

Abstract
Chimeric antigen receptors (CARs) are synthetic receptors that target T cells to cell-surface antigens and augment T-cell function and persistence. Mesothelin is a cell-surface antigen implicated in tumor invasion, which is highly expressed in mesothelioma, lung, pancreas, breast, ovarian, and other cancers. Its low-level expression in mesothelia however commands thoughtful therapeutic interventions. Encouragingly, recent clinical trials evaluating active immunization or immune-conjugates in patients with pancreatic adenocarcinoma or mesothelioma have shown responses without toxicity. Altogether, these findings and preclinical CAR therapy models using either systemic or regional T-cell delivery argue favorably for mesothelin CAR therapy in multiple solid tumors.
Keywords: cancer immunotherapy, chimeric antigen receptor (CAR), lung cancer, adoptive T-cell therapy, mesothelioma, ovarian cancer, cancer of the pancreas, triple-negative breast cancer, T-cell engineering, tumor invasion
INTRODUCTION
Adoptive cell therapy using engineered T cells is emerging as a promising strategy to rapidly establish tumor immunity and eradicate small or large tumor burdens. T cells may be engineered to target a tumor antigen through a T-cell receptor (TCR) or a chimeric antigen receptor (CAR).(1, 2) In contrast to TCRs, which are restricted to human leukocyte antigen, CARs provide direct binding to cell-surface proteins, carbohydrates or glycolipids. CARs intrinsically mediate T-cell activation as well as costimulation in the case of second-generation CARs.(3) The use of CARs specific for CD19, a B-cell activation receptor, has recently been shown to induce durable remissions in patients with relapsed, chemo-refractory B-cell malignancies, including acute lymphoblastic leukemia, chronic lymphocytic leukemia and non-Hodgkin lymphoma in multiple clinical trials.(4–6) Second-generation CARs achieve these outcomes through both targeted tumor killing and functional T-cell enhancement.(3) Given the potential high efficiency of CAR therapy, it is critical to identify appropriate antigens to tackle solid tumors, in order to achieve tumor eradication with minimal or tolerable on-target/off-tumor toxicity to healthy tissues.
SEARCHING FOR CAR TARGETS IN SOLID TUMORS
Whereas CD19 provides a nearly ideal target for B-cell malignancies, no single antigen with equivalent characteristics has yet been identified for solid tumors. CD19 is highly and relatively homogenously expressed in most B-cell malignancies, possibly including their putative tumor initiating cells. CD19 is functionally involved in B-cell activation, and may contribute to tumor survival and thus increase the likelihood of its expression in most tumor cells. Finally, in normal cells, CD19 expression is confined to the hematopoietic B-cell lineage, non-vital cells that can be dispensed (thus, the B-cell aplasia induced by CD19 CAR T cells is not lethal and clinically manageable). An ideal solid tumor CAR target would thus be overexpressed in all cancer cells, absent or very lowly expressed in non-vital normal tissue, and found in many patients. Furthermore, if expression of the target antigen is associated with tumor invasion or metastasis formation, CAR therapy may be directed to the more aggressive cancer cells and be less vulnerable to tumor relapse. Solid tumor CAR targets under investigation are altered gene products arising from genetic mutations or altered splicing (EGFRvIII),(7) altered glycosylation patterns (MUC-1),(8) cancer-testis antigen-derived peptides (MAGE),(9) differentiation antigens (CEA),(10) overexpressed antigens in tumors (PSMA, GD2, CA125, Her-2, and mesothelin [MSLN]), or tumor-associated stroma (FAP, VEGFR) (see Supplementary Table 1).(11) Examples of solid tumor antigens currently investigated in CAR T-cell clinical trials are shown in Figure 1.
Figure 1. Antigens targeted in solid tumor CAR T-cell therapy clinical trials.
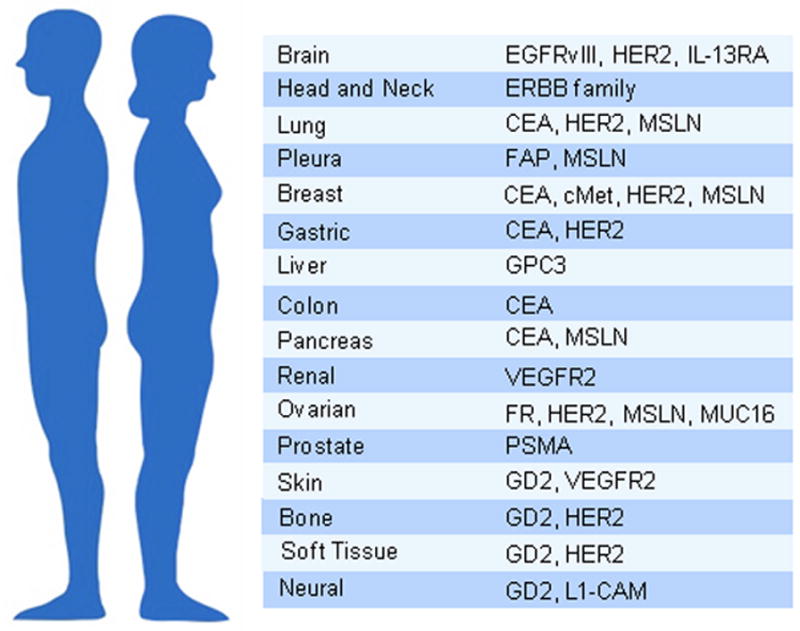
Antigens listed in the figure were compiled by search of the active clinical trials in the clincialtrials.gov. CEA: Carcinoembryonic Antigen; EGFR: Epidermal Growth Factor Receptor; FAP: Fibroblast Associated Protein; FR: Folate Receptor; GD2: Diasialoganglioside; GPC3: Glypican 3; HER2: Human Epidermal Growth Factor Receptor; L1-CAM: L1 Cell Adhesion Molecule; MSLN: Mesothelin; MUC-16: Mucin 16; VEGFR: Vascular Endothelial Growth Factor Receptor.
While overexpressed antigens are numerous and relatively frequent, they raise concerns about “on-target/off-tumor” side effects due to the high sensitivity of T cells for low-level antigen expression, which is greater than that of monoclonal antibodies.(12) For instance, the use of ERbB2 CAR T cells, administered at a high cell dose, has resulted in a fatal adverse event, attributed in part to low-level ERbB2 expression in healthy lung epithelial and cardiovascular cells.(13) Thus, an optimal solid-tumor antigen target is one whose expression is either restricted to tumor cells or only occurs at very low levels in expendable normal tissues. EGFRvIII and chondroitin sulfate proteoglycan-4 (CSPG-4) have been suggested to be examples for such favorable scenarios.(14, 15)
Mesothelin (MSLN) is emerging as an attractive target for cancer immunotherapy, considering its low expression on normal mesothelial cells and high expression in a broad spectrum of solid tumors. The MSLN-targeted immunotherapies reported to date support a favorable safety profile.(16, 17) MSLN is a potential CAR target in a number of common solid tumors (Figure 2 and Supplementary Table 2).
Figure 2. Frequency and distribution pattern of the mesothelin protein in solid malignancies.

Mesothelin is expressed in a wide range of solid tumors. For most cancers, MSLN expression is homogeneously distributed on the cell-surface, with low cytoplasmic expression. For stomach and lung cancers, MSLN is frequently expressed in the cytoplasm, as well as on the cell-surface. The distribution of MSLN in cytoplasm and/or cell-surface was represented in the figure, where data was available. The frequency and distribution of MSLN were calculated from the published literature (Supplementary Table 2).
DISCOVERY AND EARLY CHARACTERIZATION OF MSLN
In searching for targets for monoclonal antibody therapy for solid tumors, Ira Pastan and Mark Willingham discovered the MSLN protein, which they found to be specifically expressed on ovarian cancer cells but not on normal human tissues with the exception of mesothelial cells.(18) MSLN is a glycoprotein anchored to the plasma membrane by a glycophosphatidyl inositol (GPI) domain. It is initially synthesized as a 69 kDa cell-surface protein. After cleavage of the amino terminus by the furin protease, a 40-kDa C-terminal fragment remains attached to the membrane and a soluble 32-kDa N-terminal fragment, named MPF (megakaryotic potentiating factor), is released.(17) A soluble form of MSLN has also been detected in the sera of patients with solid tumors, which is referred to as soluble MSLN-related protein (SMRP). SMRP is generated either by alternative splicing or by proteolytic cleavage of the MSLN mature form, induced by the TNF-α–converting enzyme ADAM 17.(19)
MSLN FUNCTION
The biological function of MSLN seems to be nonessential in normal tissues, given that MSLN knockout mice exhibit normal development, reproduction, and blood cell count.(20) In contrast, preclinical and clinical studies increasingly show that aberrant MSLN expression plays an active role in both malignant transformation of tumors and tumor aggressiveness by promoting cancer cell proliferation, contributing to local invasion and metastasis, and conferring resistance to apoptosis induced by cytotoxic agents.(21–24) MSLN can act bidirectionally, either by directly activating intracellular pathways via its GPI domain or by interacting with its receptor, CA125/MUC16. Overexpression of MSLN alone is sufficient to constitutively activate the NF-κB, MAPK, and PI3-kinase intracellular pathways promoting cell proliferation and resistance to apoptosis.(25) Several preclinical studies, including ours, support the finding that MSLN over-expression promotes cell migration and invasion by inducing activation and expression of the matrix metalloproteases MMP-7(26) and MMP-9.(21) In addition, the high-affinity interaction between MSLN and CA125 leads to heterotypic cell adhesion, which facilitates metastasis of ovarian cancer cell lines.(27) These observations correlate with clinical observations showing that MSLN expression, as well as elevated serum SMRP levels, are associated with progressing tumor burden, increasing stage, and poor overall survival.(22–24, 28, 29) Cancer cells that possess an invasive phenotype express high amounts of membranous MSLN, rather than the cytoplasmic form.(30, 31) Our group(22) and others(29) have reported that in patients with lung adenocarcinoma, MSLN is expressed at metastatic sites, and correlates with tumor aggressiveness and KRAS mutation. These discoveries strengthen the rationale for targeting MSLN-expressing cancer cells with CARs.
MSLN EXPRESSION IN SOLID TUMORS
Physiologically, MSLN is expressed on mesothelial cells of the peritoneal and pleural cavities, and pericardium; it is expressed minimally on the epithelial cell-surface of the trachea, ovaries, rete testis, tonsil, and fallopian tubes.(32) Overexpression of MSLN was initially observed in mesothelioma and ovarian cancer, and subsequently in lung, esophageal, pancreatic, gastric, biliary, endometrial, thymic, colon, and breast cancers.(17, 21–24) MSLN overexpression thus has an estimated incidence of 340,000 patients and prevalence of 2 million patients a year (Supplementary Tables 3 & 4) in the U.S. alone. The frequency and distribution patterns of MSLN expression differ for each tumor subtype, as summarized in Figure 2 and Supplementary Table 2. Using the 5B2 MSLN-specific antibody, we developed a MSLN expression score integrating MSLN intensity and distribution.(21) In our series, MSLN expression was found in 90% of epitheloid malignant pleural mesothelioma (n=139),(21) 69% of lung adenocarcinoma (n=1209),(22) 60% of breast (n=314),(24) and 46% of esophageal cancers (n=125).(23) Furthermore, we observed that MSLN expression is more prevalent in aggressive histological subtypes of lung (KRAS+ tumors),(22) breast (triple-negative)(24) and esophageal cancers (high grade dysplasia and adenocarcinoma).(23) These findings have been corroborated in other studies.(29, 33) Within the cancer cell, MSLN expression may be luminal/membranous or cytoplasmic. In mesothelioma tumors, MSLN expression is homogeneously distributed on the cell-surface.(21) In lung adenocarcinoma, we and others have found that MSLN expression pattern is heterogeneous, with expression in the cytoplasm and on the cell-surface.(22, 29) In gastric cancer, cytoplasmic expression was found to be more prevalent than membranous expression.(30) In addition to the studies characterizing MSLN expression by IHC analysis, functional genomic mRNA profiling studies in a large cancer database (n=19,746) have reported MSLN expression in other solid tumors such as thyroid, renal, and synovial sarcoma tumors, which were not previously reported.(34)
MSLN VACCINES AND IMMUNO-CONJUGATES
Given MSLN’s distribution, protumorigenic functions, and immunogenicity (see below), various immunotherapeutic strategies have been devised, some of which have shown encouraging results in early phase clinical trials (Table 1). These strategies include the use of (1) tumor vaccines, (2) antibody-based therapies, and (3) adoptive T-cell therapy (CAR T cells) (Figure 3).
Table 1.
Phase I/II clinical trials for mesothelin-targeted immunotherapies
| Agent | Phase | Intervention | Cancer/s targeted | Status/Results | Clinicaltrials.gov Identifier | References |
|---|---|---|---|---|---|---|
| CRS-207 | II | GVAX and cyclosphosphamide with or without CRS207 | Metastatic pancreatic cancer | 31% SD, 51% PD | NCT01417000 | (36) |
| II B | CRS-207 alone or/plus GVAX therapy and cyclophosphamide | Metastatic pancreatic cancer | Recruiting | NCT02004262 | ||
| I B | CRS-207 plus pemetrexed and cisplatin w/ and w/o cyclophosphamide | Mesothelioma | Recruiting | NCT01675765 | ||
| I | CRS 207 alone | Mesothelioma, Pancreatic adenocarcinoma, NSCLC, Ovarian adenocarcinoma | Dose-dependent evidence of immune activation | NCT00585845 | (35) | |
| Immunotoxin SS1P | I | SS1P plus pemetrexed and cisplatin | Mesothelioma | 77% PR, 8% SD, 15% PD | NCT01445392 | (42) |
| I | SS1P plus pentostatin and cyclophosphamide | Mesothelioma | 30% PR, 30% SD, 40% PD | NCT01362790 | (41) | |
| I | SS1P treatment plus carboplatin, paclitaxel and bevacizumab | NSCLC | Closed | NCT01051934 | ||
| I | SS1P alone, continuous infusion | Ovarian, Pancreatic, Mesothelioma | 4% PR, 50% SD, 46% PD | NCT00006981 | (89) | |
| I | SS1P alone, bolus infusion | Ovarian, Pancreatic, Mesothelioma | 12% PR, 58% SD, 30% PD | NCT00066651 | (40) | |
| Amatuximab (Morab-009) | II | Amatuximab plus pemetrexed and cisplatin | Mesothelioma | 40% PR, 51% SD | NCT00738582 | (45) |
| II | Amatuximab plus gemcitabine | Pancreatic cancer | No objective response | NCT00570713 | ||
| I | Amatuximab alone | Colorectal, Pancreatic, Head and Neck, Mesothelioma | 18% SD, 82% PD | NCT01018784 | (90) | |
| I | Amatuximab alone | Pancreatic, Mesothelioma, Ovarian | 55% SD, 45% PD | NCT00325494 | (91) | |
| BAY94-9343 | I | BAY94-9343 alone | Invasive ovarian cancer, Primary peritoneal, Fallopian tube cancer, Mesothelioma, and other cancers | Recruiting |
NCT01439152 NCT02485119 |
|
| CAR T cells | I/II | CAR T cells plus fludarabine, cyclophosphamide and aldeslekin | Metastatic cancers, Pancreatic Cancer, Mesothelioma, Ovarian | Recruiting | NCT01583686 | |
| I | CAR T cells plus cyclophosphamide | Metastatic pancreatic ductal adenocarcinoma, Epithelial Ovarian cancer, Mesothelioma | Recruiting | NCT02159716 | ||
| I | CAR T cells alone | Metastatic pancreatic ductal adenocarcinoma | Completed | NCT01897415 | ||
| I | CAR T cells alone | Mesothelioma | Completed | NCT01355965 | ||
| I | CAR T cells plus cyclophosphamide plus CD19-CAR T cells | Pancreatic cancer | Recruiting | NCT02465983 | ||
| I | CAR T cells plus cyclophosphamide | Mesothelioma, Metastases lung and breast cancers | Recruiting | NCT02414269 |
NSCLC: Non Small Cell Lung Cancer; PR: Partial Response, CR: Complete Response, PD: Progressive Disease, SD: Stable Disease, CAR: Chimeric Antigen Receptor, MPM: Malignant Pleural Mesothelioma
Figure 3. Mesothelin-targeted immunotherapy strategies.

Several therapeutic strategies have been designed for targeting mesothelin on tumor cells (1) tumor vaccine strategy (2) antibody-based therapies (3) adoptive CAR T-cell therapy. These therapies are being evaluated in phase I and/or phase II clinical trials.
Phase I and II clinical studies have been conducted at Johns Hopkins University with CRS-207, an attenuated form of Listeria monocytogenes vector that overexpresses human MSLN, either alone(35) or in combination with cyclophosphamide and GVAX (irradiated allogeneic cell line–secreting GM-CSF).(36, 37) Although no objective responses were reported, MSLN-specific CD8 T-cell responses were induced following cyclophosphamide, GVAX, and CRS-207 administration, along with a modest increase in survival.(36) Significantly, no toxicities were observed in the patients. In addition to CD4+ and CD8+ T-cell responses,(38) MSLN-specific antibody immune responses(39) were observed in patients with ovarian and pancreatic cancer, confirming the immunogenicity of MSLN and further supporting the safety of its immunotherapeutic targeting.
Phase I studies with SS1P, an anti-MSLN immunotoxin engineered by fusing a murine anti-MSLN variable antibody fragment to PE38 to a portion of Pseudomonas exotoxin, enrolled patients with advanced mesothelioma, ovarian cancer, and pancreatic cancer.(40) As a single agent, SS1P exhibits moderate antitumor efficacy. Impressive antitumor responses were seen in patients with mesothelioma who received SS1P, together with pentostatin and cyclophosphamide, to deplete T and B cells.(41) Leveraging the knowledge that chemotherapies act in concert by disrupting the tumor structure, thereby allowing better penetration of the SS1P molecule, SS1P in combination with cisplatin and pemetrexed has been investigated and resulted in partial responses in 77% of 13 patients with mesothelioma.(42) A limitation of the strategies that use SS1P immunotoxin is the development of neutralizing antibodies specific for the toxin portion of the construct and possibly the chimeric SS1 antibody as well. Fully human anti-MSLN monoclonal antibodies have been evaluated in preclinical setting,(43, 44) with the goal of identifying agents with a lower immunogenicity profile—an important concern in the development of CARs as well.
Another therapeutic strategy based on the MSLN antibody uses Amatuximab (also called Morab-009).(45) Amatuximab binds to MSLN, thereby inhibiting adhesion between cell lines expressing CA125, and it elicits antibody-dependent cell-mediated cytotoxicity (ADCC). Phase II clinical trials have been conducted with Amatuximab treatment alone or in combination with pemetrexed and cisplatin. Combination treatment is well-tolerated; objective tumor response and stable disease were achieved in 40% and 51% of patients with nonresectable pleural mesothelioma, respectively (n=89).(45)
Therapeutic agents have been linked to anti-MSLN antibody, with the idea that the drugs will be released into the cytoplasm following internalization of the antibody: (1) duocarmycin, a DNA alkylating agent, which led to the development of MDX-1382 (Medarex); and (2) DM4, a tubulin polymerase inhibitor, which led to the development of BAY 94-9343.(46) Interestingly, in vitro, BAY 94-9343 is able to induce a bystander killing effect on neighboring MSLN-negative cancer cells without affecting nonproliferating cells, an observation that is of particular interest in the context of heterogeneous antigen-expressing tumors.(46) A phase I clinical trial investigating the safety of BAY 94-9343 is currently under way. Taken together, these reports demonstrate the safety and feasibility of MSLN as a target for CAR T-cell immunotherapy.
MSLN CAR DESIGN AND PRECLINICAL EVALUATION
CARs consist of an ectodomain (commonly derived from a single-chain variable fragment [scFv]), a hinge, a transmembrane domain, and an endodomain (typically signaling domains derived from CD3ζ and costimulatory receptors) (Figure 4A). Several MSLN-specific scFv’s have been reported, including the murine SS1 scFv (47–50) and two fully human scFv’s,(51, 52) spanning all three generations of CAR design based on their signaling domains (Figure 4B). First-generation CARs contain the CD3ζ cytoplasmic domain, which is sufficient to initiate T-cell activation and enable T-cell-mediated cytotoxicity.(3, 52, 53) Second-generation CARs further enhance T-cell function and persistence through the incorporation of signaling domains that rescue and amplify the sole activation signal provided by the CD3ζ cytoplasmic domain. Costimulatory elements may be derived from receptors such as 4-1BB,(33, 48, 54, 55) CD28,(47, 51, 52) or ICOS.(50) Dual signaling prevents T-cell anergy and increases persistence and function by augmenting T-cell proliferation and cytokine production (IFN-γ and IL-2), and reducing AICD (activation-induced cell death) through the recruitment of the PI3-kinase, TRAF, and/or other pathways.(3, 47, 52) Third-generation CARs comprise three signaling domains, typically encompassing those of CD3ζ and two costimulatory domains, for example CD28 and 4-1BB or CD28 and OX40.(47, 56) Compared to second-generation CARs, third-generation CARs have shown inconsistent antitumor activity in vivo.(47, 57) A recently described MSLN CAR construct was generated to provide the DAP12 killer immunoglobulin-like receptor with signaling activation that includes the ITAM domain.(58) Preclinical experiments demonstrated that T cells engineered with this CAR displayed increased potency in vivo associated with retention of CAR expression, compared to second-generation MSLN CARs comprising either CD28 or 4-1BB signaling domains.(58) Other costimulatory domains have been tested in combination with other antigens, including FcRγ, OX40, DAP10, and CD27.(3, 56, 59) Choosing an appropriate costimulation domain is essential to sustain CAR T-cell activity and calibrate T-cell persistence. However, the ideal costimulation domain may depend on context, as CAR function depends on multiple extraneous factors such as antigen density,(60, 61) CAR stoichiometry,(61) CAR affinity,(62–64) and the immunological features of the tumor microenvironment.(65–68)
Figure 4. CAR T-cell design. (A) Structure of the CAR.

The CAR typically contains a single chain fragment variable (scFv) binding domain specific for mesothelin fused to a transmembrane domain and intracellular signaling domain (CD3ζ and CD28 or 4-1BB). The CAR expressed into patient’s own T cells after transduction provides both specificity and effector function activation (B) Different generations of the MSLN CAR. Three generations of CAR T cells differing by their signaling domains have been designed to increase the activation strength of T cells.
A particular concern regarding mesothelin CARs is interference from soluble MSLN, which in principle could occupy and block the scFv portion. Reassuringly, mesothelin CAR T-cell activation (cytokine secretion and cytotoxic activity) is dependent on MSLN expression on the cell-surface.(47, 52) Significantly, presence of serum SMRP does not alter MSLN CAR T-cell efficiency in vitro or in vivo, even at high levels(51, 52). Similar findings have been reported with CEA(69) and HER2–targeted CARs.(70) The lack of CAR blockade by serum protein may be explained by the avidity of CAR T cells for membranous target antigen, which may be increased by interactions between adhesion molecules and other accessory molecules present on the surface of the T-cell and tumor cells.(71)
The efficiency of MSLN CAR T-cell therapy has been investigated in subcutaneous or orthotopic mouse models of mesothelioma, ovarian cancer and lung cancer.(47, 51, 52) We established a clinically relevant orthotopic mouse model of malignant pleural mesothelioma in which the tumor is aggressive loco-regionally with extensive lymphangiogenesis and mediastinal lymph node metastases mimicking human pleural mesothelioma.(21, 72, 73) In this model, we administered MSLN CAR T cells systemically or intrapleurally.(52) Intrapleurally delivery resulted in greater T-cell proliferation, T-cell redistribution to extra-thoracic metastatic sites, tumor eradication and survival, than a 30-fold higher T-cell dose administered systemically.(52) Systemically administered CAR T cells are sequestered in the lungs prior to tumor infiltration. Regional administration of MSLN CAR T cells facilitated earlier antigen encounter and T-cell activation, cytokine secretion and effector function of CD4 CAR T cells, which was associated with increased CD8 CAR T-cell proliferation and function. Furthermore, intrapleurally administered MSLN CAR T cells persisted long-term, and eliminated a tumor re-challenge 200 days after the initial tumor eradication. On the basis of these findings, we are now initiating a phase I study to investigate MSLN CAR T cells administered regionally to subjects with mesothelioma, lung cancer, and breast cancer with pleural metastases (NCT02414269, Table 1).
MSLN CARs IN CLINICAL TRIALS
With more than 150,000 individuals diagnosed with primary and metastatic pleural disease each year in the US alone (mostly from lung and breast cancer),(52) a treatment that is effective against these diseases has the potential to make a significant impact. Multiple phase I clinical trials are currently being initiated to determine the safety and the maximum tolerated dose of MSLN CAR T cell therapy. The risk of on-target/off-tumor toxicity has led to different strategies to address this safety concern.
One such strategy is based on the transfection of mRNA that encodes the MSLN CAR, which results in transient CAR expression for only a few days. In preclinical models, this approach has shown promise; multiple infusions of mRNA CAR T cells have produced a robust antitumor effect in vivo.(49) However, transient expression of the CAR may limit the long-term efficacy of the therapy. A clinical trial conducted at the University of Pennsylvania administered autologous T cells electroporated with the mRNA encoding for a second-generation MSLN CAR (SS1-4-1BB CAR).(74, 75) In this study, 4 patients with advanced mesothelioma or pancreatic tumors were treated with MSLN CAR T cells infused intravenously or intratumorally. Multiple MSLN CAR T-cell infusions were well-tolerated, with no off-target toxicities observed (pleuritis, pericarditis, or peritonitis). Encouragingly, moderate clinical responses were observed in this phase I study, and MSLN CAR T cells were detected in the tumor. The antitumoral activity of MSLN CAR T cells in vivo was established by the transient elevation of inflammatory cytokines in the sera, including IL-12, IL-6, G-CSF, MIP-1β, MCP-1, IL1RA, and RANTES.(74, 75) No severe cytokine release syndrome (CRS) was reported with MSLN CARs.(74, 75) Interestingly, the clinical evidence also highlights the capacity of CAR T-cell therapy to elicit a systemic antitumor cellular and humoral immune response by favoring epitope spreading.(74) The detection of a polyclonal IgG antibody response is consistent with the classical process of epitope spreading, where tumor lysis and inflammation induced by MSLN CAR T cells lead to the release of multiple antigens that are cross-presented on dendritic cells and activate the host immune response. This observation underscores the indirect mechanism present in MSLN CAR T-cell therapy to potentiate a broad antitumor immune response. A serious adverse event was subsequently reported by Maus et al., as one study subject developed severe anaphylaxis and cardiac arrest after the third infusion of MSLN CAR T cells.(72) This reaction was related to the high production of IgE antibodies directed against the MSLN CAR,(75) which included the murine SS1 scFv. This effect may be attributable to the multiple injections of the CAR T cells, which may have resulted in an effective prime/boost regimen to stimulate the host humoral immune response. The use of a fully human MSLN CAR (51, 52) will hopefully abrogate or at least reduce the risk of developing such an anti-CAR antibody response.
Another approach to increase T-cell safety is to utilize a suicide gene to eliminate T cells in the event of an emerging adverse event. CAR T cells can be eliminated by drug-induced activation of a suicide gene, such as herpes simplex thymidine kinase (HSV-tk) gene,(76) inducible Caspase-9(77) or EGFRΔ gene.(78) Unlike HSV-tk, the latter two are human proteins with a minimal immunogenic potential. These “safety-switch systems” have been successful in clinical investigation (77) and can rapidly deplete CAR T cells if required. The clinical-grade construct that incorporates an iCaspase-9 safety switch—which we will use in upcoming clinical trial of intrapleural MSLN-targeted CAR T-cell therapy (NCT02414269, Table 1), has been shown to be safe in preclinical experiments wherein a single dose of the AP1903 small molecule eliminated intrapleurally administered MSLN CAR T cells at the peak of their proliferation within 4 hours.
FUTURE CARs AND THEIR PATHS
The solid-tumor microenvironment poses several obstacles for MSLN CAR T cells that may limit their antitumor efficacy.(79) To optimize the efficiency of CAR T cells, numerous approaches are under evaluation to tame the host tumor microenvironment or generate “armored” CAR T cells that can overcome immune barriers. Such strategies include (1) promoting CAR T-cell infiltration, (2) augmenting the functional persistence of CAR T cells, (3) enhancing CAR T cells to overcome inhibitory signals encountered in the tumor microenvironment, and (4) improving safety by preventing on-target/off-tumor toxicity (Table 2A).
Table 2.
Genetic engineering strategies and combinational therapies potentiating CAR T-cell efficacy
| (A) Genetic engineering strategies potentiating CAR T cells | |||||
|---|---|---|---|---|---|
| Transgene Effects | Antigen targeted | Tumor targeted | References | ||
| Improve infiltration/migration | CCR2 | Promotes CAR T-cell trafficking to the tumor following systemic administration | MSLN GD2 |
Mesothelioma Neuroblastoma |
(54, 80) |
| CCR4 | Promotes CAR T-cell trafficking to the tumor following systemic administration | CD30 | Lymphoma | (92) | |
| Heparanase | Degrades the extracellular matrix, thereby improving CAR T-cell tumor infiltration and efficacy | CSPG4 GD2 |
Melanoma Neuroblastoma |
(93) | |
| Improve CAR T-cell effector function | Active akt | The constitutive Akt expression improves CAR T-cell survival, proliferation, cytokine secretion and renders them resistant to Treg suppression | GD2 | Neuroblastoma | (82) |
| IL-12 | Increases effector cytokine secretion, renders CAR T cells resistant to Treg-mediated inhibition, and induces host innate immune response | CD19 CD30 MUC-16 CEA VEGFR |
Leukemia Lymphoma Ovarian Colon melanoma, sarcoma, and colon cancer stroma |
(94–97) | |
| IL-15 | Improves T-cell expansion and reduces PD-1 expression | CD19 | Leukemia | (98, 99) | |
| IL-7 or IL-7R | Increases proliferation, survival and effector function of CAR T cells even in the presence of Tregs | CD19 GD2 |
Leukemia Neuroblastoma |
(99, 100) | |
| IL-21 | Increases CAR T-cell proliferation and cytotoxic efficacy | CD19 | Leukemia | (99, 101) | |
| CD80 or 4-1BBL | Trans-/autocostimulation between CAR T cells enhancing effector functions | PSMA | Prostate | (102) | |
| CD40L | Enhances tumor cell immunogenicity, stimulates moDC and increases CAR T-cell cytotoxic efficacy | CD19 | Leukemia | (103) | |
| 4αβ chimeric cytokine receptor | 4αβ generated by the fusion of IL-4R ectodomain and IL-2R and IL-15R subunit enhances CAR T-cell long term proliferation and cytotoxicity | MUC1 PSMA ERBBR |
Breast Prostate Head and Neck |
(104) | |
| Counteract immunosuppression | shRNA CTLA4 | Decreased CTLA4 expression enhances CAR T-cell proliferation and antitumor activity | CD19 | Leukemia | (105) |
| Improve specificity and safety | iCAR ‘safety switch’ | iCAR with PD-1 or CTLA-4 inhibitory intracellular domain linked to secondary antigen constrains CAR T-cell specificity to cancer cells expressing the primary antigen | PSMA | Prostate | (106) |
| (B) Preclinical investigation of combinational therapies potentiating CAR T-cell efficacy | |||||
|---|---|---|---|---|---|
| Agents Effects | Antigen targeted | Tumor targeted | References | ||
| Preconditioning | Radiotherapy | Total body irradiation (TBI)-induced lymphodepletion in the host promotes CAR T-cell efficacy | EGFRvIII | Glioblastoma | (107) |
| Flutamide | Fludamide-induced androgen ablation acts in additive with CAR T cells in vitro | MUC-1 | Prostate | (108) | |
| Valproate | Sodium Valproate-induced upregulation of tumor cell-surface NKG2DL expression enhances the immune recognition of CAR T cells in vitro | NKG2DL | Ovarian | (109) | |
| Monoclonal antibodies | PD-L1 or PD-1 immune checkpoint blockade | Blocking the PD-1 immunosuppressive signaling enhances CAR T-cell proliferation, cytotoxicity and cytokine secretion | CEA MSLN HER2 |
Liver metastases from colon cancer Mesothelioma Sarcoma |
(48, 65, 110) |
| Bispecific antibodies EGFR/cMet or EGFR/Epcam | Bispecific antibodies link EGFR-transduced CAR T cells to antigen-expressing tumor cells enhancing CAR T-cell recruitment/retention and cytotoxicity | CEA cMet Epcam |
Colon | (111) | |
| Anti GM-CSF or Gr-1 | Reduction of Myeloid-derived suppressor cells population (MDSC) | CEA | Liver metastases from colon cancer | (65) | |
| Small specific inhibitory drug | ABT-737 | Improves CAR T-cell killing by restoring apoptosis pathway in tumor cells | CD19 | Leukemia | (112) |
| Rapamycin | Inhibition of mTor kinase decreases the expression of anti-apoptotic molecules and others (VEGF, PD-L1, IL-10) leading to a superior antitumor effect of CAR T cells engineered with mTor resistance | CD19 | Leukemia | (113) | |
| BRAFi/MEKi | Inhibition of MAPK pathway blocks tumor cell growth and enhances apoptotic killing by CAR T cells in vitro | GD2 | Melanoma | (114) | |
| Oncolytic virus | Adenovirus vector expressing Rantes and IL-15 | Adenovirus vector-mediated Rantes and IL-15 expression in the tumor enhances CAR T-cell infiltration and persistence | GD2 | Neuroblastoma | (115) |
| Whole cell vaccine | Irradiated K562 cells expressing CD40L and OX40L | Vaccination boosts antitumor efficacy of CAR T cells | GD2 | Lung Neuroblastoma |
(116) |
To enhance trafficking to solid tumors, MSLN CAR T cells have been engineered to overexpress the chemokine receptor CCR2b, since mesothelioma cells highly express its chemokine ligand, CCL2, and the T cells express a minimal amount of CCR2b.(54) CRR2b overexpression significantly improves specific homing of MSLN CAR T cells to the tumor microenvironment, as well as the efficiency of the therapy following systemic administration. Potentiation of trafficking by cotransduction of chemokine receptors, such as CCR2 or CCR4, have been demonstrated previously in other preclinical models for T-cell therapy engineered with CARs (80) as well as TCRs.(81) This strategy is particularly relevant for MSLN CAR T-cell therapy since solid tumors overexpress CCR2 and CCR4 chemokine ligands.
Upon T-cell activation following tumor infiltration, multiple intracellular factors, such as diacyl-glycerol kinase (dgk), impair T-cell effector functions and promote T-cell anergy. Riese et al. demonstrated that genetic deletion of dgkζ significantly increased the antitumor activity of MSLN CAR T cells, as shown by the enhancement of effector cytokine secretion, FasL and TRAIL expression, and the cytotoxic functions in vitro.(55) In addition to the strategies that investigated the CAR T-cell intracellular pathways such as dgkζ or Akt,(82) other genetic engineering strategies to enhance CAR T-cell effector function have been described (IL-12, IL-7, and IL-15 secretion) in other models which are detailed in Table 2A.
Within the solid tumor, CAR T cells are confronted with a tumor-induced immunosuppressive microenvironment that can limit CAR T-cell potency. Tumor cells and associated-stroma cells, including Tregs and myeloid-derived suppressor cells, overexpress inhibitory molecules, such as TGF-β, IDO, and PD-L1, which limiting CAR T-cell efficacy.(48, 55, 65, 67, 68) Although second-generation CARs are relatively efficient in the immunosuppressive microenvironment, as shown by us and other investigators,(83) costimulation alone is not sufficient.(66) To potentiate CAR T cells in an immunosuppressive environment, multiple approaches have been investigated, including antibody-based therapy and genetic approaches, such as engineering T cells, to express a dominant negative TGF-β receptor that restores T-cell effector functions in an immunocompetent mouse melanoma model.(84) This strategy, which is currently being investigated in a clinical trial utilizing CAR T cells targeting HER2+ antigen (clinicalTrials.gov, NCT00889954), could readily be adapted for use with MSLN CAR T-cell therapy, as TGF-β is an immunosuppressive factor in lung cancer, ovarian cancer, and mesothelioma. Overexpression of PD-L1 by tumor cells has been shown to induce MSLN CAR T-cell exhaustion.(48, 65) The immunosuppressive effect of the PD-L1/PD-1 pathway can be reverted by the addition of PD-1/PD-L1–blocking antibody,(48) a PD1/CD28 converter(85, 86) or by the cointroduction of a PD-1 dominant negative receptor. Our results demonstrate that coexpression of a PD-1 dominant negative receptor together with a MSLN CAR potentiates long-term eradication of mesothelioma tumors.(87)
To improve the specificity and safety of CAR T cells, a trans-signaling strategy was developed where CD3ζ signaling is physically dissociated to the costimulatory signal through the transduction of two CARs specific for different antigens (Figure 4B); these dual-CAR T cells eliminate only cancer cells that coexpress the two targeted antigens.(53, 88) In one such strategy, T cells are engineered to express an MSLN-specific CAR containing a CD3ζ domain and a folate receptor–specific CAR containing a CD28 costimulation domain (Figure 4B). Cotransduced T cells possess superior antitumor activity against cancer cells expressing both antigens, compared with first-generation CAR T cells, and equivalent activity, compared with second-generation CAR T cells. Thus, this study demonstrates the ability to manage on-target toxicity on normal tissue, as well as the ability to combine MSLN CARs with another cancer-specific antigen to improve the safety, specificity, and efficacy of MSLN CAR T-cell therapy.
POTENTIAL COMBINATION THERAPIES
In addition to the genetic engineering strategies described above, rational combinatorial approaches with therapeutic agents that are already in clinical practice are being investigated to enhance therapy response by improving T-cell engraftment, sensitizing tumor cells to apoptosis, and stimulating the host immune system. Examples of such combinations investigated in preclinical studies potentiating CAR T-cell efficacy are shown in Table 2B. Preconditioning to achieve host lymphodepletion by use of cyclophosphamide, fludarabine, or radiotherapy is been commonly used to promote engraftment of adoptively transferred T cells. Other promising approaches include combining CAR T-cell therapy with small molecule inhibitors, monoclonal antibodies, oncolytic viruses, or whole cell vaccines. Although these studies are conducted in tumor models, such as melanoma or leukemia, some of these may be applicable to solid tumors including MSLN-expressing solid tumors.
CONCLUSION
CAR therapy using second-generation CARs has rapidly translated to clinical impact in CD19+ malignancies, paving the way for unprecedented enthusiasm for adoptive cell therapy and engineered T cells. Having such a powerful technology at hand, one important future direction for CAR research is the identification of suitable targets for tackling solid tumors. Mesothelin offers exciting prospects, based on its high expression in a variety of cancers and low level expression in normal tissues. The latter commands a thoughtful targeting strategy, noting that mesothelin-targeted immunotherapies have been very well tolerated. These clinical outcomes, combined with the preclinical data obtained with mesothelin CARs, argue favorably for a series of clinical trials targeting breast, lung, mesothelioma, ovarian and pancreatic cancer, which will soon be performed at multiple centers (NCT01355965, NCT01897415, NCT011583686, NCT02159716, NCT02414269, and NCT02465983).
Supplementary Material
Significance.
Recent success obtained with adoptive transfer of CAR T cells targeting CD19 in patients with refractory hematological malignancies have generated much enthusiasm for T-cell engineering and raise the prospect of implementing similar strategies for solid tumors. Mesothelin is expressed in a wide range and a high percentage of solid tumors, which we review here in detail. Mesothelin CAR therapy has the potential to treat multiple solid malignancies.
Acknowledgments
Funding support: This work is supported by grants from the National Institutes of Health (R21 CA164568-01A1, R21 CA164585-01A1, U54 CA137788, P50 CA086438-13, and P30 CA008748), the U.S. Department of Defense (LC110202, BC132124), the Lake Road Foundation, and Stand Up To Cancer—Cancer Research Institute Cancer Immunology Translational Cancer Research Grant [SU2C-AACR-DT1012]. Stand Up To Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research.
We thank David Sewell and Alex Torres of the MSK Thoracic Surgery Service for their editorial assistance.
Footnotes
Conflict of Interests: All authors have no conflicts of interest to disclose. P.S.A. and M.S. have submitted a patent application on MSLN CARs.
References
- 1.Ho WY, Blattman JN, Dossett ML, Yee C, Greenberg PD. Adoptive immunotherapy: engineering T cell responses as biologic weapons for tumor mass destruction. Cancer Cell. 2003;3:431–7. doi: 10.1016/s1535-6108(03)00113-2. [DOI] [PubMed] [Google Scholar]
- 2.Sadelain M, Riviere I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer. 2003;3:35–45. doi: 10.1038/nrc971. [DOI] [PubMed] [Google Scholar]
- 3.van der Stegen SJ, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov. 2015;14:499–509. doi: 10.1038/nrd4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra25. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33:540–9. doi: 10.1200/JCO.2014.56.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–18. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA, et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther. 2012;23:1043–53. doi: 10.1089/hum.2012.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilkie S, Picco G, Foster J, Davies DM, Julien S, Cooper L, et al. Retargeting of human T cells to tumor-associated MUC1: the evolution of a chimeric antigen receptor. J Immunol. 2008;180:4901–9. doi: 10.4049/jimmunol.180.7.4901. [DOI] [PubMed] [Google Scholar]
- 9.Willemsen RA, Debets R, Hart E, Hoogenboom HR, Bolhuis RL, Chames P. A phage display selected fab fragment with MHC class I-restricted specificity for MAGE-A1 allows for retargeting of primary human T lymphocytes. Gene Ther. 2001;8:1601–8. doi: 10.1038/sj.gt.3301570. [DOI] [PubMed] [Google Scholar]
- 10.Emtage PC, Lo AS, Gomes EM, Liu DL, Gonzalo-Daganzo RM, Junghans RP. Second-generation anti-carcinoembryonic antigen designer T cells resist activation-induced cell death, proliferate on tumor contact, secrete cytokines, and exhibit superior antitumor activity in vivo: a preclinical evaluation. Clin Cancer Res. 2008;14:8112–22. doi: 10.1158/1078-0432.CCR-07-4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kakarla S, Gottschalk S. CAR T cells for solid tumors: armed and ready to go? Cancer J. 2014;20:151–5. doi: 10.1097/PPO.0000000000000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahmed N, Salsman VS, Yvon E, Louis CU, Perlaky L, Wels WS, et al. Immunotherapy for osteosarcoma: genetic modification of T cells overcomes low levels of tumor antigen expression. Mol Ther. 2009;17:1779–87. doi: 10.1038/mt.2009.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–51. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beard RE, Zheng Z, Lagisetty KH, Burns WR, Tran E, Hewitt SM, et al. Multiple chimeric antigen receptors successfully target chondroitin sulfate proteoglycan 4 in several different cancer histologies and cancer stem cells. J Immunother Cancer. 2014;2:25. doi: 10.1186/2051-1426-2-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med. 2015;7:275ra22. doi: 10.1126/scitranslmed.aaa4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Villena-Vargas J, Adusumilli PS. Mesothelin-targeted immunotherapies for malignant pleural mesothelioma. Ann Cardiothorac Surg. 2012;1:466–71. doi: 10.3978/j.issn.2225-319X.2012.10.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pastan I, Hassan R. Discovery of mesothelin and exploiting it as a target for immunotherapy. Cancer Res. 2014;74:2907–12. doi: 10.1158/0008-5472.CAN-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang K, Pai LH, Batra JK, Pastan I, Willingham MC. Characterization of the antigen (CAK1) recognized by monoclonal antibody K1 present on ovarian cancers and normal mesothelium. Cancer Res. 1992;52:181–6. [PubMed] [Google Scholar]
- 19.Sapede C, Gauvrit A, Barbieux I, Padieu M, Cellerin L, Sagan C, et al. Aberrant splicing and protease involvement in mesothelin release from epithelioid mesothelioma cells. Cancer Sci. 2008;99:590–4. doi: 10.1111/j.1349-7006.2007.00715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bera TK, Pastan I. Mesothelin is not required for normal mouse development or reproduction. Mol Cell Biol. 2000;20:2902–6. doi: 10.1128/mcb.20.8.2902-2906.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Servais EL, Colovos C, Rodriguez L, Bograd AJ, Nitadori J, Sima C, et al. Mesothelin overexpression promotes mesothelioma cell invasion and MMP-9 secretion in an orthotopic mouse model and in epithelioid pleural mesothelioma patients. Clin Cancer Res. 2012;18:2478–89. doi: 10.1158/1078-0432.CCR-11-2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kachala SS, Bograd AJ, Villena-Vargas J, Suzuki K, Servais EL, Kadota K, et al. Mesothelin overexpression is a marker of tumor aggressiveness and is associated with reduced recurrence-free and overall survival in early-stage lung adenocarcinoma. Clin Cancer Res. 2014;20:1020–8. doi: 10.1158/1078-0432.CCR-13-1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rizk NP, Servais EL, Tang LH, Sima CS, Gerdes H, Fleisher M, et al. Tissue and serum mesothelin are potential markers of neoplastic progression in Barrett’s associated esophageal adenocarcinoma. Cancer Epidemiol Biomarkers Prev. 2012;21:482–6. doi: 10.1158/1055-9965.EPI-11-0993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tozbikian G, Brogi E, Kadota K, Catalano J, Akram M, Patil S, et al. Mesothelin expression in triple negative breast carcinomas correlates significantly with basal-like phenotype, distant metastases and decreased survival. PLoS One. 2014;9:e114900. doi: 10.1371/journal.pone.0114900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bharadwaj U, Marin-Muller C, Li M, Chen C, Yao Q. Mesothelin confers pancreatic cancer cell resistance to TNF-alpha-induced apoptosis through Akt/PI3K/NF-kappaB activation and IL-6/Mcl-1 overexpression. Mol Cancer. 2011;10:106. doi: 10.1186/1476-4598-10-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen SH, Hung WC, Wang P, Paul C, Konstantopoulos K. Mesothelin binding to CA125/MUC16 promotes pancreatic cancer cell motility and invasion via MMP-7 activation. Sci Rep. 2013;3:1870. doi: 10.1038/srep01870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rump A, Morikawa Y, Tanaka M, Minami S, Umesaki N, Takeuchi M, et al. Binding of ovarian cancer antigen CA125/MUC16 to mesothelin mediates cell adhesion. J Biol Chem. 2004;279:9190–8. doi: 10.1074/jbc.M312372200. [DOI] [PubMed] [Google Scholar]
- 28.Robinson BW, Creaney J, Lake R, Nowak A, Musk AW, de Klerk N, et al. Soluble mesothelin-related protein--a blood test for mesothelioma. Lung Cancer. 2005;49(Suppl 1):S109–11. doi: 10.1016/j.lungcan.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 29.Thomas A, Chen Y, Steinberg SM, Luo J, Pack S, Raffeld M, et al. High mesothelin expression in advanced lung adenocarcinoma is associated with KRAS mutations and a poor prognosis. Oncotarget. 2015;6:11694–703. doi: 10.18632/oncotarget.3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Einama T, Homma S, Kamachi H, Kawamata F, Takahashi K, Takahashi N, et al. Luminal membrane expression of mesothelin is a prominent poor prognostic factor for gastric cancer. Br J Cancer. 2012;107:137–42. doi: 10.1038/bjc.2012.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kawamata F, Homma S, Kamachi H, Einama T, Kato Y, Tsuda M, et al. C-ERC/mesothelin provokes lymphatic invasion of colorectal adenocarcinoma. J Gastroenterol. 2014;49:81–92. doi: 10.1007/s00535-013-0773-6. [DOI] [PubMed] [Google Scholar]
- 32.Ordonez NG. Application of mesothelin immunostaining in tumor diagnosis. Am J Surg Pathol. 2003;27:1418–28. doi: 10.1097/00000478-200311000-00003. [DOI] [PubMed] [Google Scholar]
- 33.Tchou J, Wang LC, Selven B, Zhang H, Conejo-Garcia J, Borghaei H, et al. Mesothelin, a novel immunotherapy target for triple negative breast cancer. Breast Cancer Res Treat. 2012;133:799–804. doi: 10.1007/s10549-012-2018-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lamberts LE, de Groot DJ, Bense RD, de Vries EG, Fehrmann RS. Functional genomic mRNA Profiling of a large cancer data base demonstrates mesothelin overexpression in a broad range of tumor types. Oncotarget. 2015 doi: 10.18632/oncotarget.4461. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Le DT, Brockstedt DG, Nir-Paz R, Hampl J, Mathur S, Nemunaitis J, et al. A live-attenuated Listeria vaccine (ANZ-100) and a live-attenuated Listeria vaccine expressing mesothelin (CRS-207) for advanced cancers: phase I studies of safety and immune induction. Clin Cancer Res. 2012;18:858–68. doi: 10.1158/1078-0432.CCR-11-2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Le DT, Wang-Gillam A, Picozzi V, Greten TF, Crocenzi T, Springett G, et al. Safety and Survival With GVAX Pancreas Prime and Listeria Monocytogenes-Expressing Mesothelin (CRS-207) Boost Vaccines for Metastatic Pancreatic Cancer. J Clin Oncol. 2015;33:1325–33. doi: 10.1200/JCO.2014.57.4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lutz ER, Wu AA, Bigelow E, Sharma R, Mo G, Soares K, et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol Res. 2014;2:616–31. doi: 10.1158/2326-6066.CIR-14-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen Y, Ayaru L, Mathew S, Morris E, Pereira SP, Behboudi S. Expansion of anti-mesothelin specific CD4+ and CD8+ T cell responses in patients with pancreatic carcinoma. PLoS One. 2014;9:e88133. doi: 10.1371/journal.pone.0088133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ho M, Hassan R, Zhang J, Wang QC, Onda M, Bera T, et al. Humoral immune response to mesothelin in mesothelioma and ovarian cancer patients. Clin Cancer Res. 2005;11:3814–20. doi: 10.1158/1078-0432.CCR-04-2304. [DOI] [PubMed] [Google Scholar]
- 40.Hassan R, Bullock S, Premkumar A, Kreitman RJ, Kindler H, Willingham MC, et al. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin Cancer Res. 2007;13:5144–9. doi: 10.1158/1078-0432.CCR-07-0869. [DOI] [PubMed] [Google Scholar]
- 41.Hassan R, Miller AC, Sharon E, Thomas A, Reynolds JC, Ling A, et al. Major cancer regressions in mesothelioma after treatment with an anti-mesothelin immunotoxin and immune suppression. Sci Transl Med. 2013;5:208ra147. doi: 10.1126/scitranslmed.3006941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hassan R, Sharon E, Thomas A, Zhang J, Ling A, Miettinen M, et al. Phase 1 study of the antimesothelin immunotoxin SS1P in combination with pemetrexed and cisplatin for front-line therapy of pleural mesothelioma and correlation of tumor response with serum mesothelin, megakaryocyte potentiating factor, and cancer antigen 125. Cancer. 2014;120:3311–9. doi: 10.1002/cncr.28875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ho M, Feng M, Fisher RJ, Rader C, Pastan I. A novel high-affinity human monoclonal antibody to mesothelin. Int J Cancer. 2011;128:2020–30. doi: 10.1002/ijc.25557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feng Y, Xiao X, Zhu Z, Streaker E, Ho M, Pastan I, et al. A novel human monoclonal antibody that binds with high affinity to mesothelin-expressing cells and kills them by antibody-dependent cell-mediated cytotoxicity. Mol Cancer Ther. 2009;8:1113–8. doi: 10.1158/1535-7163.MCT-08-0945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hassan R, Kindler HL, Jahan T, Bazhenova L, Reck M, Thomas A, et al. Phase II clinical trial of amatuximab, a chimeric anti-mesothelin antibody with pemetrexed and cisplatin in advanced unresectable pleural mesothelioma. Clin Cancer Res. 2014;20:5927–36. doi: 10.1158/1078-0432.CCR-14-0804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Golfier S, Kopitz C, Kahnert A, Heisler I, Schatz CA, Stelte-Ludwig B, et al. Anetumab ravtansine: a novel mesothelin-targeting antibody-drug conjugate cures tumors with heterogeneous target expression favored by bystander effect. Mol Cancer Ther. 2014;13:1537–48. doi: 10.1158/1535-7163.MCT-13-0926. [DOI] [PubMed] [Google Scholar]
- 47.Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A. 2009;106:3360–5. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moon EK, Wang LC, Dolfi DV, Wilson CB, Ranganathan R, Sun J, et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res. 2014;20:4262–73. doi: 10.1158/1078-0432.CCR-13-2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao Y, Moon E, Carpenito C, Paulos CM, Liu X, Brennan AL, et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 2010;70:9053–61. doi: 10.1158/0008-5472.CAN-10-2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guedan S, Chen X, Madar A, Carpenito C, McGettigan SE, Frigault MJ, et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood. 2014;124:1070–80. doi: 10.1182/blood-2013-10-535245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lanitis E, Poussin M, Hagemann IS, Coukos G, Sandaltzopoulos R, Scholler N, et al. Redirected antitumor activity of primary human lymphocytes transduced with a fully human anti-mesothelin chimeric receptor. Mol Ther. 2012;20:633–43. doi: 10.1038/mt.2011.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med. 2014;6:261ra151. doi: 10.1126/scitranslmed.3010162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lanitis E, Poussin M, Klattenhoff AW, Song D, Sandaltzopoulos R, June CH, et al. Chimeric antigen receptor T cells with dissociated signaling domains exhibit focused anti-tumor activity with reduced potential for toxicity. Cancer Immunol Res. 2013;1:43–53. doi: 10.1158/2326-6066.CIR-13-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moon EK, Carpenito C, Sun J, Wang LC, Kapoor V, Predina J, et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res. 2011;17:4719–30. doi: 10.1158/1078-0432.CCR-11-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Riese MJ, Wang LC, Moon EK, Joshi RP, Ranganathan A, June CH, et al. Enhanced effector responses in activated CD8+ T cells deficient in diacylglycerol kinases. Cancer Res. 2013;73:3566–77. doi: 10.1158/0008-5472.CAN-12-3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hombach AA, Abken H. Costimulation by chimeric antigen receptors revisited the T cell antitumor response benefits from combined CD28-OX40 signalling. Int J Cancer. 2011;129:2935–44. doi: 10.1002/ijc.25960. [DOI] [PubMed] [Google Scholar]
- 57.Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4–1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther. 2010;18:413–20. doi: 10.1038/mt.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang E, Wang LC, Tsai CY, Bhoj V, Gershenson Z, Moon EK, et al. Generation of Potent T cell Immunotherapy for Cancer using DAP12-based, Multichain, Chimeric Immunoreceptors. Cancer Immunol Res. 2015;3:815–26. doi: 10.1158/2326-6066.CIR-15-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sentman CL, Meehan KR. NKG2D CARs as cell therapy for cancer. Cancer J. 2014;20:156–9. doi: 10.1097/PPO.0000000000000029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Watanabe K, Terakura S, Martens AC, van Meerten T, Uchiyama S, Imai M, et al. Target antigen density governs the efficacy of anti-CD20-CD28-CD3 zeta chimeric antigen receptor-modified effector CD8+ T cells. J Immunol. 2015;194:911–20. doi: 10.4049/jimmunol.1402346. [DOI] [PubMed] [Google Scholar]
- 61.Weijtens ME, Hart EH, Bolhuis RL. Functional balance between T cell chimeric receptor density and tumor associated antigen density: CTL mediated cytolysis and lymphokine production. Gene Ther. 2000;7:35–42. doi: 10.1038/sj.gt.3301051. [DOI] [PubMed] [Google Scholar]
- 62.Song DG, Ye Q, Poussin M, Liu L, Figini M, Powell DJ., Jr A fully human chimeric antigen receptor with potent activity against cancer cells but reduced risk for off-tumor toxicity. Oncotarget. 2015 doi: 10.18632/oncotarget.4071. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chmielewski M, Hombach A, Heuser C, Adams GP, Abken H. T cell activation by antibody-like immunoreceptors: increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. J Immunol. 2004;173:7647–53. doi: 10.4049/jimmunol.173.12.7647. [DOI] [PubMed] [Google Scholar]
- 64.Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, et al. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin Cancer Res. 2013;19:3153–64. doi: 10.1158/1078-0432.CCR-13-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Burga RA, Thorn M, Point GR, Guha P, Nguyen CT, Licata LA, et al. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol Immunother. 2015;64:817–29. doi: 10.1007/s00262-015-1692-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kofler DM, Chmielewski M, Rappl G, Hombach A, Riet T, Schmidt A, et al. CD28 costimulation Impairs the efficacy of a redirected t-cell antitumor attack in the presence of regulatory t cells which can be overcome by preventing Lck activation. Mol Ther. 2011;19:760–7. doi: 10.1038/mt.2011.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee JC, Hayman E, Pegram HJ, Santos E, Heller G, Sadelain M, et al. In vivo inhibition of human CD19-targeted effector T cells by natural T regulatory cells in a xenotransplant murine model of B cell malignancy. Cancer Res. 2011;71:2871–81. doi: 10.1158/0008-5472.CAN-10-0552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ninomiya S, Narala N, Huye L, Yagyu S, Savoldo B, Dotti G, et al. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood. 2015;125:3905–16. doi: 10.1182/blood-2015-01-621474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nolan KF, Yun CO, Akamatsu Y, Murphy JC, Leung SO, Beecham EJ, et al. Bypassing immunization: optimized design of “designer T cells” against carcinoembryonic antigen (CEA)-expressing tumors, and lack of suppression by soluble CEA. Clin Cancer Res. 1999;5:3928–41. [PubMed] [Google Scholar]
- 70.Beecham EJ, Ortiz-Pujols S, Junghans RP. Dynamics of tumor cell killing by human T lymphocytes armed with an anti-carcinoembryonic antigen chimeric immunoglobulin T-cell receptor. J Immunother. 2000;23:332–43. doi: 10.1097/00002371-200005000-00006. [DOI] [PubMed] [Google Scholar]
- 71.Gonzalez PA, Carreno LJ, Cespedes PF, Bueno SM, Riedel CA, Kalergis AM. Modulation of tumor immunity by soluble and membrane-bound molecules at the immunological synapse. Clin Dev Immunol. 2013;2013:450291. doi: 10.1155/2013/450291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Servais EL, Colovos C, Kachala SS, Adusumilli PS. Pre-clinical mouse models of primary and metastatic pleural cancers of the lung and breast and the use of bioluminescent imaging to monitor pleural tumor burden. Curr Protoc Pharmacol. 2011;Chapter 14(Unit14):21. doi: 10.1002/0471141755.ph1421s54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Servais EL, Suzuki K, Colovos C, Rodriguez L, Sima C, Fleisher M, et al. An in vivo platform for tumor biomarker assessment. PLoS One. 2011;6:e26722. doi: 10.1371/journal.pone.0026722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. 2014;2:112–20. doi: 10.1158/2326-6066.CIR-13-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Maus MV, Haas AR, Beatty GL, Albelda SM, Levine BL, Liu X, et al. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol Res. 2013;1:26–31. doi: 10.1158/2326-6066.CIR-13-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cooper LJ, Ausubel L, Gutierrez M, Stephan S, Shakeley R, Olivares S, et al. Manufacturing of gene-modified cytotoxic T lymphocytes for autologous cellular therapy for lymphoma. Cytotherapy. 2006;8:105–17. doi: 10.1080/14653240600620176. [DOI] [PubMed] [Google Scholar]
- 77.Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365:1673–83. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang X, Chang WC, Wong CW, Colcher D, Sherman M, Ostberg JR, et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011;118:1255–63. doi: 10.1182/blood-2011-02-337360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Adusumilli PS. Translational immunotherapeutics: Chemoimmunotherapy for malignant pleural mesothelioma. Cancer. 2014;120:3268–71. doi: 10.1002/cncr.28883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. 2010;33:780–8. doi: 10.1097/CJI.0b013e3181ee6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Asai H, Fujiwara H, An J, Ochi T, Miyazaki Y, Nagai K, et al. Co-introduced functional CCR2 potentiates in vivo anti-lung cancer functionality mediated by T cells double gene-modified to express WT1-specific T-cell receptor. PLoS One. 2013;8:e56820. doi: 10.1371/journal.pone.0056820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sun J, Dotti G, Huye LE, Foster AE, Savoldo B, Gramatges MM, et al. T cells expressing constitutively active Akt resist multiple tumor-associated inhibitory mechanisms. Mol Ther. 2010;18:2006–17. doi: 10.1038/mt.2010.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Loskog A, Giandomenico V, Rossig C, Pule M, Dotti G, Brenner MK. Addition of the CD28 signaling domain to chimeric T-cell receptors enhances chimeric T-cell resistance to T regulatory cells. Leukemia. 2006;20:1819–28. doi: 10.1038/sj.leu.2404366. [DOI] [PubMed] [Google Scholar]
- 84.Zhang L, Yu Z, Muranski P, Palmer DC, Restifo NP, Rosenberg SA, et al. Inhibition of TGF-beta signaling in genetically engineered tumor antigen-reactive T cells significantly enhances tumor treatment efficacy. Gene Therapy. 2013;20:575–80. doi: 10.1038/gt.2012.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ankri C, Shamalov K, Horovitz-Fried M, Mauer S, Cohen CJ. Human T cells engineered to express a programmed death 1/28 costimulatory retargeting molecule display enhanced antitumor activity. J Immunol. 2013;191:4121–9. doi: 10.4049/jimmunol.1203085. [DOI] [PubMed] [Google Scholar]
- 86.Prosser ME, Brown CE, Shami AF, Forman SJ, Jensen MC. Tumor PD-L1 co-stimulates primary human CD8(+) cytotoxic T cells modified to express a PD1:CD28 chimeric receptor. Mol Immunol. 2012;51:263–72. doi: 10.1016/j.molimm.2012.03.023. [DOI] [PubMed] [Google Scholar]
- 87.Cherkassky L, Villena-Vargas J, Morello A, Rusch V, Sadelain M, Adusumilli P. Mesothelin-targeted T Cells Gene-Engineered with 4–1BB Costimulation Overcome Tumor-Mediated Immunoinhibition and Eradicate Established Solid Tumors. J Am Coll Surg. 2014;219:S134. [Google Scholar]
- 88.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31:71–5. doi: 10.1038/nbt.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kreitman RJ, Hassan R, Fitzgerald DJ, Pastan I. Phase I trial of continuous infusion anti-mesothelin recombinant immunotoxin SS1P. Clin Cancer Res. 2009;15:5274–9. doi: 10.1158/1078-0432.CCR-09-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fujisaka Y, Kurata T, Tanaka K, Kudo T, Okamoto K, Tsurutani J, et al. Phase I study of amatuximab, a novel monoclonal antibody to mesothelin, in Japanese patients with advanced solid tumors. Invest New Drugs. 2015;33:380–8. doi: 10.1007/s10637-014-0196-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hassan R, Cohen SJ, Phillips M, Pastan I, Sharon E, Kelly RJ, et al. Phase I clinical trial of the chimeric anti-mesothelin monoclonal antibody MORAb-009 in patients with mesothelin-expressing cancers. Clin Cancer Res. 2010;16:6132–8. doi: 10.1158/1078-0432.CCR-10-2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 2009;113:6392–402. doi: 10.1182/blood-2009-03-209650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. 2015;21:524–9. doi: 10.1038/nm.3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119:4133–41. doi: 10.1182/blood-2011-12-400044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors. Oncoimmunology. 2015;4:e994446. doi: 10.4161/2162402X.2014.994446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chinnasamy D, Yu Z, Kerkar SP, Zhang L, Morgan RA, Restifo NP, et al. Local delivery of interleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin Cancer Res. 2012;18:1672–83. doi: 10.1158/1078-0432.CCR-11-3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chmielewski M, Kopecky C, Hombach AA, Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011;71:5697–706. doi: 10.1158/0008-5472.CAN-11-0103. [DOI] [PubMed] [Google Scholar]
- 98.Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. 2010;24:1160–70. doi: 10.1038/leu.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Markley JC, Sadelain M. IL-7 and IL-21 are superior to IL-2 and IL-15 in promoting human T cell-mediated rejection of systemic lymphoma in immunodeficient mice. Blood. 2010;115:3508–19. doi: 10.1182/blood-2009-09-241398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Perna SK, Pagliara D, Mahendravada A, Liu H, Brenner MK, Savoldo B, et al. Interleukin-7 mediates selective expansion of tumor-redirected cytotoxic T lymphocytes (CTLs) without enhancement of regulatory T-cell inhibition. Clin Cancer Res. 2014;20:131–9. doi: 10.1158/1078-0432.CCR-13-1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Singh H, Figliola MJ, Dawson MJ, Huls H, Olivares S, Switzer K, et al. Reprogramming CD19-specific T cells with IL-21 signaling can improve adoptive immunotherapy of B-lineage malignancies. Cancer Res. 2011;71:3516–27. doi: 10.1158/0008-5472.CAN-10-3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Stephan MT, Ponomarev V, Brentjens RJ, Chang AH, Dobrenkov KV, Heller G, et al. T cell-encoded CD80 and 4–1BBL induce auto- and transcostimulation, resulting in potent tumor rejection. Nat Med. 2007;13:1440–9. doi: 10.1038/nm1676. [DOI] [PubMed] [Google Scholar]
- 103.Curran KJ, Seinstra BA, Nikhamin Y, Yeh R, Usachenko Y, van Leeuwen DG, et al. Enhancing antitumor efficacy of chimeric antigen receptor T cells through constitutive CD40L expression. Mol Ther. 2015;23:769–78. doi: 10.1038/mt.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wilkie S, Burbridge SE, Chiapero-Stanke L, Pereira AC, Cleary S, van der Stegen SJ, et al. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J Biol Chem. 2010;285:25538–44. doi: 10.1074/jbc.M110.127951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Condomines M, Arnason J, Benjamin R, Gunset G, Plotkin J, Sadelain M. Tumor-Targeted Human T Cells Expressing CD28-Based Chimeric Antigen Receptors Circumvent CTLA-4 Inhibition. PLoS One. 2015;10:e0130518. doi: 10.1371/journal.pone.0130518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. 2013;5:215ra172. doi: 10.1126/scitranslmed.3006597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sampson JH, Choi BD, Sanchez-Perez L, Suryadevara CM, Snyder DJ, Flores CT, et al. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin Cancer Res. 2014;20:972–84. doi: 10.1158/1078-0432.CCR-13-0709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sanchez C, Chan R, Bajgain P, Rambally S, Palapattu G, Mims M, et al. Combining T-cell immunotherapy and anti-androgen therapy for prostate cancer. Prostate Cancer Prostatic Dis. 2013;16:123–31. S1. doi: 10.1038/pcan.2012.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Song DG, Ye Q, Santoro S, Fang C, Best A, Powell DJ., Jr Chimeric NKG2D CAR-expressing T cell-mediated attack of human ovarian cancer is enhanced by histone deacetylase inhibition. Hum Gene Ther. 2013;24:295–305. doi: 10.1089/hum.2012.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. 2013;19:5636–46. doi: 10.1158/1078-0432.CCR-13-0458. [DOI] [PubMed] [Google Scholar]
- 111.Kobold S, Steffen J, Chaloupka M, Grassmann S, Henkel J, Castoldi R, et al. Selective bispecific T cell recruiting antibody and antitumor activity of adoptive T cell transfer. J Natl Cancer Inst. 2015;107:364. doi: 10.1093/jnci/dju364. [DOI] [PubMed] [Google Scholar]
- 112.Karlsson H, Lindqvist AC, Fransson M, Paul-Wetterberg G, Nilsson B, Essand M, et al. Combining CAR T cells and the Bcl-2 family apoptosis inhibitor ABT-737 for treating B-cell malignancy. Cancer Gene Ther. 2013;20:386–93. doi: 10.1038/cgt.2013.35. [DOI] [PubMed] [Google Scholar]
- 113.Huye LE, Nakazawa Y, Patel MP, Yvon E, Sun J, Savoldo B, et al. Combining mTor inhibitors with rapamycin-resistant T cells: a two-pronged approach to tumor elimination. Mol Ther. 2011;19:2239–48. doi: 10.1038/mt.2011.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gargett T, Fraser CK, Dotti G, Yvon ES, Brown MP. BRAF and MEK inhibition variably affect GD2-specific chimeric antigen receptor (CAR) T-cell function in vitro. J Immunother. 2015;38:12–23. doi: 10.1097/CJI.0000000000000061. [DOI] [PubMed] [Google Scholar]
- 115.Nishio N, Diaconu I, Liu H, Cerullo V, Caruana I, Hoyos V, et al. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. 2014;74:5195–205. doi: 10.1158/0008-5472.CAN-14-0697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Caruana I, Weber G, Ballard BC, Wood MS, Savoldo B, Dotti G. K562-Derived Whole-Cell Vaccine Enhances Antitumor Responses of CAR-Redirected Virus-Specific Cytotoxic T Lymphocytes In Vivo. Clin Cancer Res. 2015;21:2952–62. doi: 10.1158/1078-0432.CCR-14-2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.