557–570. doi: 10.1016/0306-4522(92)90226-r. [DOI] [PubMed] [Google Scholar]
- Klein J. Functions and pathophysiological roles of phospholipase D in the brain. J Neurochem. 2005;94:1473–1487. doi: 10.1111/j.1471-4159.2005.03315.x. [DOI] [PubMed] [Google Scholar]
- Klein J, Iovino M, Vakil M, Shinozaki H, Loffelholz K. Ontogenetic and pharmacological studies on metabotropic glutamate receptors coupled to phospholipase D activation. Neuropharmacology. 1997;36:305–311. doi: 10.1016/s0028-3908(97)00024-5. [DOI] [PubMed] [Google Scholar]
- Krishnan B, Genzer KM, Pollandt SW, Liu J, Gallagher JP, Shinnick-Gallagher P. Dopamine-induced plasticity, phospholipase D (PLD) activity and cocaine-cue behavior depend on PLD-linked metabotropic glutamate receptors in amygdala. PLoS One. 2011;6:e25639. doi: 10.1371/journal.pone.0025639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBar KS, Gatenby JC, Gore JC, LeDoux JE, Phelps EA. Human amygdala activation during conditioned fear acquisition and extinction: a mixed-trial fMRI study. Neuron. 1998;20:937–945. doi: 10.1016/s0896-6273(00)80475-4. [DOI] [PubMed] [Google Scholar]
- Lamprecht R, Farb CR, Rodrigues SM, LeDoux JE. Fear conditioning drives profilin into amygdala dendritic spines. Nat Neurosci. 2006;9:481–483. doi: 10.1038/nn1672. [DOI] [PubMed] [Google Scholar]
- LeDoux JE. Emotion circuits in the brain. Annu Rev Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- LeDoux JE, Cicchetti P, Xagoraris A, Romanski LM. The lateral amygdaloid nucleus: sensory interface of the amygdala in fear conditioning. J Neurosci. 1990a;10:1062–1069. doi: 10.1523/JNEUROSCI.10-04-01062.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux JE, Farb C, Ruggiero DA. Topographic organization of neurons in the acoustic thalamus that project to the amygdala. J Neurosci. 1990b;10:1043–1054. doi: 10.1523/JNEUROSCI.10-04-01043.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux JE, Farb CR. Neurons of the acoustic thalamus that project to the amygdala contain glutamate. Neurosci Lett. 1991;134:145–149. doi: 10.1016/0304-3940(91)90527-z. [DOI] [PubMed] [Google Scholar]
- Lee O, Lee CJ, Choi S. Induction mechanisms for L-LTP at thalamic input synapses to the lateral amygdala: requirement of mGluR5 activation. Neuroreport. 2002;13:685–691. doi: 10.1097/00001756-200204160-00030. [DOI] [PubMed] [Google Scholar]
- Lorez M, Humbel U, Pflimlin MC, Kew JN. Group III metabotropic glutamate receptors as autoreceptors in the cerebellar cortex. Br J Pharmacol. 2003;138:614–625. doi: 10.1038/sj.bjp.0705099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Malinow R, Miller JP. Postsynaptic hyperpolarization during conditioning reversibly blocks induction of long-term potentiation. Nature. 1986;320:529–530. doi: 10.1038/320529a0. [DOI] [PubMed] [Google Scholar]
- Manabe T, Wyllie DJ, Perkel DJ, Nicoll RA. Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J Neurophysiol. 1993;70:1451–1459. doi: 10.1152/jn.1993.70.4.1451. [DOI] [PubMed] [Google Scholar]
- Maren S. Synaptic transmission and plasticity in the amygdala. An emerging physiology of fear conditioning circuits. Mol Neurobiol. 1996;13:1–22. doi: 10.1007/BF02740749. [DOI] [PubMed] [Google Scholar]
- Maren S. The amygdala, synaptic plasticity, and fear memory. Ann N Y Acad Sci. 2003;985:106–113. doi: 10.1111/j.1749-6632.2003.tb07075.x. [DOI] [PubMed] [Google Scholar]
- Martin R, Durroux T, Ciruela F, Torres M, Pin JP, Sanchez-Prieto J. The metabotropic glutamate receptor mGlu7 activates phospholipase C, translocates munc-13-1 protein, and potentiates glutamate release at cerebrocortical nerve terminals. J Biol Chem. 2010;285:17907–17917. doi: 10.1074/jbc.M109.080838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez A, Dalfo E, Muntane G, Ferrer I. Glycolitic enzymes are targets of oxidation in aged human frontal cortex and oxidative damage of these proteins is increased in progressive supranuclear palsy. J Neural Transm (Vienna) 2008;115:59–66. doi: 10.1007/s00702-007-0800-y. [DOI] [PubMed] [Google Scholar]
- Masugi M, Yokoi M, Shigemoto R, Muguruma K, Watanabe Y, Sansig G, van der Putten H, Nakanishi S. Metabotropic glutamate receptor subtype 7 ablation causes deficit in fear response and conditioned taste aversion. J Neurosci. 1999;19:955–963. doi: 10.1523/JNEUROSCI.19-03-00955.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott M, Wakelam MJ, Morris AJ. Phospholipase D. Biochem Cell Biol. 2004;82:225–253. doi: 10.1139/o03-079. [DOI] [PubMed] [Google Scholar]
- McDonald AJ. Cortical pathways to the mammalian amygdala. Prog Neurobiol. 1998;55:257–332. doi: 10.1016/s0301-0082(98)00003-3. [DOI] [PubMed] [Google Scholar]
- McKernan MG, Shinnick-Gallagher P. Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature. 1997;390:607–611. doi: 10.1038/37605. [DOI] [PubMed] [Google Scholar]
- Mochida S. Activity-dependent regulation of synaptic vesicle exocytosis and presynaptic short-term plasticity. Neurosci Res. 2011;70:16–23. doi: 10.1016/j.neures.2011.03.005. [DOI] [PubMed] [Google Scholar]
- Monfils MH, Cowansage KK, Klann E, LeDoux JE. Extinction-reconsolidation boundaries: key to persistent attenuation of fear memories. Science. 2009;324:951–955. doi: 10.1126/science.1167975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mor F, Izak M, Cohen IR. Identification of aldolase as a target antigen in Alzheimer’s disease. J Immunol. 2005;175:3439–3445. doi: 10.4049/jimmunol.175.5.3439. [DOI] [PubMed] [Google Scholar]
- Muller J, Corodimas KP, Fridel Z, LeDoux JE. Functional inactivation of the lateral and basal nuclei of the amygdala by muscimol infusion prevents fear conditioning to an explicit conditioned stimulus and to contextual stimuli. Behav Neurosci. 1997;111:683–691. doi: 10.1037//0735-7044.111.4.683. [DOI] [PubMed] [Google Scholar]
- Nader K, Hardt O. A single standard for memory: the case for reconsolidation. Nat Rev Neurosci. 2009;10:224–234. doi: 10.1038/nrn2590. [DOI] [PubMed] [Google Scholar]
- Nader K, Majidishad P, Amorapanth P, LeDoux JE. Damage to the lateral and central, but not other, amygdaloid nuclei prevents the acquisition of auditory fear conditioning. Learn Mem. 2001;8:156–163. doi: 10.1101/lm.38101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima Y, Mochida S, Okawa K, Nakanishi S. Ca2+-dependent release of Munc18-1 from presynaptic mGluRs in short-term facilitation. Proc Natl Acad Sci U S A. 2009;106:18385–18389. doi: 10.1073/pnas.0910088106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima Y, Yamamoto T, Nakayama T, Nakanishi S. A relationship between protein kinase C phosphorylation and calmodulin binding to the metabotropic glutamate receptor subtype 7. J Biol Chem. 1999;274:27573–27577. doi: 10.1074/jbc.274.39.27573. [DOI] [PubMed] [Google Scholar]
- Nicoletti F, Bockaert J, Collingridge GL, Conn PJ, Ferraguti F, Schoepp DD, Wroblewski JT, Pin JP. Metabotropic glutamate receptors: from the workbench to the bedside. Neuropharmacology. 2011;60:1017–1041. doi: 10.1016/j.neuropharm.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoletti F, Bruno V, Ngomba RT, Gradini R, Battaglia G. Metabotropic glutamate receptors as drug targets: what’s new? Curr Opin Pharmacol. 2015;20:89–94. doi: 10.1016/j.coph.2014.12.002. [DOI] [PubMed] [Google Scholar]
- Nielsen KS, Macphail EM, Riedel G. Class I mGlu receptor antagonist 1-aminoindan-1,5-dicarboxylic acid blocks contextual but not cue conditioning in rats. Eur J Pharmacol. 1997;326:105–108. doi: 10.1016/s0014-2999(97)85402-7. [DOI] [PubMed] [Google Scholar]
- Oertner TG, Sabatini BL, Nimchinsky EA, Svoboda K. Facilitation at single synapses probed with optical quantal analysis. Nat Neurosci. 2002;5:657–664. doi: 10.1038/nn867. [DOI] [PubMed] [Google Scholar]
- Parsons RG, Davis M. Temporary disruption of fear-potentiated startle following PKMzeta inhibition in the amygdala. Nat Neurosci. 2011;14:295–296. doi: 10.1038/nn.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkey KA, Lavezzari G, Racca C, Roche KW, McBain CJ. mGluR7 is a metaplastic switch controlling bidirectional plasticity of feedforward inhibition. Neuron. 2005;46:89–102. doi: 10.1016/j.neuron.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Pellegrini-Giampietro DE, Torregrossa SA, Moroni F. Pharmacological characterization of metabotropic glutamate receptors coupled to phospholipase D in the rat hippocampus. Br J Pharmacol. 1996;118:1035–1043. doi: 10.1111/j.1476-5381.1996.tb15503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicciari R, Marinozzi M, Costantino G, Natalini B, Moroni F, Pellegrini-Giampietro D. (2R,1′S,2′R,3′S)-2-(2′-Carboxy-3′-phenylcyclopropyl)glycine (PCCG-13), the first potent and selective competitive antagonist of phospholipase D-coupled metabotropic glutamate receptors: asymmetric synthesis and preliminary biological properties. J Med Chem. 1999;42:2716–2720. doi: 10.1021/jm990128v. [DOI] [PubMed] [Google Scholar]
- Penhoet EE, Kochman M, Rutter WJ. Molecular and catalytic properties of aldolase C. Biochemistry. 1969;8:4396–4402. doi: 10.1021/bi00839a026. [DOI] [PubMed] [Google Scholar]
- Perroy J, Prezeau L, De Waard M, Shigemoto R, Bockaert J, Fagni L. Selective blockade of P/Q-type calcium channels by the metabotropic glutamate receptor type 7 involves a phospholipase C pathway in neurons. J Neurosci. 2000;20:7896–7904. doi: 10.1523/JNEUROSCI.20-21-07896.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkanen A, Savander V, LeDoux JE. Organization of intra-amygdaloid circuitries in the rat: an emerging framework for understanding functions of the amygdala. Trends Neurosci. 1997;20:517–523. doi: 10.1016/s0166-2236(97)01125-9. [DOI] [PubMed] [Google Scholar]
- Ribeiro FM, Paquet M, Cregan SP, Ferguson SS. Group I metabotropic glutamate receptor signalling and its implication in neurological disease. CNS Neurol Disord Drug Targets. 2010;9:574–595. doi: 10.2174/187152710793361612. [DOI] [PubMed] [Google Scholar]
- Rico MJ, Merlin LR. Evidence that phospholipase D activation prevents group I mGluR-induced persistent prolongation of epileptiform bursts. J Neurophysiol. 2004;91:2385–2388. doi: 10.1152/jn.01140.2003. [DOI] [PubMed] [Google Scholar]
- Riedel G, Casabona G, Platt B, Macphail EM, Nicoletti F. Fear conditioning-induced time- and subregion-specific increase in expression of mGlu5 receptor protein in rat hippocampus. Neuropharmacology. 2000;39:1943–1951. doi: 10.1016/s0028-3908(00)00037-x. [DOI] [PubMed] [Google Scholar]
- Rizo J, Sudhof TC. Snares and Munc18 in synaptic vesicle fusion. Nat Rev Neurosci. 2002;3:641–653. doi: 10.1038/nrn898. [DOI] [PubMed] [Google Scholar]
- Rodrigues SM, Bauer EP, Farb CR, Schafe GE, LeDoux JE. The group I metabotropic glutamate receptor mGluR5 is required for fear memory formation and long-term potentiation in the lateral amygdala. J Neurosci. 2002;22:5219–5229. doi: 10.1523/JNEUROSCI.22-12-05219.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues SM, Schafe GE, LeDoux JE. Molecular mechanisms underlying emotional learning and memory in the lateral amygdala. Neuron. 2004;44:75–91. doi: 10.1016/j.neuron.2004.09.014. [DOI] [PubMed] [Google Scholar]
- Rogan MT, Staubli UV, LeDoux JE. Fear conditioning induces associative long-term potentiation in the amygdala. Nature. 1997;390:604–607. doi: 10.1038/37601. [DOI] [PubMed] [Google Scholar]
- Rumpel S, LeDoux J, Zador A, Malinow R. Postsynaptic receptor trafficking underlying a form of associative learning. Science. 2005;308:83–88. doi: 10.1126/science.1103944. [DOI] [PubMed] [Google Scholar]
- Sananes CB, Davis M. N-methyl-D-aspartate lesions of the lateral and basolateral nuclei of the amygdala block fear-potentiated startle and shock sensitization of startle. Behav Neurosci. 1992;106:72–80. doi: 10.1037//0735-7044.106.1.72. [DOI] [PubMed] [Google Scholar]
- Schafe GE, Doyere V, LeDoux JE. Tracking the fear engram: the lateral amygdala is an essential locus of fear memory storage. J Neurosci. 2005;25:10010–10014. doi: 10.1523/JNEUROSCI.3307-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafe GE, LeDoux JE. Memory consolidation of auditory pavlovian fear conditioning requires protein synthesis and protein kinase A in the amygdala. J Neurosci. 2000;20:Rc96. doi: 10.1523/JNEUROSCI.20-18-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz A, Grillon C, Avenevoli S, Cui L, Merikangas KR. Developmental investigation of fear-potentiated startle across puberty. Biol Psychol. 2014;97:15–21. doi: 10.1016/j.biopsycho.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder BW, Shinnick-Gallagher P. Fear memories induce a switch in stimulus response and signaling mechanisms for long-term potentiation in the lateral amygdala. Eur J Neurosci. 2004;20:549–556. doi: 10.1111/j.1460-9568.2004.03517.x. [DOI] [PubMed] [Google Scholar]
- Scott MT, Shinnick-Gallagher P. Internuclear synapse in the amygdala is not facilitated in fear conditioning. Synapse. 2005;55:67–70. doi: 10.1002/syn.20092. [DOI] [PubMed] [Google Scholar]
- Shen J, Tareste DC, Paumet F, Rothman JE, Melia TJ. Selective activation of cognate SNAREpins by Sec1/Munc18 proteins. Cell. 2007;128:183–195. doi: 10.1016/j.cell.2006.12.016. [DOI] [PubMed] [Google Scholar]
- Shigemoto R, Kinoshita A, Wada E, Nomura S, Ohishi H, Takada M, Flor PJ, Neki A, Abe T, Nakanishi S, Mizuno N. Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J Neurosci. 1997;17:7503–7522. doi: 10.1523/JNEUROSCI.17-19-07503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemoto R, Kulik A, Roberts JD, Ohishi H, Nusser Z, Kaneko T, Somogyi P. Target-cell-specific concentration of a metabotropic glutamate receptor in the presynaptic active zone. Nature. 1996;381:523–525. doi: 10.1038/381523a0. [DOI] [PubMed] [Google Scholar]
- Shigemoto R, Nomura S, Ohishi H, Sugihara H, Nakanishi S, Mizuno N. Immunohistochemical localization of a metabotropic glutamate receptor, mGluR5, in the rat brain. Neurosci Lett. 1993;163:53–57. doi: 10.1016/0304-3940(93)90227-c. [DOI] [PubMed] [Google Scholar]
- Shin RM, Tully K, Li Y, Cho JH, Higuchi M, Suhara T, Bolshakov VY. Hierarchical order of coexisting pre- and postsynaptic forms of long-term potentiation at synapses in amygdala. Proc Natl Acad Sci U S A. 2010;107:19073–19078. doi: 10.1073/pnas.1009803107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinnick-Gallagher P, McKernan MG, Xie J, Zinebi F. L-type voltage-gated calcium channels are involved in the in vivo and in vitro expression of fear conditioning. Ann N Y Acad Sci. 2003;985:135–149. doi: 10.1111/j.1749-6632.2003.tb07078.x. [DOI] [PubMed] [Google Scholar]
- Shinomura T, del Rio E, Breen KC, Downes CP, McLaughlin M. Activation of phospholipase D by metabotropic glutamate receptor agonists in rat cerebrocortical synaptosomes. Br J Pharmacol. 2000;131:1011–1018. doi: 10.1038/sj.bjp.0703651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegl S, Flor PJ, Fendt M. Amygdaloid metabotropic glutamate receptor subtype 7 is involved in the acquisition of conditioned fear. Neuroreport. 2008;19:1147–1150. doi: 10.1097/WNR.0b013e328307f295. [DOI] [PubMed] [Google Scholar]
- Sigurdsson T, Doyère V, Cain CK, LeDoux JE. Long-term potentiation in the amygdala: A cellular mechanism of fear learning and memory. Neuropharmacology. 2007;52:215–227. doi: 10.1016/j.neuropharm.2006.06.022. [DOI] [PubMed] [Google Scholar]
- Straub C, Pazdrak K, Young TW, Stafford SJ, Wu Z, Wiktorowicz JE, Haag AM, English RD, Soman KV, Kurosky A. Toward the Proteome of the Human Peripheral Blood Eosinophil. Proteomics Clin Appl. 2009;3:1151–1173. doi: 10.1002/prca.200900043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh YH, Park JY, Park S, Jou I, Roche PA, Roche KW. Regulation of metabotropic glutamate receptor 7 (mGluR7) internalization and surface expression by Ser/Thr protein phosphatase 1. J Biol Chem. 2013;288:17544–17551. doi: 10.1074/jbc.M112.439513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana R, Boyd-Kimball D, Cai J, Pierce WM, Klein JB, Merchant M, Butterfield DA. Proteomics analysis of the Alzheimer’s disease hippocampal proteome. J Alzheimers Dis. 2007;11:153–164. doi: 10.3233/jad-2007-11203. [DOI] [PubMed] [Google Scholar]
- Tsvetkov E, Carlezon WA, Jr, Benes FM, Kandel ER, Bolshakov VY. Fear Conditioning Occludes LTP-Induced Presynaptic Enhancement of Synaptic Transmission in the Cortical Pathway to the Lateral Amygdala. Neuron. 2002;34:289–300. doi: 10.1016/s0896-6273(02)00645-1. [DOI] [PubMed] [Google Scholar]
- Turski WA, Herrling PL, Do KQ. Effects of L-cysteine-sulphinate and L-aspartate, mixed excitatory amino acid agonists, on the membrane potential of cat caudate neurons. Brain Res. 1987;414:330–338. doi: 10.1016/0006-8993(87)90014-x. [DOI] [PubMed] [Google Scholar]
- Vitale N, Caumont AS, Chasserot-Golaz S, Du G, Wu S, Sciorra VA, Morris AJ, Frohman MA, Bader MF. Phospholipase D1: a key factor for the exocytotic machinery in neuroendocrine cells. EMBO J. 2001;20:2424–2434. doi: 10.1093/emboj/20.10.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DL, Davis M. Quantifying fear potentiated startle using absolute versus proportional increase scoring methods: implications for the neurocircuitry of fear and anxiety. Psychopharmacology (Berl) 2002;164:318–328. doi: 10.1007/s00213-002-1213-0. [DOI] [PubMed] [Google Scholar]
- Waring M, Drappatz J, Weichel O, Seimetz P, Sarri E, Bockmann I, Kempter U, Valeva A, Klein J. Modulation of neuronal phospholipase D activity under depolarizing conditions. FEBS Lett. 1999;464:21–24. doi: 10.1016/s0014-5793(99)01669-5. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Yamazaki M, Miyazaki H, Arikawa C, Itoh K, Sasaki T, Maehama T, Frohman MA, Kanaho Y. Phospholipase D2 functions as a downstream signaling molecule of MAP kinase pathway in L1-stimulated neurite outgrowth of cerebellar granule neurons. J Neurochem. 2004a;89:142–151. doi: 10.1111/j.1471-4159.2004.02308.x. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Yokozeki T, Yamazaki M, Miyazaki H, Sasaki T, Maehama T, Itoh K, Frohman MA, Kanaho Y. Essential role for phospholipase D2 activation downstream of ERK MAP kinase in nerve growth factor-stimulated neurite outgrowth from PC12 cells. J Biol Chem. 2004b;279:37870–37877. doi: 10.1074/jbc.M402610200. [DOI] [PubMed] [Google Scholar]
- Weisskopf MG, Bauer EP, LeDoux JE. L-type voltage-gated calcium channels mediate NMDA-independent associative long-term potentiation at thalamic input synapses to the amygdala. J Neurosci. 1999;19:10512–10519. doi: 10.1523/JNEUROSCI.19-23-10512.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilensky AE, Schafe GE, LeDoux JE. Functional inactivation of the amygdala before but not after auditory fear conditioning prevents memory formation. J Neurosci. 1999;19:Rc48. doi: 10.1523/JNEUROSCI.19-24-j0006.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh SH, Mao SC, Lin HC, Gean PW. Synaptic expression of glutamate receptor after encoding of fear memory in the rat amygdala. Mol Pharmacol. 2006;69:299–308. doi: 10.1124/mol.105.017194. [DOI] [PubMed] [Google Scholar]
- Zhang W, Yu L, Zhang Y, Wang X. Phospholipase D in the signaling networks of plant response to abscisic acid and reactive oxygen species. Biochim Biophys Acta. 2005;1736:1–9. doi: 10.1016/j.bbalip.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Zinebi F, McKernan M, Shinnick-Gallagher P. Expression of fear-conditioning is accompanied by increased paired-pulse depression within the amygdala. Pharmacol Biochem Behav. 2002;71:393–400. doi: 10.1016/s0091-3057(01)00684-0. [DOI] [PubMed] [Google Scholar]
- Zinebi F, Russell RT, McKernan M, Shinnick-Gallagher P. Comparison of paired-pulse facilitation of AMPA and NMDA synaptic currents in the lateral amygdala. Synapse. 2001;42:115–127. doi: 10.1002/syn.1107. [DOI] [PubMed] [Google Scholar]
- Zinebi F, Xie J, Liu J, Russell RT, Gallagher JP, McKernan MG, Shinnick-Gallagher P. NMDA currents and receptor protein are downregulated in the amygdala during maintenance of fear memory. J Neurosci. 2003;23:10283–10291. doi: 10.1523/JNEUROSCI.23-32-10283.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]