

Abstract
Immune responses against cancer cells are often hindered by immunosuppressive mechanisms that are developed in the tumor microenvironment. Induction of a hyporesponsive state in tumor antigen-specific T cells is one of the major events responsible for the inability of the adaptive immune system to mount an efficient anti-tumor responses and frequently contributes to lessen the efficacy of immunotherapeutic approaches. Treatment of localized tumors by focused ultrasound is a minimally invasive therapy that uses a range of input energy for in situ tumor ablation through the generation of thermal and cavitation effect. Using a murine B16 melanoma tumor model, we show that a variant of focused ultrasound that delivers a reduced level of energy at the focal point and generates mild mechanical and thermal stress in target cells, has the ability to increase immunogenic presentation of tumor antigens, which results in reversal of tumor-induced T cell tolerance. Furthermore, we show that the combination of non-ablative low energy focused ultrasound with an ablative hypofractionated radiation therapy results in synergistic control of primary tumors and leads to a dramatic reduction in spontaneous pulmonary metastases while prolonging recurrence free survival only in immunocompetent mice.
Introduction
Immune responses against cancer cells are frequently hampered by the immunosuppressive nature of the tumor microenvironment, which is also responsible for hindering the efficacy of cancer immunotherapy (1, 2). Several mechanisms have been identified underlying the ability of tumors to generate an immunosuppressive environment, including secretion of cytokines or other factors with inhibitory activity (3–5), recruitment of regulatory T cells and myeloid-derived suppressor cells (6–9), increased expression of ligands for co-inhibitory receptors (10–13) or inhibition of dendritic cell maturation (14, 15). As a consequence of those mechanisms T cells are often rendered unresponsive to tumor antigens (15). Induction of a hyporesponsive state to tumor antigens occurs both in CD4+ and CD8+ T cell populations and is often responsible for the inability of the adaptive immune system to mount an efficient anti-tumor response (16–18). Decreased T cell responses to tumor antigens occur in both solid and hematological tumors and appear to be caused by inefficient presentation of antigens by dendritic cells, which results in the preferential activation of tolerogenic programs of gene expression that are dependent on the transcription factors NFAT and Egr2 (18–21). The important role of this process of tumor-induced T cell hyporesponsiveness is underscored by the fact that genetic mouse models where the induction of this tolerogenic gene expression program is prevented result in enhanced anti-tumor T cell responses and control of tumor growth (19, 21).
Treatment of localized tumors by focused ultrasound (FUS) is an image guided minimally invasive therapy that uses a range of input energy for in situ tumor ablation (22, 23). The application of FUS to biological tissues is associated with the generation of thermal and cavitation effects, causing changes in target cell physiology, depending on the energy delivered. High intensity focused ultrasound (HIFU) has been used clinically to thermally ablate localized tumors (23–26). The substantial thermal energy generated by that modality of FUS treatment causes rapid coagulative necrosis of the tissue at the targeted focal spots. Though several studies have reported some immunomodulatory effects, including increased lymphocyte infiltration, generation of IFNγ producing tumor-specific T cells in lymphoid organs and dendritic cell maturation and migration into tumors (26–29), the thermally induced coagulative necrosis resulting from HIFU treatment can also attenuate the release of immunostimulatory molecules within the tumor microenvironment. Thus, although able to halt the progression of established primary tumors, HIFU might fail to protect against local and distant metastases arising from the surviving tumor cells.
In this report, we designed a variant of FUS that delivers a reduced level of energy at the focal point for a short time with limited tissue temperature elevation (LOFU). Using a murine B16 melanoma tumor model, we show that LOFU treatment is capable of inducing a stress response in the tumor, which increases the expression of immunomodulatory factors enhancing efficient tumor antigen presentation and leading to reversal of tumor-induced tolerance, with increased effector cytokine production in tumor-antigen specific CD4+ T cells. Furthermore, the combination of LOFU with an ablative hypofractionated Cone Beam CT image-guided radiation therapy (IGRT) results in synergistic control of primary tumors and a marked reduction in spontaneous pulmonary metastases with prolonged recurrence free survival only in immunocompetent mice.
Methods
Mice
6–8 week old C57BL/6, B6.Cg-Rag1tm1MomTyrp1B-wTg(TcraTcrb)9Rest/J (Tyrp1) and B6.Cg-Tg(TcraTcrb)425Cbn/J (OT-II) mouse strains were purchased from The Jackson Laboratory. BALBc/Nude mice were obtained from National Cancer Institute, distributed through Charles River. All mice were housed and maintained in pathogen-free facilities.
Culture of B16 cell lines and primary CD4+ T cells
B16-F1 and B16-F10 melanoma cell lines were purchased from the American Type Culture Collection (ATCC). A highly aggressive subclone of B16-F10 (B16-M1) was generated by isolating and expanding a metastatic clone that arose in a C57BL/6 mouse 6 weeks after surgical removal of an established primary tumor. The B16-OVA melanoma cell line was kindly provided by E.M. Lord (University of Rochester Medical Center, Rochester, NY). The expression of OVA by B16-OVA cells was confirmed by real time PCR. All melanoma cells were cultured in DMEM (Thermo Scientific) supplemented with 10% heat inactivated FBS, 2mM L-Glutamine and 250 IU of penicillin/streptomycin.
CD4+ T cells were isolated using anti-CD4 conjugated magnetic Dynabeads (Life Technologies) according to the manufacturer’s protocol. Where indicated, CD4+ T cells were differentiated into TH1 helper cells by activation with plate-bound anti-CD3ε (clone 2C11; 0.25 μg/mL) and anti-CD28 (clone 37.51; 0.25 μg/mL) antibodies (BD Biosciences) and cultured for six days in DMEM supplemented with 10% heat inactivated FBS, 2 mM L-glutamine, 50 μM 2-mercaptoethanol, nonessential amino acids and essential vitamins (Cambrex), in the presence of murine IL-12 (10 ng/mL) (eBioscience), anti-mouse IL-4 antibody (clone 11C.11; 10 μg/ml) and 10 U/mL recombinant human IL-2 (Biological Resources Branch of the National Cancer Institute).
Generation of Dendritic cells from bone marrow derived cells
Bone marrow derived cells were harvested from the femur and tibia of C57Bl/6 mice and were cultured in RPMI 1640 medium supplemented with 10% FBS in presence of 20 ng/ml murine GM-CSF (Peprotech). Loosely adherent immature dendritic cells were harvested after 7 days of culture. If necessary, murine TNFα (Peprotech) at a concentration of 100 ng/ml was added to the culture for the last 24 hours to induce dendritic cell maturation.
Tumor models
3×105 B16-F1 melanoma cells suspended in Hanks’ Balanced Salt Solution (Invitrogen) were injected s.c. in the lumbar flanks of mice. Melanoma tumors were induced in the footpads by injecting 2×105 B16-M1 cells in the dorsum of the right hind limb.
Tumor growth monitoring
Primary B16-M1 melanoma dorsal hind limb tumors were measured three times per week with vernier calipers. Tumor volume was calculated using an ellipsoid formula: V = (π/6 × length × width × height). Primary dorsal hind limb tumors exhibit Gompertzian growth, with a phase I volume of 30–50 mm3, phase II volume of 90–150 mm3, and phase III volume of 300–500 mm3. Therefore, treatment efficacy was determined by determining the tumor growth delay (TGD) to 90–150 mm3, in which the tumor is in the exponential phase II. Tumors that reach 300–500 mm3 begin to enter phase III due to anatomical and vascular limitations. Consequently, below-the-knee amputations were performed on mice with tumors ≥300–500 mm3, in accordance with IACUC approved protocol.
ELISA
1.5 to 2.5×104 T cells were left rested or stimulated with either anti-CD3ε+anti-CD28 antibodies, T cell depleted OVA peptide 323-339 (OVA323-339)-loaded splenocytes at a 1:5 T cell:splenocyte ratio, or CD11c+ purified dendritic cells (using CD11c-beads; Miltenyi Biotech) loaded with OVA323-339 or melanoma tumor lysates at a 1:3 dendritic cell:T cell ratio. Culture supernatants were typically harvested 24 hours after stimulation, and IL-2 or IFNγ levels were measured by a sandwich ELISA (BD Biosciences).
Tumor lysates
Tumors were resected from tumor bearing mice, cut into 1–2 mm pieces and passed through 40 μm nylon meshes. Cells were washed in PBS and resuspended in serum-free DMEM. Cell suspensions were then snap frozen in liquid nitrogen, and thawed at 37°C for five cycles with visual confirmation of complete lysis by light microscopy. The lysates were spun at 10,000g for 15 minutes at 4°C, and the pellets with cellular debris were discarded. The supernatant was used along with purified dendritic cells to stimulate T cells.
Immunofluorescence staining
Tumor tissue was isolated, washed in PBS and embedded in OCT compound (Electron Microscopy Sciences). Tissue sections (5μm) were prepared and permeabilized with acetone for 5 min and incubated with goat serum for 30 min to block non-specific protein-protein interactions. Tissue sections were incubated overnight with the following antibodies: anti-Calreticulin (Pierce, PA5-25922), anti-Trp1 (Abcam, ab3312; clone TA99) and anti-Hsp70 (Novus Biologicals, NBP1-77455). Appropriate secondary antibodies were used for 30 min at room temperature. DAPI (Invitrogen) was used to detect nuclei. At least 10 fields/sample were blindly analyzed with an Inverted Olympus IX81 fluorescence microscope.
Focused ultrasound therapy system
A therapy and imaging probe system (TIPS, Philips Research North America, Briarcliff Manor, NY, USA) was utilized for all ultrasound exposures. The system is capable of delivering focused and spatiotemporally controlled ultrasound energy and consists of a therapy control workstation, RF generators and control electronics, an 8-element spherical shell annular array ultrasound transducer (80 mm radius of curvature, 80 mm aperture), as well as a motion stage to allow for in-plane transducer movement and accurate positioning perpendicular to ultrasound beam axis. The focused ultrasound beam can also be steered approximately ±15 mm out-of-plane using electronic deflection of the focal point. The ultrasound beam propagates vertically into the target through a thin (25 μm) circular plastic membrane, with acoustic coupling provided by degassed water. During therapy, the system allows adjustments of acoustic output power, ultrasound exposure duration, duty cycle, and ultrasound frequency.
In vivo focused ultrasound (FUS) therapy
Mice were anesthetized with a continuous flow 1.5 liters/minute of 1.5% isoflurane in pure oxygen. To ensure proper acoustic coupling, the tumor- bearing leg or lumbar flank were carefully shaved. Once the animal was positioned for therapy, the tumor was acoustically coupled to the TIPS system using degassed water and ultrasound gel. The center of the tumor was then placed at the focal length of 80 mm from the transducer. Ultrasound exposures were delivered to the tumor using a 1 mm grid pattern extending over the entire tumor volume. Two layers of grid points (spaced 5 mm apart) were performed in each tumor, resulting in approximately 160 discrete foci and 5 min exposure duration per tumor. The ultrasound transducer was operated at 1.0 MHz, resulting in an ellipsoid focal spot approximately 1.5 mm in diameter and 12 mm in length (−6 dB of pressure), as measured along the ellipsoid axes. Ultrasound exposures were delivered to the tumor using a 1 mm grid pattern extending over the entire tumor volume. Prior to therapy, the tumor volume was measured to calculate the grid size for the particular treatment. The duration of ultrasound exposure at each grid point was 1.5 s, after which the transducer was automatically positioned over the next grid point and the procedure repeated until the entire tumor volume was covered. Two layers of grid points were performed in each tumor. The therapeutic ultrasound device was operated in continuous wave mode at a specific acoustic power/pressure regimen: acoustic power 3W, peak negative pressure=2.93 MPa (80 mm focal length)/3.81 MPa (85 mm focal length); to provide non-ablative low-energy FUS (LOFU). The resulting in situ intensity (Ispta) at the focus was estimated to be 550 W/cm2 at a depth of 4 mm in tissue. Total energy deposition to a tumor was approximately 900 J. For the ablative HIFU regimen, a similar setup was used with the following changes: 5.42 MPa peak negative pressure, 1.2 mm spacing between grid points, 4 seconds duration at each grid point at 75% duty cycle, generating an acoustic power of 12.5 W. Unlike the LOFU protocol, the HIFU was carried out at a single focal length of 80 mm.
In vivo hypofractionated cone beam CT image-guided Radiation Therapy (IGRT)
All radiation was delivered using Xstrahl Limited’s Small Animal Radiation Research Platform (SARRP) to deliver a 10Gy dose to a target tumor in 341 seconds. Anesthetized animals were placed on stage attached to a motorized platform and the tumor-bearing right hind limb was extended, elevated, and secured to a 1.5 cm adhesive platform to minimize extraneous tissue exposure. Once secure, a cone beam CT (CBCT) was performed and the data opened in 3D Slicer for tissue segmentation and treatment planning. 10 Gy each was delivered for three successive days for a total hypofractionated dose of 30 Gy. In the combination therapy groups, LOFU was performed 2–4 hours prior to CBCT.
Pulmonary Metastasis Evaluation
Lungs were isolated from animals that died spontaneously, were euthanized or were sacrificed at the end of the 8-week experiment. 1 mL of Fekete’s solution (Ethanol, Glacial acetic acid and formaldehyde based bleaching fixative) was injected to insufflate the lungs. The trachea was then clamped, and the entire lungs and heart removed en bloc and washed with PBS. The lungs were then placed in Fekete’s solution and allowed to bleach for 48 hours prior to analysis. The left lung and the 4 lobes of the right lung were isolated and nodules counted with the aid of a dissecting microscope. Indistinct or fused nodules cannot be reliably enumerated; therefore, the lung was labeled as too numerous to account and assigned an arbitrary metastasis count of 250. Statistical analysis was performed using the non-parametric Kruskal-Wallis test, followed by the Dunn’s posttest for multiple comparisons.
Recurrence Free Survival
The following events were scored as positive events in our recurrence free survival analysis: spontaneous death with necropsy validation of tumor involvement, euthanasia due to extensive local metastasis to the draining popliteal or inguinal lymph nodes, or euthanasia due to moribund appearance indicating extensive systemic tumor burden. The following non-tumor-dependent deaths were processed as censored data: death within 24–48 hours of amputation or sacrifice of any animals at the end of the 8-week experiment. In order to prevent selective sacrifice of control or treated animals, cages were labeled using an alphanumeric code such that animal institute veterinarians were blinded from treatment and control groups. Recurrence free survival was analyzed using a Mantel-Cox test, with statistical significance defined as P< 0.05.
Real time PCR
Total RNA was extracted from cells using RNeasy Micro kit (Qiagen), and cDNA was synthesized using qScript cDNA supermix (Quanta Biosciences). The cDNA samples were subjected to real time PCR using PowerSYBR (Applied Biosystems) as the reporter dye on a StepOnePlus real time PCR system (Applied Biosystems). Expression of the transcripts studied was normalized to beta actin. The primer sets used are the following:
actinb: F-GTGACGTTGACATCCGTAAAGA, R-GCCGGACTCATCGTACTCC;
Cblb: F-GCAGCATCATTGACCCTTTCA, R-ATGTGACTGGTGAGTTCTGCC;
Rnf128: F-ATGCAAGAGCTCAAAGCAGGAAGC, R-GTGCGCAGCTGAAGCTTTCCAATA;
Ikaros: F-GCTGGCTCTCGGAGGAG, R-CGCACTTGTACACCTTCAGC;
Casp3: F-ACGCGCACAAGCTAGAATTT, R-CTTTGCGTGGAAAGTGGAGT;
Egr2: F-TCAGTGGTTTTATGCACCAGC, R- GAAGCTACTCGGATACGGGAG;
Tle4: F-TCACTCAAGTTTGCCCACTG, R-CACAGCTAAGCACCGATGAG;
Itch: F-GTGTGGAGTCACCAGACCCT, R-GCTTCTACTTGCAGCCCATC;
Foxp3: F-GGCCCTTCTCCAGGACAGA; R-GCTGATCATGGCTGGGTTGT.
Flow cytometry
Cells were pre-blocked with Fc block (CD16/CD32) antibody prior to immunostaining. The following fluorochrome conjugated antibodies were used: anti CD80, anti CD86, anti MHC-I, anti Gr1, anti CD11b, anti CD11c, anti CD8 and anti CD45 (all from eBioscience). Anti CD83 and anti CD4 were purchased from Biolegend. Anti Hsp70 was from Novus Biologicals and anti-TRP1 was purchased from Abcam. Dead cells were detected by using a UV LIVE/DEAD® Fixable Dead Cell Stain Kit (Invitrogen). The immunostained cells were analyzed on an LSR-II Flow Cytometer (Becton Dickinson), and post-acquisition analyses were carried out using the FlowJo software.
Animal study approval
All animal work and protocols were performed under an approved protocol designed following the guidelines set by the Institutional Animal Care and Use Committee (IACUC) of the Albert Einstein College of Medicine.
Results
Treatment of primary B16 melanoma with LOFU overcomes tumor-induced tolerance in CD4+ T cells
To determine how melanoma cells may modulate tumor induced effector CD4+ T cell responses, we used three different mouse models. First, B16-F1 melanoma tumors were induced in C57Bl/6J mice by subcutaneous injection of B16 cells in the lumbar flanks. Tumors were allowed to grow to a size of 70–80 mm3 and CD4+ T cells were then isolated from both the ipsilateral inguinal draining lymph nodes (DLN) and distal-contralateral non-draining cervical lymph nodes (NDLN). T cells were also obtained from control mice that did not harbor any tumors. Supporting previous reports of tumor-antigen specific T cell tolerance in murine melanoma (18, 21), CD4+ T cells isolated from the tumor DLN produced significantly less IL-2 than cells isolated from the distal contralateral NDLN of the same mice, or from lymph nodes of control tumor-free mice, when stimulated ex vivo with anti-CD3 and anti-CD28 antibodies (Fig 1A). To confirm these data, a B16-F1 melanoma cell line that had been stably transfected to express OVA as a surrogate tumor antigen was used. These cells were subcutaneously injected into OT-II mice, a mouse strain with T cells expressing a transgenic MHC class II-restricted TCR that recognizes the OVA323-339 peptide. T cells were collected from these mice as described above, and stimulated ex vivo using splenocytes loaded with OVA323-339 peptide. CD4+ T cells from the ipsilateral DLN again produced significantly reduced amounts of IL-2 compared to cells from the contralateral NDLN or from tumor free mice (Fig 1B). These results were further corroborated in a third model using Tyrp1 mice, which are deficient in tyrosinase-related protein 1 and bear T cells expressing a MHC class II-restricted TCR specific for the TRP-1113-127 peptide of this endogenous melanocyte differentiation antigen. Those mice were injected with B16-F1 cells. As in the previous two models, IL-2 production by CD4+T cells harvested from the ipsilateral DLN was significantly reduced compared to cells harvested from contralateral NDLN or from tumor-free mice. (Fig 1C). Similar but less pronounced effects were also observed when IFNγ expression was analyzed (Fig. 1D–F). Altogether, these results support that melanoma tumors induce hyporesponsiveness in tumor antigen-specific CD4+ T cells, which translates in a reduced capacity to produce effector cytokines upon re-stimulation.
Figure 1. Treatment of melanoma tumors with LOFU overcomes tumor induced CD4+ T cell tolerance.

A: C57Bl/6 mice were challenged in the lumbar flanks with 3×105 B16-F1 melanoma cells. Tumors were allowed to grow to 70–80 mm3 in size. CD4+T cells were isolated from the tumor DLN and distal contralateral NDLN, and stimulated with anti-CD3 and anti-CD28 antibodies. IL-2 was measured by ELISA. CD4+T cells from tumor-free mice were used as controls. B: OTII mice were challenged with 3×105 B16-F1-OVA melanoma cells as described above. T cells were stimulated with OVA323-339 peptide-loaded splenocytes and IL-2 production measured by ELISA. C. B16-F1 cells were used to induce tumors in Tyrp1 mice as described above. Isolated CD4+ T cells were stimulated with anti-CD3 and anti-CD28 antibodies and IL-2 production determined by ELISA. (D–F) IFNγ production was also determined by ELISA in CD4+ T cells isolated from mice treated as in (A), (B) or (C), respectively. Graphs show mean+SEM from 4 (A–B) or 3 (C–F) independent experiments. Results are shown as mean+SEM from 3–5 mice for each experiment. Data were analyzed using ANOVA with a Tukey post-test (***P<0.01; **P<0.01; *P<0.05). G–H. Tumors were induced in C57Bl/6 mice by s.c. injection of 3×105 B16-F1 melanoma cells in the lumbar flank. Tumors were left untreated or treated with LOFU. Thirty-six hours after LOFU treatment, CD4+ T cells were isolated from tumor DLN or NDLNs and stimulated with anti-CD3 and anti-CD28 antibodies. IL-2 and IFNγ production was assessed by ELISA. The results (total cytokine production and ratio of the levels of cytokines produced by T cells from NDLN and DLN in each group) are presented as mean+SEM from 3 different mice per condition. Differences between cytokine production of DLN T cells in untreated or treated mice were analyzed using a 2-tailed t test (*P<0.05).
HIFU is currently being used to predominantly cause tumor ablation through the generation of high amounts of heat inside the tumor tissue leading to coagulative necrosis. Although HIFU is a very effective, noninvasive ablative procedure to achieve local tumor control, it destroys the vasculature and tissue infrastructure almost instantaneously, thereby possibly limiting the ability of different immune cells to support antigen presentation and recognition. We, therefore, hypothesized that instead of HIFU, administering LOFU would induce a non-lethal thermal/mechanical stress in the tumor tissue that could generate novel tumor antigens and/or induce the expression of stress-induced proteins, which could increase the immunogenicity of the tumor and overcome tumor-induced tolerance of CD4+ T cells. In order to examine this possibility, we treated primary B16-F1 melanoma tumors grown on separate groups of C57Bl/6J mice that were either left untreated or treated with LOFU. Thirty six hours after LOFU treatment, DLN and NDLN resident CD4+ T cells were isolated from both groups of mice, and re-stimulated ex-vivo with antiCD3 and antiCD28 antibodies. CD4+ T cells from the DLN of the LOFU-treated mice produced significantly more IL-2 compared to the cells obtained from the group of mice bearing untreated tumors. In contrast, T cells from the corresponding NDLN produced comparable amounts of IL-2 in treated and untreated mice (Fig 1G). Interestingly while LOFU treatment led to increased capacity of CD4+ T cells to produce IL-2, T cells isolated from HIFU-treated tumors did not support a significant increase in IL-2 production compared with T cells isolated from untreated tumor-bearing mice (Suppl. Fig. 1). We also observed a similar, but less pronounced effect, on IFNγ production in LOFU-treated mice (Fig 1H). Overall, these results indicated that LOFU treatments of B16 melanoma tumors appear to overcome the hyporesponsive state induced by the melanoma tumor microenvironment.
LOFU treatment prevents the induction of anergy-associated genes in tumor specific CD4+ T cells
We have previously shown that melanoma tumors can induce an NFAT1-dependent program of gene expression that leads to the production of a set of proteins which interfere with TCR signaling and directly inhibit expression of cytokine genes, resulting in the establishment of functional anergy in CD4+ T cells (21). Based on the ability of LOFU to prevent tumor-induced T cell tolerance, we assessed the possibility that LOFU treatment could inhibit tumor-induced T cell tolerance by preventing anergy induction, which would account for the increased cytokine expression observed in the DLN resident CD4+ T cells following treatment with LOFU. We first monitored the expression of anergy-associated genes in CD4+ T cells isolated from the DLN of mice bearing B16 tumors and compared it with the expression of those genes in T cells harvested from NDLN of the same mice. As we had observed previously, T cells from the DLN of tumor-bearing mice expressed higher levels of anergy-associated genes, including those encoding for the E3 ubiquitin ligases Rnf128 (encoding Grail), Cblb and Itch and the transcription factor Egr2 (Fig. 2A). We did not observe, however, any difference in the expression of Foxp3 between the DLN and NDLN T cells in tumor bearing mice, suggesting that an increased presence of regulatory T cells was not likely contributing to the decreased CD4+ T cell responses under the conditions used in this study (Fig 2A). We then determined if treatment of B16 melanomas with LOFU would have an effect on the expression of those anergy-associated genes in T cells. To assess responses induced by endogenous tumor antigens, B16 tumors growing on Tyrp1 mice were either left untreated or treated with LOFU. CD4+ T cells were isolated from the DLN and NDLN and the expression of several anergy-associated genes was assessed. T cells derived from the DLN showed varying degrees of upregulation of 6 of the 7 anergy genes analyzed, including Rnf128, Itch, and Cblb and Egr2, as well as Tle4 and Casp3 (encoding for Caspase 3) (Fig 2B–H). Another transcription factor, Ikaros, which is also upregulated in several in vitro and in vivo T cell anergy models, was not upregulated in this melanoma model of tumor-induced anergy, and its levels remained largely similar in both the DLN and NDLN derived T cells (Fig 2G). Interestingly, when the tumors were treated with LOFU, the expression of 4 of those genes, Grail, Itch, Cblb and Grg4 in T cells isolated from the DLN was significantly lower than in the T cells isolated from untreated mice (Fig 2B–F), supporting that LOFU treatment prevented the induction of the expression of anergy-inducing genes in tumor antigen-specific CD4+ T cells.
Figure 2. Treatment of melanoma tumors with LOFU prevents expression of anergy-associated genes in tumor-specific CD4+ T cells.
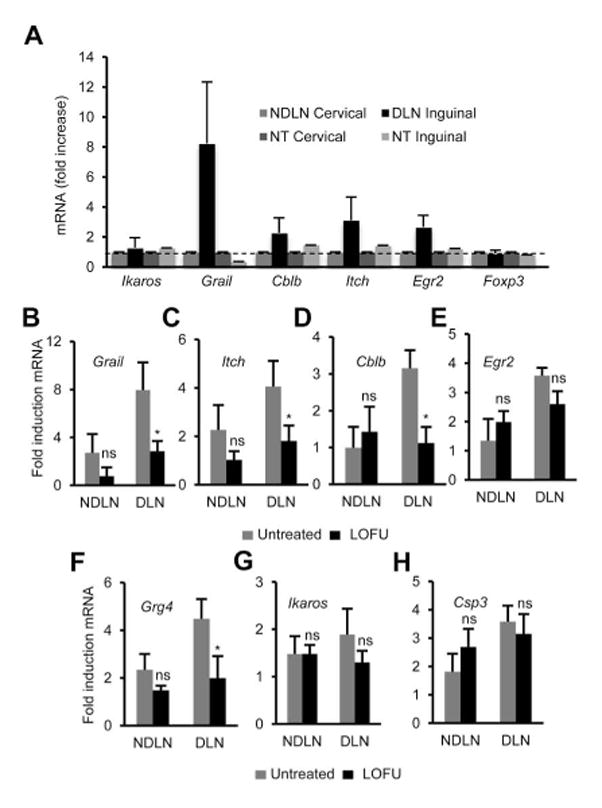
A. C57Bl/6 mice were challenged with 3×105 B16 melanoma cells to induce tumors. Following tumor development total RNA samples were extracted from CD4+T cells isolated from the DLN and NDLN of tumor-bearing mice, and tumor-free control mice. Expression of anergy-associated genes was measured by quantitative RT-PCR. The results are shown as fold induction of gene expression in the DLN or NDLN resident T cells in tumor bearing mice compared to T cells isolated from tumor-free mice. The data represent mean+SEM from 3 independent experiments. B–H. B16-F1 melanoma tumors were induced in Tyrp1 mice that were then left untreated or treated with LOFU. The expression of different anergy-associated genes was measured by RT-PCR in CD4+ T cells isolated from the DLNs and NDLNs. Expression of the anergy-associated genes is presented as fold induction (mean+SEM from 5 independent experiments. *P<0.05 between untreated and LOFU-treated conditions. t test) over the values obtained in T cells from Tyrp1 mice bearing no tumor.
LOFU treatment of melanoma tumors potentiates dendritic cell-mediated activation of tumor-specific CD4+ T cells
To determine the possibility that the ability of LOFU treatment to prevent tumor-induced T cell tolerance may result in the production of immunomodulatory molecules that could increase the stimulatory capacity of dendritic cells, we directly tested if lysates prepared from LOFU treated tumors could elicit enhanced priming of antigen specific T cells and consequently generate a more robust effector response. For this experiment, B16-F1-OVA melanoma cells were used to induce tumors in C57BL/6 mice. Lysates were prepared from untreated and LOFU treated tumors. Splenic dendritic cells and responder naïve CD4+T cells were isolated from C57BL/6 and OT-II tumor-free mice, respectively, and were co-cultured in the presence or absence of the different tumor lysates described above. Though the OVA containing tumor lysates could act as a source of tumor antigen to prime responder T cells, we also added exogenous OVA323-339 peptide to ensure uniform loading of dendritic cells with this peptide in all conditions and more accurately determine the tolerogenic or activating nature of the different tumor lysates. As expected, control responder OT-II T cells, upon activation with dendritic cells loaded with OVA323-339 peptide, showed a strong response with elevated levels of IL-2 production. However, lysates obtained from untreated tumors markedly inhibited OT-II responses and resulted in a profound decrease in IL-2 production, even though exogenous OVA323-339 peptide was added to the culture (Fig. 3A). Interestingly, lysates derived from LOFU treated tumors did not only have no negative effect on the responses of OT-II cells to OVA323-339 but were also able to elicit a strong activation of OT-II responder T cells even in the absence of exogenous peptide (Fig. 3A). Since the lysates prepared from LOFU-treated tumors seemingly exhibited a greater priming ability by dendritic cells of T cells, we proceeded to isolate bone marrow derived cells containing the monocyte and dendritic cell progenitor population and differentiated them to dendritic cells. Immature dendritic cells thus obtained were then treated with lysates prepared from untreated or LOFU-treated tumors. Whereas B16-F1 lysates prepared from untreated tumors were able to increase CD86 surface expression in dendritic cells as much as lysates prepared from LOFU-treated tumors, only the latter were also able to induce a significant increase in the expression of CD80, MHC-II and CD83 on dendritic cells (Fig. 3B). These results extended further support to our observation that LOFU treatment of B16 melanoma tumors prevents the negative effect on the T cell priming capacity of dendritic cells that normally occurs in the tumor microenvironment.
Figure 3. LOFU treatment of melanoma tumors potentiate dendritic cell-mediated priming of CD4+ T cells.
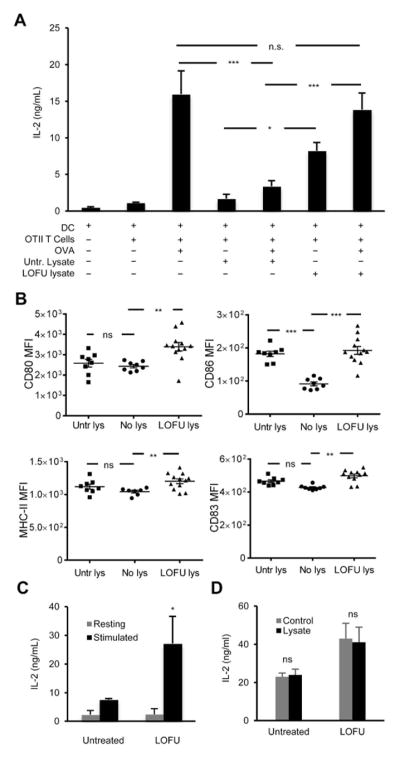
A. CD11c+ splenic dendritic cells were purified from C57Bl/6 mice and co-cultured with responder naïve CD4+ T cells isolated from OT-II mice. B16-F1-OVA melanoma tumor lysates were prepared from untreated or LOFU treated tumor-bearing mice and added to the respective cultures to drive dendritic cell mediated T cell stimulation. In separate samples exogenous OVA323-339 peptide was also added along with tumor lysates. Supernatants were collected after 24 hours, and IL-2 production was assessed by ELISA. The results are shown as mean+SEM from 4 independent experiments and analyzed with one-way ANOVA followed by a Tukey posttest (*P< 0.05; ***P<0.001; n.s., not significant). B. Bone marrow cells were isolated from the femur and tibia bones of C57Bl/6 mice and differentiated in vitro into immature dendritic cells. Cells were further cultured for 24 more hours in presence of either tumor lysate-free complete DMEM (No lys), lysates from untreated melanoma tumors (Untr lys), or lysates from LOFU-treated melanoma tumors (LOFU lys). CD11c+ cells were analyzed by flow cytometry for expressions of dendritic cell-activation markers CD80, CD83, CD86, and also for MHC-II molecules. Results are shown as average of mean fluorescence intensity (MFI) ± SEM and analyzed by one-way ANOVA with Tukey posttest. (**P< 0.01; ***P<0.001; n.s., not significant). C: B16-F1 melanoma tumors were left untreated or treated with LOFU. Tumor DLN were isolated and depleted of T cells. DLN cells were then co-cultured with naïve Tyrp1 CD4+ T cells and stimulated with B16 melanoma tumor lysates. Supernatants were collected 24 hours later and analyzed for IL-2 levels by ELISA. The data is shown as mean+SEM from 3 independent experiments. Differences between cytokine production in cultures using DLN cells from untreated or LOFU-treated mice were analyzed using a 2-tailed t test (*P<0.05). D: CD4+T cells were isolated from the tumor DLNs of untreated or LOFU-treated B16-F1 melanoma tumor-bearing C57Bl/6 mice, and stimulated with anti-CD3/anti-CD28 antibodies in presence or absence of tumor lysates prepared from untreated B16-F1 melanoma tumors. IL-2 outputs were measured by ELISA. Results are presented as mean+SEM from 3 different mice per condition.
Our results prompted us to directly determine whether tumor DLN resident antigen presenting cells would be functionally more efficient at activating target T cells following LOFU treatment of melanoma tumors. To that effect, B16-F1 melanomas were induced on C57BL/6 mice and were either left untreated or treated with LOFU. DLN cell suspensions were depleted of T cells and used to test the capacity and DLN antigen presenting cells to activate tumor antigen specific T cells. T cell-depleted DLN cells were co-cultured for 24 hours with naïve Tyrp1-TCR CD4+ T cells and lysates prepared from B16-F1 tumors. IL-2 production was measured by ELISA to monitor responder T cell priming. Cells isolated from the DLN of LOFU treated tumor-bearing mice showed a significantly increased ability to activate Tyrp1 CD4+ T cells compared with cells isolated from untreated mice. (Fig 3C), supporting that LOFU treatment of B16 melanoma results in the generation of antigen presenting cells that are functionally more efficient at activating tumor-antigen responder T cells. The effects of LOFU on tumors appeared to primarily target dendritic cells and not directly T cells. As expected (see Fig. 1) when T cells isolated from the DLN of LOFU-treated B16-bearing mice were activated with plate-bound antiCD3 and antiCD28 antibodies they produced higher amounts of IL-2 that cells obtained from untreated mice, however, this difference was not altered by co-incubation with lysates prepared from untreated B16 tumors (Fig. 3D).
Activation of melanoma-specific T cells by dendritic cells is a crucial event in determining their fate. A successful antigen presentation event that is able to elicit an effector T cell response is critically dependent on the activation state of dendritic cells that would otherwise deliver tolerogenic stimuli. Our results indicate that treatment of melanoma tumors with LOFU resulted in increased CD4+ T cell activation as consequence of inducing increased stimulatory activity on antigen presenting cells and thus hindering tumor-induced T cell tolerance. We did not observe any significant differences in the levels of infiltrating CD11b+CD11c+ dendritic cells or CD11b+Gr1+ myeloid-derived suppressor cells (MDSC) in LOFU-treated B16 melanomas, suggesting that the effect of LOFU was not being exerted on dendritic cell trafficking but likely on dendritic cell activation and antigen presentation (Fig. 4A). Trafficking of tumor antigens by molecular chaperones, including calreticulin and Hsp70, is crucial for the subsequent productive presentation of antigens to T cells (30–33). We, therefore, employed both in vivo and in vitro approaches to detect changes in the expression of calreticulin and Hsp70 in untreated and LOFU treated B16 melanoma tumors. Tumors, either left untreated or treated with LOFU, were harvested from tumor bearing mice, made into single cell suspensions and stained with a live/dead marker to assess cell viability. We did not observe changes in cell viability in response to LOFU treatment, supporting this low energy form of FUS was not directly inducing tumor cell death (Fig. 4B). When B16 melanoma tumors were left untreated or exposed to LOFU treatment and tumor tissue sections were put on slides, LOFU treated cells showed a change in the distribution of calreticulin, compared to untreated cells, which appeared to accumulate in discrete regions of the plasma membrane on B16 cells (Fig. 4C). Immunofluorescence analyses of LOFU treated melanomas also indicated that LOFU induced increased expression of Hsp70 (Fig. 4D). To determine if the increased Hsp70 expression also correlated with increased presence in the membrane of this protein, non-permeabilized CD45−TRP1+ B16 melanoma cells were stained for Hsp70 and cell surface expression following LOFU treatment assessed by FACS. This analysis confirmed that LOFU treatment of B16 melanomas caused increased membrane presence of Hsp-70 in tumor cells (Fig 4E). B16 melanoma cells express very low levels of MHC-I molecules on their surfaces, leading to a very inefficient direct tumor antigen presentation by these cells to cytotoxic T lymphocytes, which further reduces the chance of elimination of malignant cells by the adaptive immune system. Treatment of melanoma tumors by LOFU also increased cell surface MHC-I expression nearly two-fold, and interestingly also led to a similar enhancement of the expression of the tumor-specific antigen TRP1 (Fig 4F).
Figure 4. LOFU treatment of melanoma tumors enhances expression of immunogenic markers in tumor cell.

A. Tumor cells isolated from untreated (Ctrl) or LOFU-treated mice were analyzed by flow cytometry for the presence and percentage representation of Gr1+CD11bhigh cells and CD11b+CD11c+dendritic cells. Results are shown as mean+SEM from 3–5 mice for each experimental condition B. Representative FACS dot plot of B16 tumor cell suspension obtained from untreated or LOFU treated mice were stained with a viability marker (Live/dead Mk). Relative quantification of dead cells is reported. Box and arrow indicate dead cells (Live/dead MK+). C–D. Immunofluorescence staining of B16-F1 tumor tissues isolated from untreated mice or from mice treated with LOFU. Tissue sections were stained with antibodies to detect calreticulin (C) or Hsp70 (D) (green) and TRP1 (red). Nuclei were stained with DAPI. Magnification 60x. E–F. Cells from tumors of LOFU treated mice and untreated mice were stained for CD45 and for the expression of TRP1. CD45-TRP1+ B16 cells were then analyzed for the expression of Hsp70 (E) or MHC-I (F). A representative histogram is shown. Gates and arrows indicate the selected population for the analysis. Data (mean+SEM from 3 mice) was analyzed using a 2-tailed t test (*P<0.05)
Lysates from LOFU-treated B16 tumors allow dendritic cells to reactivate anergic tumor antigen-specific T cells
Our results supported that tumor-induced T cell tolerance could be overcome following LOFU treatment. This observation was substantiated by the fact that following LOFU treatment of the tumor site, the expression of several anergy-associated genes in T cells from tumor DLN was not increased compared to control T cells isolated from distal NDLN (Fig. 2), whereas activation-induced cytokine expression was also restored to levels close to those detected in T cells isolated from distal NDLN or in T cells isolated from control non-tumor bearing mice (Fig. 1). FACS analyses revealed that those differences in the expression of anergy-associated genes did not result from differences in the populations of T cells present in DLN or in the tumor infiltrating lymphocyte populations (Fig. 5A–B). This indicated that tumor-induced anergy did not result in decreased migration of T cells or in decreased number of contacts with antigen presenting cells, which should elicit expression of the early activation marker CD69 even under conditions that would induce anergy in T cells (34). Rather, these data supported the existence of differences in the quality on the interaction between T cells and tumor-antigen presenting dendritic cells that would induce anergy in untreated tumors but prevent the establishment of T helper cell tolerance in LOFU-treated mice. Further supporting this hypothesis, similar results were obtained when mice treated with HIFU, which did not prevent the induction of tolerance (Suppl. Fig. 1), were analyzed (Fig. 5). Of note, there was a significant increase in the presence of CD45+ cells in the HIFU-treated tumors, although it did not correlate with increased presence of T cells, suggesting that an influx of other inflammatory cells might occur in response to the extensive tissue damage caused by HIFU.
Figure 5. Distribution of T cell populations in DLN and tumors in FUS-treated B16-bearing mice.
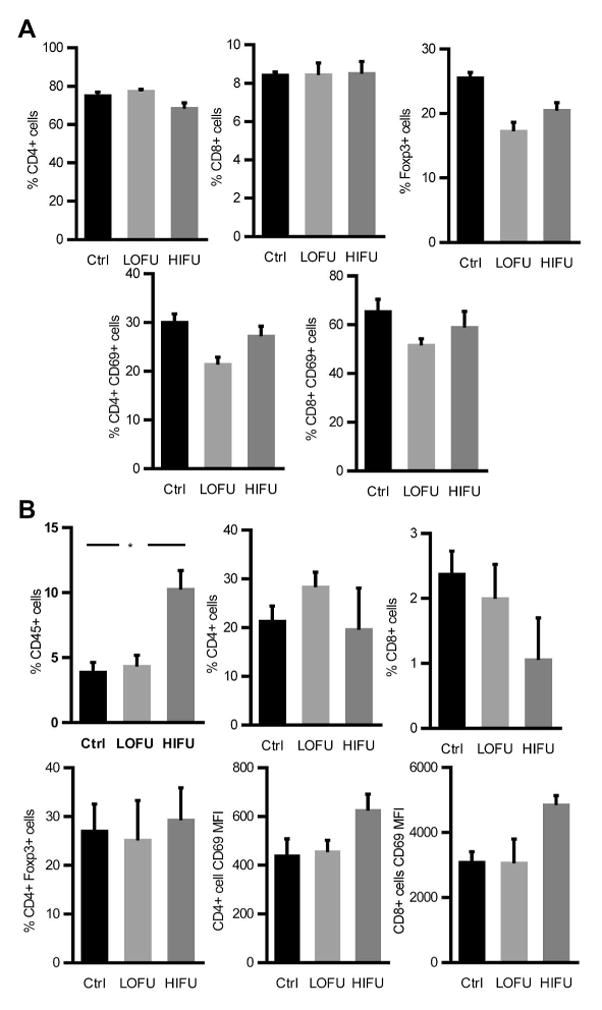
A–B. B16-F1 tumors were injected in the hind leg of mice and either left untreated (Ctrl) or treated with LOFU or HIFU. Thirty six hours following FUS treatment, tumors and respective DLNs were collected. Percentage of CD8+, CD4+ and CD4+Foxp3+ T cells as well as expression of CD69 in CD8+ and CD4+ cells were analyzed by flow cytometry in the DLNs (A) and tumors (B). The data, shown as mean+SEM of 3–4 mice per condition, was analyzed by ANOVA with Tukey posttest (*p=0.05).
This prompted us to investigate if LOFU might not only prevent the induction of tumor-antigen specific T cell anergy but also reverse established anergy and allow dendritic cells to generate a productive effector response even in previously tolerized T cells. To address this question, naïve CD4+ T cells were isolated from spleen and lymph nodes of Tyrp1 mice, in vitro differentiated into TH1 cells and anergized by activating them through partial stimulation using with anti-CD3 antibodies in the absence of co-stimulation. As expected, T cells became hyporesponsive and showed a profound decrease in IL-2 production upon re-stimulation with anti-CD3 and anti-CD28 antibodies (Fig 6A). These anergic cells were then re-activated with dendritic cells loaded with lysates derived from either untreated or LOFU treated melanoma tumors. As expected, anergic Tyrp1 T cells stimulated with dendritic cells loaded with tumor lysates from untreated B16-F1 melanoma produced negligible amounts of IL-2. However, when dendritic cells were loaded with tumor lysates prepared from LOFU-treated tumors, previously anergized T cells produced significantly more IL-2 than those activated with untreated lysates (Fig 6B). These results support that LOFU treatment of melanoma tumors might result in the generation of immunogenic molecules that may enable dendritic cells to deliver activating signals that can breach tolerance, allowing otherwise anergic T cells to respond to antigen re-encounter to generate a productive response.
Figure 6. Lysates from LOFU-treated B16-F1 melanoma tumors can reverse the hyporesponsive state of anergic T cells.
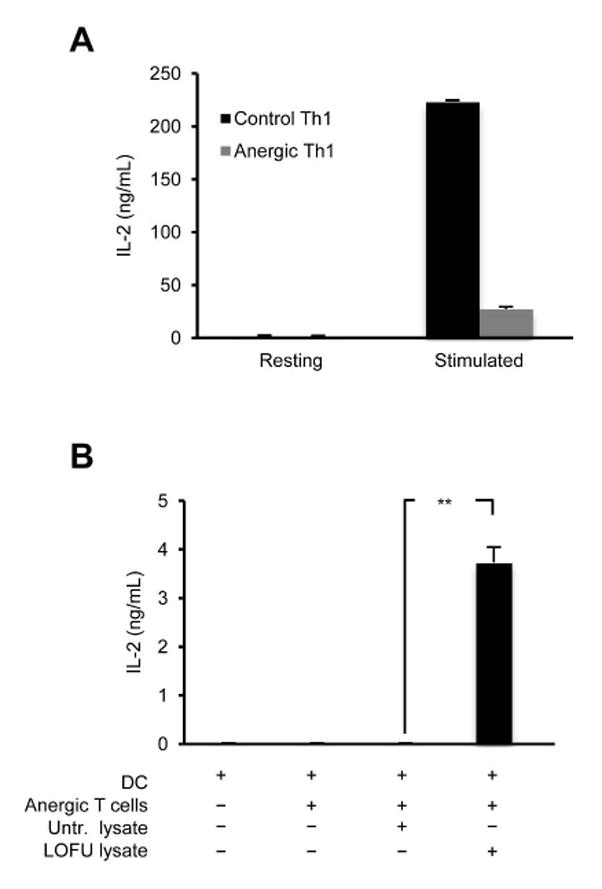
A. Naïve CD4+T cells were isolated from spleens and lymph nodes of Tyrp1 mice, and differentiated into TH1 cells. Cells were then either left untreated or treated with anti-CD3 alone for 16 hours to induce anergy. Cells were then rested for 72 hours in strict absence of IL-2 and re-stimulated with anti-CD3 and anti-CD28 antibodies. IL-2 levels were measured by ELISA. The results are shown as mean+SEM from 2 independent experiments. B. CD11c+ dendritic cells were isolated from spleens of tumor-free Tyrp1 mice. Anergic TH1 cells generated from Tyrp1 mouse-derived CD4+T cells as described in (A) were co-cultured with the dendritic cells and tumor lysates derived from untreated or LOFU-treated B16-F1 melanoma tumors. Supernatants were collected after 24 hours and assayed for IL-2 by ELISA. Results are shown as mean+SEM from 2 independent experiments with 3 independent sets of tumor lysates used in each experiment. Data were analyzed using ANOVA with a Tukey post-test (**P<0.01).
LOFU followed by ablation of tumor by hypofractionated IGRT results in enhanced T-cell mediated control of primary melanoma lesions
In order to further determine the consequences of our observation that LOFU therapy can modulate tumor immunogenicity and enhance anti-tumor immune responses, we performed a series of in vivo treatment strategies evaluating primary tumor control using a combination of LOFU with or without tumor ablation using daily 10 Gy hypofractionated IGRT to a total dose of 30 Gy per mouse with established B16-M1 tumors located subcutaneously in the right dorsal hind limb. Treatment was initiated for all mice when tumor volume reached ~50mm3. Tumor volumes in each group were then measured three times a week for up to 62 days (Fig. 7A). Untreated C57BL/6 or mice treated with LOFU alone continued to experience rapid primary tumor growth, reaching a volume of ≥300 mm3 within 10 days of treatment, at which point a below-the-knee amputation (BKA) was performed (Fig. 7A). In contrast, mice within the hypofractionated IGRT or LOFU+IGRT groups experienced significant growth delay for up to 3-weeks following treatment after which mice treated with IGRT alone began to exhibit primary tumor regrowth, reaching a volume ≥300 mm3 at approximately 5-weeks. Remarkably, the mice in the LOFU+IGRT group had a sustained response, with limited tumor growth for more than 6-weeks following treatment. The reduction in tumor volume in the group receiving LOFU+IGRT or IGRT when compared with the untreated or LOFU alone groups was statistically significant by day 25 (P<0.05). Moreover, the reduction in tumor volume in the LOFU+IGRT group compared with IGRT alone was statistically significant by day 35 and remained statistically significant (P<0.05) for the duration of the experiment. (Fig. 7A). In addition, mice in the LOFU+IGRT group demonstrated regression of tumors from their baseline measurements and a complete tumor-free response was seen in 4 out of 5 mice.
Figure 7. LOFU followed by hypofractionated IGRT results in T-cell mediated long term primary tumor control and reduced distal metastases.
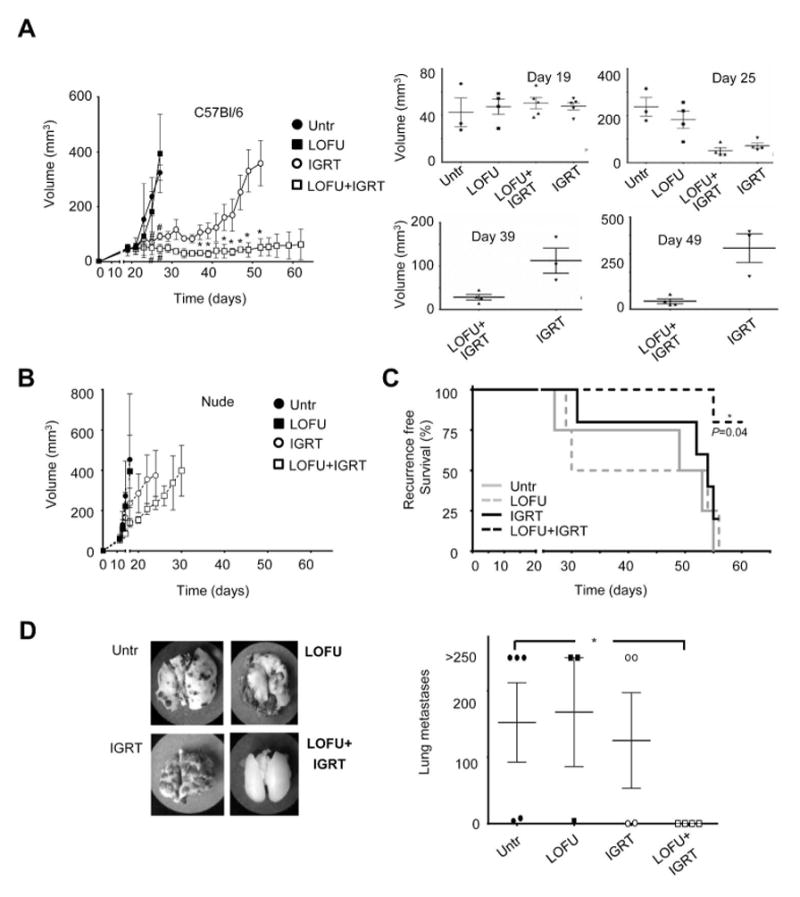
A C57Bl/6 mice with 50mm3 subcutaneous dorsal right hind limb tumors were separated into one of four treatment groups: untreated, LOFU, hypofractionated IGRT, or LOFU+IGRT and tumor growth monitored for 62 days or until primary tumor grew beyond 300 mm3. Graph shows mean±SEM of tumor volume from one of two representative experiments (3–5 mice per group). Data were analyzed with either one-way ANOVA followed by a Bonferroni correction posttest (before day 29) or by 2-tailed student t test (after day 29). Significant differences (defined as P<0.05) between untreated or LOFU-treated mice and IGRT or LOFU+IGRT treated mice (#) occurred after day 25, and between IGRT treated and LOFU+IGRT treated mice (*) after day 35. Individual graphs showing the distribution of tumor size at specific days are also shown. B. Similar experiments as the ones described in A were performed in BALB/c nude mice. No significant differences were observed among the different groups at any time point. C. C57Bl/6 mice were monitored for primary tumor progression/recurrence, defined as either recurrence reaching a volume of 150 mm3 or the development of local metastasis to the popliteal or inguinal lymph nodes. In addition, animals that died spontaneously were scored as having recurrence or progression of disease. Recurrence free survival data was analyzed using the Mantel-Cox test. D. Lungs were harvested from animals that either died spontaneously, required euthanasia due to overwhelming tumor burden, or were sacrificed at the end of a two month long experiment. Lung metastasis were then measured. Lungs with nodules that fuse into plaques, or exceed 250 were deemed too numerous to count and assigned a maximal value of 250. A representative specimen is shown for each treatment group. The results are shown as mean± SEM, with n=3–5 mice per group, analyzed with a Kruskal-Wallis test, followed by Dunn’s posttest. *P < 0.05.
In a separate set of experiments, we combined IGRT with LOFU or HIFU to assess their comparative effects. All tumors were induced subcutaneously in the right dorsal hind limb. To compare the effects of LOFU+IGRT or HIFU+IGRT on tumor growth in a more unbiased way, and to minimize exposure of normal non-melanoma tissue to high temperatures generated by HIFU, we started FUS treatment with tumor sizes equaling or exceeding 75mm3. HIFU treatment led to quick thermal destruction of tissue. In nearly all animals tested, HIFU treatment of melanoma caused not only the loss of primary tumors but also of the tumor-bearing limb. Thus, only one-time 10 Gy doses of IGRT could be administered to HIFU-treated tumors. HIFU+IGRT administration ablated the tumors in the respective cohort within 48 hours of treatment. From this point onwards, appearance and growth of secondary tumors in the two experimental groups were monitored. Twenty days following treatment and ablation of primary tumors, secondary tumors exceeding 500–1000 mm3 in volume necessitated sacrifice of several mice in the HIFU+IGRT group (Suppl. Fig. 1B). Some of the mice in the LOFU+IGRT group of mice also started to show palpable secondary tumors in the popliteal lymph nodes but continued to live well past the 40 day mark post treatment, with two out of 5 mice showing no apparent tumor growth in spite of having started off with a much larger primary tumor load than in the experiments shown in Fig 7A.
To corroborate the immunomodulatory effect of LOFU, we performed experiments using the immunocompromised BALB/c nude model. In these mice B16-M1 tumors grew much more rapidly, reaching ≥300 mm3 approximately 1-week earlier than C57BL/6 mice. The overall treatment response was similar, with untreated and LOFU alone resulting in no significant primary tumor control, while IGRT and LOFU+IGRT induced some delay in primary tumor growth (Fig. 7B), in both the IGRT and LOFU+IGRT treatments, primary control was short lived. In fact, BKA was required in the IGRT group less than 2-weeks after starting treatment and in less than 3-weeks in the LOFU+IGRT group. Additionally, LOFU+IGRT in immunocompromised mice failed to result in statistically significant primary tumor control when compared to IGRT alone (Fig. 7B).
LOFU followed by hypofractionated IGRT results in prolonged recurrence free survival and reduced pulmonary metastasis
Based on our data, we hypothesized that LOFU-induced enhanced anti-tumor responses might augment therapeutic IGRT not only achieving better control of local disease, but also of microscopic disease and distant metastases. As B16-F10 is an aggressive cell line that rapidly grows to an unacceptable size if not treated, mice with primary tumors >300mm3 required BKA. Of note, by the time a BKA was performed, cells from the primary tumor had already spread to the draining popliteal LN (data not shown). Within the subsequent weeks, the draining popliteal LN grew rapidly and became visibly enlarged, while the more distal inguinal LN became clearly palpable. When the tumors reached this point, there are no procedures that can be performed to alleviate discomfort and these mice were euthanized. Consequently, overall survival cannot be adequately assessed in mice with massive tumor burden. Therefore, we decided to assess two other parameters: recurrence free survival, where spontaneous death or euthanized animals with excessive local recurrence tumor burden were scored as positive events; and the development of lung metastases. The combination of LOFU+IGRT provided a statistically significant (P=0.04) recurrence free survival advantage over either treatment alone in C57BL/6 mice (Fig. 7C). Notably, in all groups except C57BL/6 LOFU+IGRT, local metastasis to the draining popliteal or inguinal LN frequently necessitated the use of euthanasia. Furthermore, while mice that were treated with LOFU+IGRT showed a strict control of lung metastases, in the other three groups, even animals with relatively little local recurrence ultimately died due to overwhelming lung metastasis (Fig. 7D).
Discussion
The adaptive immune system constantly surveys for malignantly transformed cells. This is largely achieved by recognition of tumor-associated antigens that prime the appropriate T cell repertoire to mount antitumor immune responses. However, tumors also employ diverse mechanisms to evade the adaptive immune system and thwart antitumor T cell responses (1). As a result, successful immunotherapy against cancer has to overcome the major obstacle of tumor-induced tolerance (35). Several mechanisms have been described to explain how tumors induce tolerance in different T cell subtypes, including defective presentation of tumor antigens and inadequate activation of antigen presenting cells, signaling through co-inhibitory receptors, immunosuppression by factors released within the tumor microenvironment and local recruitment of suppressor cells (9, 15–17, 20, 36–38). In order for the immune response to become activated in a tumor antigen-specific way, a successful therapeutic approach must promote immunogenic cell death (ICD). The hallmarks of ICD include the release of damage-associated molecular patterns, translocation of certain chaperone complexes to the cell surface, and increased susceptibility to dendritic cell-mediated cross-presentation of tumor-associated antigens (39). In this study we sought to investigate if novel treatment for melanoma using non-ablative LOFU would result in in enhanced anti-tumor immune responses and prevention of tumor-induced tolerance.
Thermally ablative HIFU, while able to control primary tumors, is not always effective at preventing micro-metastatic invasions in surrounding or distant tissues, suggesting that cell death caused by this FUS modality might fail to adequately prime an adaptive anti-tumor immune response. High thermal stress induce by HIFU is likely to destroy the vasculature and tissue infrastructure, limiting the ability of immune cells to reach the tumor site and support antigen presentation and/or recognition. Local or distal micro-metastatic invasions should be prevented or ameliorated by an adequately primed immune system that could eliminate the relatively small tumor load of cells that escape initial ablative treatment. In this study, using a B16 murine melanoma model, we show that the use of non-ablative LOFU treatment enhances priming activity of dendritic cells, which potentiates CD4+ T cell effector responses by overcoming the tolerizing effects of the tumor microenvironment and prevents local recurrence and distal metastases when administered prior to an ablative treatment.
Development of T cell hyporesponsiveness to tumor antigens has been described in T cells in several mouse tumor models and in human cancers (15, 18, 40). We have previously reported that tumor-antigen specific CD4+ T cells become anergic in tumor bearing mice and express a series of anergy-associated genes that have been shown to hinder their ability to proliferate and produce effector cytokines (21, 41). Furthermore, prevention of the expression of those genes in mice that lack NFAT1 or Egr2, two transcription factors responsible for the expression of anergy-inducing genes (42–44), leads to inhibition of tumor-antigen specific T cell hyporesponsiveness and improved control of local tumor growth (19, 21). Using two different B16 mouse melanoma models, our data confirm that resident tumor antigen specific CD4+ T cells in the tumor DLN upregulate the expression of anergy associated genes, including Grail, Itch, Cblb, Grg4, and Egr2. The activation of this program of gene expression was well correlated with a reduced ability to produce cytokines following ex-vivo restimulation, supporting that B16 melanoma induces an intrinsic state of hyporesponsiveness in tumor antigen specific CD4+ T cells. Importantly, treatment of the primary tumor with LOFU resulted in an increased ability of those tumor-antigen specific CD4+ T cells to produce cytokines upon re-stimulation. LOFU-induced restoration of the responsiveness to TCR engagement, in otherwise anergic cells, was accompanied by a reduction, to varying extents, of the expression of most anergy-inducing genes. The absence of changes in Foxp3 transcripts in DLN resident CD4+ T cells in tumor bearing mice suggests, however, that LOFU did not affect Foxp3+ Treg migration or differentiation and supports that LOFU prevented tumor-induced-tolerance by inhibiting the induction of T cell anergy.
Initial studies on tumor induced T cell anergy identified the key role that antigen presenting cells played in this process and defective dendritic cell maturation has been defined as a major determinant of inefficient priming of tumor-antigen specific T cells (20, 45). Recently, it has been shown that unstable immunological synapses formed between T cells and dendritic cells presenting tumor antigens result in delayed nuclear export of NFAT and the likely activation of a tolerogenic NFAT-dependent program of gene expression that includes Egr2 (46). Increased T cell activation that follows LOFU treatment of B16 melanomas would potentially result from several different phenomena. First, treatment of the tumors with non-ablative LOFU delivers both thermal and mechanical stress to the tumor cells. This stress could help generate novel unique “non-self” tumor antigens that, in turn, could make the tumor more immunogenic and less able to induce tolerance. Alternatively, the release of stress-induced danger signals by tumor cells could generate a tumor microenvironment that targeting dendritic cells would find less conducive to induction of tolerance in T cells. Stress associated molecular chaperones, including heat shock proteins and calreticulin, have been implicated in dendritic cell maturation and enhanced anti-tumor immunity (30, 47–50). There is evidence that primary tumor lysates are rich in heat shock proteins that can trigger maturation signals in dendritic cells (51). Importantly, heat shock proteins are also capable of associating with and delivering antigenic peptides from tumor cells to dendritic cells, furthermore their presence in the tumor cell plasma membrane has been associated with increased immune responses (52–54). Calreticulin has also been described to play an important role in antitumor response and its translocation to the surface of tumor cells has been associated with increased phagocytosis of the cell by dendritic cells and immune activation (55, 56). Previous studies using HIFU in murine adenocarcinoma models showed that this treatment significantly increased expression of co-stimulatory molecules on dendritic cells, which also produced higher levels of IL-12 and resulted in increased CTL activity (27, 28). Our data show that LOFU induces a redistribution of calreticulin in B16 cells and an increase in the expression of the inducible heat shock protein Hsp70, suggesting that cellular stress mediated by LOFU is capable of inducing changes in the expression of those stress-induced proteins. We also detected increased expression of MHC-II and B7 proteins, supporting that LOFU may leads to changes in the state of maturation or activation of dendritic cells that contribute to the potentiation of the efficient presentation of tumor antigens that is likely responsible for the enhanced tumor immunogenicity and for promoting T cell activation over anergy.
T cell tolerance induced by tumor antigens remains a major challenge in cancer immunotherapy. Effective reversal of tolerance in tumor-specific T cells could be a key goal in clinical antitumor strategies. Our data find that pre-established anergy in T cells can be reversed by the activating effect on dendritic cells of lysates prepared from LOFU treated melanoma tumors. This observation opens up the possibility that LOFU treatment of tumors could release novel immunogenic molecules from tumor cells that would not only prevent but also reverse pre-established tumor tolerance in T cells. Signaling through the IL-2 receptor has long been known to prevent and reverse clonal anergy in T cells (57–59). However, we could not detect elevated amounts of IL-2 in any of our lysates, untreated or treated with LOFU (data not shown), making presence of IL-2 an unlikely candidate to have caused the reversal of anergy in our experiments. However, it is possible that other factors could contribute to this phenotype. In fact, T cell co-stimulation through the TNFR family member OX-40 ligand has also been shown to prevent and overcome T cell anergy in addition to increased effector response in both CD4+ and CD8+ T cells (60–62). Engagement of CD137, CD40 and blockade of PD1 have also been reported to prevent and reverse pre-established CD8+ T cell tolerance in vivo (63–65).
Whereas LOFU, a non-ablative treatment, was not able to control the growth of an established B16 melanoma, which usually has a very aggressive behavior and a fast rate of growth, pretreatment of melanoma tumors with LOFU before performing ablative therapy using hypofractionated IGRT resulted in a significant delay in tumor growth, and in several cases, complete regression of tumors was observed only in immunocompetent mice. Recurrence free survival in these mice was also markedly improved following that protocol. In addition, incidences of lung metastases were minimal in mice that received LOFU prior to tumor ablation compared to mice that received only ablative IGRT. Strikingly, LOFU failed to confer similar protection on T cell-deficient nude mice. This observation indicates that the protective effect of LOFU might not be restricted only to control primary tumor, but that it can also prevent the establishment of metastatic foci either locally or distally. This protection from metastasis may result from the enhancement of priming by dendritic cells of tumor-antigen specific T cells, causing prevention or reversal of T cell tolerance. LOFU pretreatment not only controlled tumor growth more effectively, but, as indicated before, likely resulted in the generation of strongly immunogenic IGRT-induced tumor death that provided protection from metastasis and ensured longer recurrence free survival.
Prevention of T cell tolerance to endogenous tumor antigens is of paramount importance in anticancer immunotherapy. Our work shows that treatment of primary tumors with LOFU can accomplish that, making it a candidate therapy for the development of an in situ autologous tumor vaccine. LOFU treatment of solid tumors, in combination with an ablative approach may results thus in increased efficacy of primary tumor eradication, as well as prevention of distal metastases.
Supplementary Material
Acknowledgments
This work was supported by NIH grants EB009040 (to CG) and AI059738 (to FM).
Footnotes
Conflict of interest statement
A. Partanen is an employee of Philips Healthcare, Bethesda, MD. The rest of the authors declare they have no conflicting financial interest.
References
- 1.Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–296. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dougan M, Dranoff G. Immune therapy for cancer. Annu Rev Immunol. 2009;27:83–117. doi: 10.1146/annurev.immunol.021908.132544. [DOI] [PubMed] [Google Scholar]
- 3.Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, Boon T, Van den Eynde BJ. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9:1269–1274. doi: 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- 4.Thomas DA, Massague J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8:369–380. doi: 10.1016/j.ccr.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 5.Gerlini G, Tun-Kyi A, Dudli C, Burg G, Pimpinelli N, Nestle FO. Metastatic melanoma secreted IL-10 down-regulates CD1 molecules on dendritic cells in metastatic tumor lesions. Am J Pathol. 2004;165:1853–1863. doi: 10.1016/S0002-9440(10)63238-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turk MJ, Guevara-Patino JA, Rizzuto GA, Engelhorn ME, Sakaguchi S, Houghton AN. Concomitant tumor immunity to a poorly immunogenic melanoma is prevented by regulatory T cells. J Exp Med. 2004;200:771–782. doi: 10.1084/jem.20041130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, Divino CM, Chen SH. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66:1123–1131. doi: 10.1158/0008-5472.CAN-05-1299. [DOI] [PubMed] [Google Scholar]
- 8.Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest. 2007;117:1155–1166. doi: 10.1172/JCI31422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 10.van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med. 1999;190:355–366. doi: 10.1084/jem.190.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Phan GQ, Yang JC, Sherry RM, Hwu P, Topalian SL, Schwartzentruber DJ, Restifo NP, Haworth LR, Seipp CA, Freezer LJ, Morton KE, Mavroukakis SA, Duray PH, Steinberg SM, Allison JP, Davis TA, Rosenberg SA. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci USA. 2003;100:8372–8377. doi: 10.1073/pnas.1533209100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, Lennon VA, Celis E, Chen L. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 13.Rubinstein N, Alvarez M, Zwirner NW, Toscano MA, Ilarregui JM, Bravo A, Mordoh J, Fainboim L, Podhajcer OL, Rabinovich GA. Targeted inhibition of galectin-1 gene expression in tumor cells results in heightened T cell-mediated rejection; A potential mechanism of tumor-immune privilege. Cancer Cell. 2004;5:241–251. doi: 10.1016/s1535-6108(04)00024-8. [DOI] [PubMed] [Google Scholar]
- 14.Troy AJ, Summers KL, Davidson PJ, Atkinson CH, Hart DN. Minimal recruitment and activation of dendritic cells within renal cell carcinoma. Clin Cancer Res. 1998;4:585–593. [PubMed] [Google Scholar]
- 15.Staveley-O’Carroll K, Sotomayor E, Montgomery J, Borrello I, Hwang L, Fein S, Pardoll D, Levitsky H. Induction of antigen-specific T cell anergy: An early event in the course of tumor progression. Proc Natl Acad Sci USA. 1998;95:1178–1183. doi: 10.1073/pnas.95.3.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Willimsky G, Blankenstein T. Sporadic immunogenic tumours avoid destruction by inducing T-cell tolerance. Nature. 2005;437:141–146. doi: 10.1038/nature03954. [DOI] [PubMed] [Google Scholar]
- 17.Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, Heimann DM, Klebanoff CA, Yu Z, Hwang LN, Feigenbaum L, Kruisbeek AM, Rosenberg SA, Restifo NP. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cuenca A, Cheng F, Wang H, Brayer J, Horna P, Gu L, Bien H, Borrello IM, Levitsky HI, Sotomayor EM. Extra-lymphatic solid tumor growth is not immunologically ignored and results in early induction of antigen-specific T-cell anergy: dominant role of cross-tolerance to tumor antigens. Cancer Res. 2003;63:9007–9015. [PubMed] [Google Scholar]
- 19.Zheng Y, Zha Y, Driessens G, Locke F, Gajewski TF. Transcriptional regulator early growth response gene 2 (Egr2) is required for T cell anergy in vitro and in vivo. J Exp Med. 2012;209:2157–2163. doi: 10.1084/jem.20120342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sotomayor EM, Borrello I, Rattis FM, Cuenca AG, Abrams J, Staveley-O’Carroll K, Levitsky HI. Cross-presentation of tumor antigens by bone marrow-derived antigen-presenting cells is the dominant mechanism in the induction of T-cell tolerance during B-cell lymphoma progression. Blood. 2001;98:1070–1077. doi: 10.1182/blood.v98.4.1070. [DOI] [PubMed] [Google Scholar]
- 21.Abe BT, Shin DS, Mocholi E, Macian F. NFAT1 supports tumor-induced anergy of CD4(+) T cells. Cancer Res. 2012;72:4642–4651. doi: 10.1158/0008-5472.CAN-11-3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jang HJ, Lee JY, Lee DH, Kim WH, Hwang JH. Current and Future Clinical Applications of High-Intensity Focused Ultrasound (HIFU) for Pancreatic Cancer. Gut Liver. 2010;4(Suppl 1):S57–61. doi: 10.5009/gnl.2010.4.S1.S57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kennedy JE. High-intensity focused ultrasound in the treatment of solid tumours. Nat Rev Cancer. 2005;5:321–327. doi: 10.1038/nrc1591. [DOI] [PubMed] [Google Scholar]
- 24.Blana A, Walter B, Rogenhofer S, Wieland WF. High-intensity focused ultrasound for the treatment of localized prostate cancer: 5-year experience. Urology. 2004;63:297–300. doi: 10.1016/j.urology.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 25.Illing RO, Kennedy JE, Wu F, ter Haar GR, Protheroe AS, Friend PJ, Gleeson FV, Cranston DW, Phillips RR, Middleton MR. The safety and feasibility of extracorporeal high-intensity focused ultrasound (HIFU) for the treatment of liver and kidney tumours in a Western population. Br J Cancer. 2005;93:890–895. doi: 10.1038/sj.bjc.6602803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu F, Zhou L, Chen WR. Host antitumour immune responses to HIFU ablation. Int J Hyperthermia. 2007;23:165–171. doi: 10.1080/02656730701206638. [DOI] [PubMed] [Google Scholar]
- 27.Hu Z, Yang XY, Liu Y, Morse MA, Lyerly HK, Clay TM, Zhong P. Release of endogenous danger signals from HIFU-treated tumor cells and their stimulatory effects on APCs. Biochem Biophys Res Comm. 2005;335:124–131. doi: 10.1016/j.bbrc.2005.07.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu Z, Yang XY, Liu Y, Sankin GN, Pua EC, Morse MA, Lyerly HK, Clay TM, Zhong P. Investigation of HIFU-induced anti-tumor immunity in a murine tumor model. J Transl Med. 2007;5:34–34. doi: 10.1186/1479-5876-5-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu HL, Hsieh HY, Lu LA, Kang CW, Wu MF, Lin CY. Low-pressure pulsed focused ultrasound with microbubbles promotes an anticancer immunological response. J Transl Med. 2012;10:221. doi: 10.1186/1479-5876-10-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Basu S, Srivastava PK. Calreticulin, a peptide-binding chaperone of the endoplasmic reticulum, elicits tumor- and peptide-specific immunity. J Exp Med. 1999;189:797–802. doi: 10.1084/jem.189.5.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castelli C, Ciupitu AM, Rini F, Rivoltini L, Mazzocchi A, Kiessling R, Parmiani G. Human heat shock protein 70 peptide complexes specifically activate antimelanoma T cells. Cancer Res. 2001;61:222–227. [PubMed] [Google Scholar]
- 32.Haug M, Dannecker L, Schepp CP, Kwok WW, Wernet D, Buckner JH, Kalbacher H, Dannecker GE, Holzer U. The heat shock protein Hsp70 enhances antigen-specific proliferation of human CD4+ memory T cells. Eur J Immunol. 2005;35:3163–3172. doi: 10.1002/eji.200535050. [DOI] [PubMed] [Google Scholar]
- 33.Pawaria S, Binder RJ. CD91-dependent programming of T-helper cell responses following heat shock protein immunization. Nat Commun. 2011;2:521. doi: 10.1038/ncomms1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wells AD, Walsh MC, Bluestone JA, Turka LA. Signaling through CD28 and CTLA-4 controls two distinct forms of T cell anergy. J Clin Invest. 2001;108:895–903. doi: 10.1172/JCI13220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gajewski TF, Woo SR, Zha Y, Spaapen R, Zheng Y, Corrales L, Spranger S. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr Opin Immunol. 2013;25:268–276. doi: 10.1016/j.coi.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 36.Driessens G, Kline J, Gajewski TF. Costimulatory and coinhibitory receptors in anti-tumor immunity. Immunol Rev. 2009;229:126–144. doi: 10.1111/j.1600-065X.2009.00771.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 38.Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007;117:1147–1154. doi: 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nat Rev Immunol. 2009;9:353–363. doi: 10.1038/nri2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee PP, Yee C, Savage PA, Fong L, Brockstedt D, Weber JS, Johnson D, Swetter S, Thompson J, Greenberg PD, Roederer M, Davis MM. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat Med. 1999;5:677–685. doi: 10.1038/9525. [DOI] [PubMed] [Google Scholar]
- 41.Valdor R, Macian F. Induction and stability of the anergic phenotype in T cells. Semin Immunol. 2013;25:313–320. doi: 10.1016/j.smim.2013.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Macian F, Garcia-Cozar F, Im SH, Horton HF, Byrne MC, Rao A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell. 2002;109:719–731. doi: 10.1016/s0092-8674(02)00767-5. [DOI] [PubMed] [Google Scholar]
- 43.Safford M, Collins S, Lutz MA, Allen A, Huang CT, Kowalski J, Blackford A, Horton MR, Drake C, Schwartz RH, Powell JD. Egr-2 and Egr-3 are negative regulators of T cell activation. Nat Immunol. 2005;6:472–480. doi: 10.1038/ni1193. [DOI] [PubMed] [Google Scholar]
- 44.Soto-Nieves N, Puga I, Abe BT, Bandyopadhyay S, Baine I, Rao A, Macian F. Transcriptional complexes formed by NFAT dimers regulate the induction of T cell tolerance. J Exp Med. 2009;206:867–876. doi: 10.1084/jem.20082731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Enk AH, Jonuleit H, Saloga J, Knop J. Dendritic cells as mediators of tumor-induced tolerance in metastatic melanoma. Int J Cancer. 1997;73:309–316. doi: 10.1002/(sici)1097-0215(19971104)73:3<309::aid-ijc1>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 46.Marangoni F, Murooka TT, Manzo T, Kim EY, Carrizosa E, Elpek NM, Mempel TR. The transcription factor NFAT exhibits signal memory during serial T cell interactions with antigen-presenting cells. Immunity. 2013;38:237–249. doi: 10.1016/j.immuni.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Srivastava P. Interaction of heat shock proteins with peptides and antigen presenting cells: chaperoning of the innate and adaptive immune responses. Annu Rev Immunol. 2002;20:395–425. doi: 10.1146/annurev.immunol.20.100301.064801. [DOI] [PubMed] [Google Scholar]
- 48.Udono H, Srivastava PK. Heat shock protein 70-associated peptides elicit specific cancer immunity. J Exp Med. 1993;178:1391–1396. doi: 10.1084/jem.178.4.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Udono H, Srivastava PK. Comparison of tumor-specific immunogenicities of stress-induced proteins gp96, hsp90, and hsp70. J Immunol. 1994;152:5398–5403. [PubMed] [Google Scholar]
- 50.Ullrich SJ, Robinson EA, Law LW, Willingham M, Appella E. A mouse tumor-specific transplantation antigen is a heat shock-related protein. Proc Natl Acad Sci USA. 1986;83:3121–3125. doi: 10.1073/pnas.83.10.3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Somersan S, Larsson M, Fonteneau JF, Basu S, Srivastava P, Bhardwaj N. Primary tumor tissue lysates are enriched in heat shock proteins and induce the maturation of human dendritic cells. J Immunol. 2001;167:4844–4852. doi: 10.4049/jimmunol.167.9.4844. [DOI] [PubMed] [Google Scholar]
- 52.Liu B, DeFilippo AM, Li Z. Overcoming Immune Tolerance to Cancer by Heat Shock Protein Vaccines. Mol Cancer Ther. 2002;1:1147–1151. [PubMed] [Google Scholar]
- 53.Chen X, Tao Q, Yu H, Zhang L, Cao X. Tumor cell membrane-bound heat shock protein 70 elicits antitumor immunity. Imuno Lett. 2002;84:81–87. doi: 10.1016/s0165-2478(02)00042-1. [DOI] [PubMed] [Google Scholar]
- 54.Vega VL, Rodriguez-Silva M, Frey T, Gehrmann M, Diaz JC, Steinem C, Multhoff G, Arispe N, De Maio A. Hsp70 translocates into the plasma membrane after stress and is released into the extracellular environment in a membrane-associated form that activates macrophages. J Immunol. 2008;180:4299–4307. doi: 10.4049/jimmunol.180.6.4299. [DOI] [PubMed] [Google Scholar]
- 55.Basu S, Binder RJ, Suto R, Anderson KM, Srivastava PK. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int Immunol. 2000;12:1539–1546. doi: 10.1093/intimm/12.11.1539. [DOI] [PubMed] [Google Scholar]
- 56.Obeid M, Panaretakis T, Tesniere A, Joza N, Tufi R, Apetoh L, Ghiringhelli F, Zitvogel L, Kroemer G. Leveraging the immune system during chemotherapy: moving calreticulin to the cell surface converts apoptotic death from “silent” to immunogenic. Cancer Res. 2007;67:7941–7944. doi: 10.1158/0008-5472.CAN-07-1622. [DOI] [PubMed] [Google Scholar]
- 57.Boussiotis VA, Barber DL, Nakarai T, Freeman GJ, Gribben JG, Bernstein GM, D’Andrea AD, Ritz J, Nadler LM. Prevention of T cell anergy by signaling through the gamma c chain of the IL-2 receptor. Science. 1994;266:1039–1042. doi: 10.1126/science.7973657. [DOI] [PubMed] [Google Scholar]
- 58.Dure M, Macian F. IL-2 signaling prevents T cell anergy by inhibiting the expression of anergy-inducing genes. Mol Immunol. 2009;46:999–1006. doi: 10.1016/j.molimm.2008.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gao B, Kong Q, Kemp K, Zhao YS, Fang D. Analysis of sirtuin 1 expression reveals a molecular explanation of IL-2-mediated reversal of T-cell tolerance. Proc Natl Acad Sci USA. 2012;109:899–904. doi: 10.1073/pnas.1118462109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gramaglia I, Weinberg AD, Lemon M, Croft M. Ox-40 ligand: a potent costimulatory molecule for sustaining primary CD4 T cell responses. J Immunol. 1998;161:6510–6517. [PubMed] [Google Scholar]
- 61.Lathrop SK, Huddleston CA, Dullforce PA, Montfort MJ, Weinberg AD, Parker DC. A signal through OX40 (CD134) allows anergic, autoreactive T cells to acquire effector cell functions. J Immunol. 2004;172:6735–6743. doi: 10.4049/jimmunol.172.11.6735. [DOI] [PubMed] [Google Scholar]
- 62.Murata S, Ladle BH, Kim PS, Lutz ER, Wolpoe ME, Ivie SE, Smith HM, Armstrong TD, Emens LA, Jaffee EM, Reilly RT. OX40 costimulation synergizes with GM-CSF whole-cell vaccination to overcome established CD8+ T cell tolerance to an endogenous tumor antigen. J Immunol. 2006;176:974–983. doi: 10.4049/jimmunol.176.2.974. [DOI] [PubMed] [Google Scholar]
- 63.Tsushima F, Yao S, Shin T, Flies A, Flies S, Xu H, Tamada K, Pardoll DM, Chen L. Interaction between B7-H1 and PD-1 determines initiation and reversal of T-cell anergy. Blood. 2007;110:180–185. doi: 10.1182/blood-2006-11-060087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wilcox RA, Tamada K, Flies DB, Zhu G, Chapoval AI, Blazar BR, Kast WM, Chen L. Ligation of CD137 receptor prevents and reverses established anergy of CD8+ cytolytic T lymphocytes in vivo. Blood. 2004;103:177–184. doi: 10.1182/blood-2003-06-2184. [DOI] [PubMed] [Google Scholar]
- 65.Zhang L, Chen X, Liu X, Kline DE, Teague RM, Gajewski TF, Kline J. CD40 ligation reverses T cell tolerance in acute myeloid leukemia. J Clin Invest. 2013;123:1999–2010. doi: 10.1172/JCI63980. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.