

Abstract
Aims
Genetic risk scores (GRSs) have been associated with coronary heart disease (CHD) in large studies. We asked whether expanding an established 27-variant GRS (GRS27) to a 50-variant GRS (GRS50) improved CHD prediction and whether GRSs are independent of self-reported family history of CHD.
Methods and results
The association between GRSs and incident CHD was assessed in Cox models adjusting for established risk factors in 23 595 participants of the Malmö Diet and Cancer study—a prospective, population-based study. During a median follow-up of 14.4 years, 2213 participants experienced a first CHD event. After adjustment for established risk factors, both GRS27 and GRS50 were associated with incident CHD [hazard ratio (HR) = 1.70 for high (top quintile) vs. low (bottom quintile) of GRS27; 95% confidence interval (CI): 1.48–1.94; Ptrend = 1.6 × 10−15 and HR = 1.92 for GRS50; 95% CI: 1.67–2.20; Ptrend = 6.2 × 10−22]. Adding 23 single nucleotide polymorphisms (SNPs) to GRS27 improved risk prediction (P = 3 × 10−6). Further adjustment for self-reported family history did not appreciably change the risk estimates of either GRS27 (HR = 1.65; 95% CI: 1.45–1.89) or GRS50 (HR = 1.87; 95% CI: 1.63–2.14). The addition of GRS50 to established risk factors, including self-reported family history, improved discrimination (P < 0.0001) and reclassification (continuous net reclassification improvement index = 0.17, P < 0.0001). In young participants (below median age), those with high GRS50 had 2.4-fold greater risk (95% CI: 1.85–3.12) than those with low GRS50.
Conclusion
The addition of 23 SNPs to an existing GRS27 improved CHD risk prediction and was independent of self-reported family history. Coronary heart disease risk assessment by GRS could be particularly useful in young individuals.
Keywords: Coronary heart disease risk, Genetic risk scores
See page 568 for the editorial comment on this article (doi:10.1093/eurheartj/ehv545)
Clinical perspective.
Assessing risk of coronary events is integral to the prevention and treatment of cardiovascular disease. However, current risk assessment algorithms do not explicitly incorporate information about a patient's genetic risk. This large, population-based, prospective study of middle aged Europeans, found that genetic risk—as measured by a genetic risk score comprising dozens of single nucleotide polymorphisms—is independent of traditional risk factors, including family history of cardiovascular disease.
Introduction
Coronary heart disease (CHD) is the leading cause of death in the European Union as well as in the USA—an estimated ∼1.8 million Europeans and ∼400 000 Americans die of CHD annually, at an estimated annual cost of €60 billion in the European Union and $108.9 billion in the USA.1,2 Therefore, improving CHD risk prediction in order to effectively direct CHD risk prevention resources is an important public health goal. The association of established risk factors with CHD in large prospective studies has been used to develop a number of CHD risk prediction models, most recently by the European Association for Cardiovascular Prevention & Rehabilitation3 and US American College of Cardiology/American Heart Association task force on practice guidelines.4 Typically, these models incorporate information about age, sex, hypertension, blood cholesterol, smoking history, and history of diabetes to calculate the probability of CHD event in the short term (10 years) or long term. The heritability of CHD is well documented,5–7 which motivated the inclusion of family history of CHD as an option for risk assessment in patients with borderline risk.8,9 However, despite the identification of numerous genetic variants that are associated with CHD,10,11 a direct assessment of a patient's genetic risk is not typically included in accepted CHD risk prediction models.
Genetic risk scores (GRSs)—based on single nucleotide polymorphisms (SNPs) associated with CHD—have been shown to be associated with future CHD events in large case–control and prospective studies.12–16 However, it has not been fully assessed (i) whether increasing the number of genome-wide significant (GWS) SNPs in GRS-based CHD risk prediction continue to improve risk prediction, (ii) whether GRS could improve CHD risk assessment beyond self-reported family history, and (iii) whether GRS risk differs between younger and older individuals. We investigated these questions in a large population-based prospective study of middle-aged men and women.
Methods
Study participants
The Malmö Diet and Cancer (MDC) study is a community-based, prospective observational study of 30 447 participants drawn from ∼230 000 residents of Malmö, Sweden. Men aged 46–73 years and women aged 45–73 years were invited to participate and were enrolled between 1991 and 1996.17 Details of the MDC design have been previously reported.17,18 The primary endpoints of the study were time to first occurrence of CHD (composite endpoint of coronary event, cardiovascular death, and revascularizations). After exclusions, 23 595 participants were included in the current study. A detailed description of the inclusion and exclusion criteria in the current study, baseline assessment, and endpoint determination are provided in the Supplemental material online.
Modelling of genetic risk score
We assessed the association of CHD with each of two GRSs (Supplementary material online, Table S1). One GRS is the 27-SNP GRS (GRS27) described by Mega et al.19 A second GRS comprised the GRS27 SNPs and 23 additional SNPs, for a total of 50 SNPs (GRS50). Each of these 23 additional SNPs has been shown to be associated with CHD at a GWS level. The GRS of each individual in the current study was calculated as follows: the previously reported risk estimate for the modelled allele of each SNP (Supplementary material online, Table S1) was natural log transformed and multiplied by one (for heterozygotes) or two (for homozygotes); these products were then summed. The mean (2.49 for GRS27 and 3.82 for GRS50) and standard deviation (0.34 for GRS27 and 0.43 for GRS50) of the study population were used to standardize each GRS to have a mean of 0 and unit variance. Genetic risk was analysed per standard deviation of the standardized GRS as well as by comparing those with high GRS (Quintile 5), those with intermediate risk score (Quintiles 2 to 4), and those with low GRS (Quintile 1).
Results
Study population and genetic risk scores
The baseline characteristics of the 23 595 MDC participants in this study are provided in Table 1 (stratified by incident CHD event status) and Supplementary material online, Table S2 (stratified by self-reported family history). During a median follow-up of 14.4 years, 2213 participants experienced a first CHD event.
Table 1.
Baseline characteristics according to coronary heart disease event status
| Baseline characteristics | Events (n = 2213) | Non-events (n = 21 382) |
|---|---|---|
| Age (years) | 61.5 ± 6.9 | 57.7 ± 7.7 |
| Men, n (%) | 1420 (64.2) | 7553 (35.3) |
| Body mass index (kg/m2) | 26.7 ± 4.0 | 25.6 ± 4.0 |
| Current smoker, n (%) | 790 (35.7) | 5832 (27.3) |
| Systolic blood pressure (mmHg) | 150.3 ± 20.2 | 140.1 ± 19.9 |
| Diastolic blood pressure (mmHg) | 88.7 ± 9.8 | 85.2 ± 10.0 |
| Use of anti-hypertensives, n (%) | 646 (29.2) | 3407 (15.9) |
| Prevalent diabetes mellitus, n (%) | 242 (10.9) | 688 (3.2) |
| Apolipoprotein A-I (g/L) | 1.47 ± 0. 26 | 1.58 ± 0.28 |
| Apolipoprotein B (g/L) | 1.18 ± 0.26 | 1.06 ± 0.26 |
| Self-reported family history of CHD, n (%) | 998 (0.45) | 7790 (0.36) |
| GRS27 | 0.18 ± 1.01 | −0.02 ± 1.00 |
| GRS50 | 0.20 ± 1.00 | −0.02 ± 1.00 |
Data are presented as mean ± standard deviation unless indicated.
GRS27, 27-variant genetic risk score; GRS50, 50-variant genetic risk score.
We genotyped 50 SNPs reported to be associated with CHD at a GWS level (Supplementary material online, Table S1). None of the SNPs were in strong linkage disequilibrium (r2 < 0.45 for any pair of SNPs). We assessed the association of incident CHD with two GRSs: the previously described GRS2719 as well as GRS50, an expanded GRS that included all GRS27 SNPs as well as 23 additional SNPs. We calculated a weighted GRS for each participant using the literature risk estimates as weights for the risk allele of each of the SNPs. The standardized GRS means and standard deviations for those with and without events are reported in Table 1.
Genetic risk scores and incident coronary heart disease
Both GRS27 and GRS50 were associated with incident CHD [hazard ratio (HR) = 1.20 per SD; 95% confidence interval (CI): 1.15–1.25 and HR = 1.23; 95% CI: 1.18–1.28, respectively] after adjustment for established risk factors including age, sex, systolic blood pressure, hypertension treatment, smoking, apoB, apoA-I, and prevalent diabetes (Table 2). Those with high GRS50 had 1.92-fold greater risk of CHD than those with low genetic risk (95% CI: 1.67–2.20, P = 7.5 × 10−21). For GRS27, those with high genetic risk had 1.7-fold greater risk of CHD than those with low genetic risk (95% CI: 1.48–1.94). When both GRS27 and a GRS comprising the 23 SNPs present in GRS50 but not in GRS27 were included in a model that also adjusted for established risk factors, both GRS27 and GRS23 were associated with incident CHD events (P = 2 × 10−13 and 3 × 10−6, respectively). That is, CHD risk prediction by a model that included GRS27 was improved by adding 23 additional SNPs.
Table 2.
Genetic risk scores and incident coronary heart disease
| GRS risk |
Per SD | |||
|---|---|---|---|---|
| Low | Intermediate | High | ||
| GRS27 | ||||
| N (event) | 4719 (343) | 14 157 (1294) | 4719 (576) | |
| Event rate (95% CI) | 5.21 (4.67–5.79) | 6.62 (6.27–7.01) | 8.97 (8.25–9.74) | |
| HR (95% CI) | Reference | 1.26 (1.12–1.42) | 1.70 (1.48–1.94) | 1.20 (1.15–1.25) |
| P value (vs. low) | 1.1 × 10−4 | 9.2 × 10−15 (Ptrend = 1.6 × 10−15) | 5.0 × 10−18 | |
| GRS50 | ||||
| N (event) | 4719 (318) | 14 157 (1303) | 4719 (592) | |
| Event rate (95% CI) | 4.82 (4.31–5.39) | 6.67 (6.31–7.04) | 9.25 (8.52–10.03) | |
| HR (95% CI) | Reference | 1.39 (1.23–1.57) | 1.92 (1.67–2.20) | 1.23 (1.18–1.28) |
| P value (vs. low) | 1.4 × 10−7 | 7.5 × 10−21 (Ptrend = 6.2 × 10−22) | 6.8 × 10−23 | |
Risk estimates were adjusted for age, sex, systolic blood pressure, hypertension treatment, smoking, apoB, apoA-I, and prevalent diabetes. GRS27 risk boundaries (SD): low ≤− 0.8547; intermediate >− 0.8547 and ≤0.8236; high > 0.8236. GRS50 risk boundaries (SD): low ≤− 0.8517; intermediate >− 0.8517 and ≤0.8360; high > 0.8360. Event rates are per 1000 person-years.
HR, hazard ratio; CI, confidence interval; GRS, genetic risk score; GRS27, 27-variant genetic risk score; GRS50, 50-variant genetic risk score.
Discrimination and reclassification
27-Variant genetic risk score and GRS50 each improved the discrimination of a model that included established risk factors even after adding self-reported family history (P ≤ 2 × 10−16) although the magnitude of the improvement of c-statistic was modest (Supplementary material online, Table S3). Self-reported family history improved discrimination of an established risk factors model. Risk classification by a model that included established risk factors and self-reported family history was improved by both GRS27 [continuous net reclassification improvement index (cNRI) = 0.15, P < 0.0001, 7% in those without events and 8% in those with events, Supplementary material online, Table S4] and GRS50 (cNRI = 0.17, P < 0.0001, 10% in those without events and 7% in those with events). Risk classification of an established risk factor model was also improved by self-reported family history, although among patients with events, more patients were reclassified in the wrong direction (lower risk). Risk classification was not improved by GRS27 and GRS50 in a categorical analysis (above and below 7.5% 10-year risk categories4; Supplementary material online, Table S5).
Genetic risk scores and self-reported family history
27-Variant genetic risk score and GRS50 were associated with incident CHD events in participants with and without self-reported family history. In a stratified analysis that adjusted for established risk factors, the HR for CHD for a high compared with a low GRS50 was 1.75 (95% CI: 1.43–2.15) among those with family history and was 1.96 (95% CI: 1.63–2.35) among those without family history (Table 3). The association between GRS50 and CHD events did not differ according to self-reported family history status: in an analysis of the combined strata, the P for interaction was 0.33 in a model that adjusted for established risk factors and included a term for the interaction between family history and GRS50. Self-reported family history was associated with incident CHD events after adjustment for established risk factors (HR = 1.43, 95% CI: 1.31–1.56, P = 8 × 10−17). Adjustment for self-reported family history in the combined strata did not appreciably change these risk estimates (HR = 1.87; 95% CI: 1.63–2.14 for GRS50 and HR = 1.40; 95% CI: 1.29–1.53 for family history). Similar results were observed for GRS27 (Table 3). Both GRS27 and GRS50 were associated with self-reported family history of CHD (P < 0.0001); the odds of a positive self-reported family history among those with high GRS compared with having low GRS were modest; OR = 1.37 (95% CI: 1.26–1.49) for GRS27 and OR = 1.40 (95% CI: 1.29–1.53) for GRS50. The GRS distribution among participants with and without self-reported family history is shown in Supplementary material online, Figure S2.
Table 3.
Genetic risk scores and incident coronary heart disease according to self-reported family history
| Self-reported family history | GRS | Intermediate riska |
High riskb |
P intxn | ||
|---|---|---|---|---|---|---|
| HR (95% CI) | P value | HR (95% CI) | P value | |||
| Yes | GRS50 | 1.29 (1.07–1.56) | 0.007 | 1.75 (1.43–2.15) | 7.7 × 10−8 | 0.33 |
| No | GRS50 | 1.43 (1.21–1.68) | 1.9 × 10−5 | 1.96 (1.63–2.35) | 7.4 × 10−13 | |
| Yes | GRS27 | 1.26 (1.05–1.52) | 0.013 | 1.64 (1.34–2.01) | 2.1 × 10−6 | 0.38 |
| No | GRS27 | 1.23 (1.05–1.44) | 0.009 | 1.67 (1.39–1.99) | 2.1 × 10−8 | |
Risk estimates were adjusted for age, sex, systolic blood pressure, hypertension treatment, smoking, apoB, apoA-I, and prevalent diabetes. GRS27 risk boundaries (SD): low ≤− 0.8547; intermediate >− 0.8547 and ≤0.8236; high > 0.8236. GRS50 risk boundaries (SD): low ≤− 0.8517; intermediate >− 0.8517 and ≤0.8360; high > 0.8360. Pintxn: Pinteraction between GRS as a continuous variable and self-reported family history status for the incident CHD outcome.
HR, hazard ratio; CI, confidence interval; GRS, genetic risk score; GRS27, 27-variant genetic risk score; GRS50, 50-variant genetic risk score.
aIntermediate risk: Quintiles 2, 3, and 4 compared with low risk (Quintile 1).
bHigh risk: Quintile 5 compared with low risk (Quintile 1).
Genetic risk score in young and old
Since genetic risk is generally thought to be more important in young individuals, we investigated the interaction between age and GRS and found that the CHD risk associated with GRS varied with age for both GRS27 and GRS50 (Pinteraction = 0.03 for both). We further assessed the GRS in those below and above the median age of this study (57.6) and found that risk associated with either GRS50 or GRS27 differed among those below and above the median age (Pinteraction < 0.003) in models that adjusted for established risk factors (Table 4 and Supplementary material online, Table S6 for event rate by GRS category and age category). Among the young, those with high GRS had more than two-fold greater risk of CHD than those with low GRS (HR = 2.40; 95% CI: 1.84–3.12; P = 7.5 × 10−11 for GRS50 and HR = 2.24; 95% CI: 1.72–2.90; P = 1.4 × 10−9 for GRS27). We examined the event rate in those with and without self-reported family history of CHD according to their GRS status (Figure 1). We found that in the young, the cumulative incidence in the presence of competing risk among those with high GRS50 without a self-reported family history (0.065 at Year 15) was greater than the cumulative incidence among those with low GRS50 with a self-reported family history (0.041, P = 0.013 for Gray test; Figure 1A). Similar trends (0.153 and 0.128, P = 0.094) were also observed in those above the median age (Panel B). Similar results were observed for GRS27 (Supplementary material online, Figure S3). We also examined whether CHD risk prediction by GRS varied by sex and found no evidence for interaction between sex and GRS (Pinteraction > 0.3).
Table 4.
Genetic risk scores and coronary heart disease risk in young and old
| ≤Median agea |
>Median agea |
P intxn b | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Intermediate riskc |
High riskd |
Intermediate riskc |
High riskd |
||||||
| HR (95% CI) | P value | HR (95% CI) | P value | HR (95% CI) | P value | HR (95% CI) | P value | ||
| Model 1e | |||||||||
| GRS50 | 1.55 (1.22–1.98) | 0.0004 | 2.40 (1.84–3.12) | 7.5 × 10−11 | 1.33 (1.15–1.53) | 7.9 × 10−5 | 1.75 (1.49–2.06) | 6.5 × 10−12 | 0.0029 |
| GRS27 | 1.53 (1.20–1.94) | 0.0005 | 2.24 (1.72–2.90) | 1.4 × 10−9 | 1.18 (1.03–1.35) | 0.02 | 1.52 (1.30–1.78) | 1.8 × 10−7 | 0.0004 |
| Model 2f | |||||||||
| GRS50 | 1.53 (1.20–1.96) | 0.0006 | 2.35 (1.81–3.06) | 2.0 × 10−10 | 1.32 (1.14–1.52) | 0.0002 | 1.71 (1.45–2.01) | 6.5 × 10−11 | 0.0025 |
| GRS27 | 1.50 (1.18–1.91) | 0.0009 | 2.19 (1.69–2.84) | 4.1 × 10−9 | 1.17 (1.02–1.34) | 0.03 | 1.48 (1.27–1.73) | 9.6 × 10−7 | 0.0003 |
GRS27 had a mean of 2.49 and standard deviation of 0.34; GRS50 had a mean of 3.82 and standard deviation of 0.43. Risk boundaries (27-SNPs GRS): low GRS ≤− 0.8547; intermediate GRS >− 0.8547 and ≤0.8236; high GRS > 0.8236. Risk boundaries (50-SNPs GRS): low GRS ≤− 0.8517; intermediate GRS >− 0.8517 and ≤0.8360; high GRS > 0.8360.
HR, hazard ratio; CI, confidence interval; GRS, genetic risk score; GRS27, 27-variant genetic risk score; GRS50, 50-variant genetic risk score.
aMedian age: 57.6.
b P intxn: for interaction between continuous GRS and median age status for the incident CHD outcome.
cIntermediate risk: Quintiles 2, 3, and 4 compared with low risk (Quintile 1).
dHigh risk: Quintile 5 compared with low risk (Quintile 1).
eModel 1: adjusted for age, sex, systolic blood pressure, use of antihypertensive medication, smoking, apoB, apoA-I, and prevalent diabetes.
fModel 2: Model 1 and additional adjustment for family history.
Figure 1.
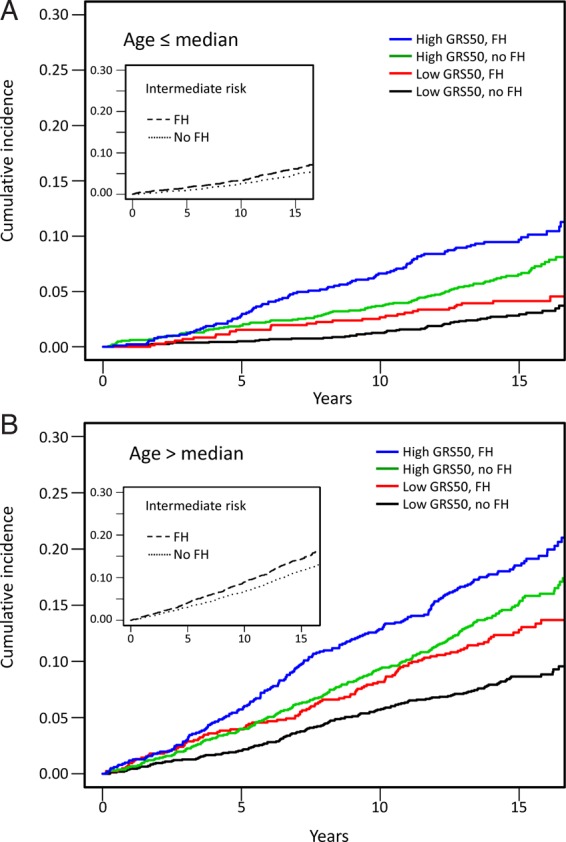
Cumulative incidence of coronary heart disease events according to self-reported family history of coronary heart disease and 50-variant genetic risk score. Blue and green: those with high 50-variant genetic risk score with (blue) or without (green) a self-reported family history. Red and black: those with low 50-variant genetic risk score with (red) or without (black) a self-reported family history. Inset: those with intermediate 50-variant genetic risk score with (dashed) or without (dotted) a self-reported family history. FH, self-reported family history. Cumulative incidence was estimated while considering non-coronary heart disease death as competing risk. (A) Participants younger than median age (≤57.6). Median age for this younger group is 51.4 (interquartile range, 48.8–54.2). (B) Participants older than median age (>57.6). Median age for this older group is 64.7 (interquartile range, 61.1–67.7).
Discussion
In a large community-based prospective study of 23 595 participants, we investigated a 27-SNP and a 50-SNP GRS for CHD and found that CHD risk assessed by both GRS is independent of self-reported family history, and for both the 27-SNP and the 50-SNP GRS, the associated CHD risk estimates were higher among younger individuals than among older individuals.
Genetic risk scores comprising SNPs that are individually associated with CHD at a GWS level have been investigated previously. In 2010, Ripatti et al.12 reported that GRS comprising 13 SNPs was associated with incident CHD in several cohorts; however, this 13-SNP GRS did not improve net reclassification when added to established traditional risk factors. Following the publication of the largest to date meta-analysis genome-wide association in 2013,11 Andrea et al.13 investigated a 46-SNP GRS in six Swedish studies comprising ∼10 000 individuals and found that this 46-SNP GRS improved net reclassification when added to established risk factors. More recently, Mega et al.19 investigated a 27-SNP GRS in randomized, placebo-controlled studies of statin therapy and found that the 27-SNP GRS could identify individuals who would benefit from statin therapy.
Since the risk associated with each individual SNP is modest, and since SNPs that have been only identified in recent large studies typically have risk estimates that are lower than SNPs that were identified in smaller, earlier studies, it was unclear whether it would be useful to include these more recently identified SNPs in a GRS. Our study investigated this question directly. We showed a model that includes 23 SNPs in addition to those in the previously reported 27-SNP GRS improves risk prediction. However, adding either GRS27 or GRS50 to established risk factors resulted in similar improvement in discrimination (c-statistic) and reclassification (cNRI).
Our study also found that subjective, self-reported family history of CHD and objectively measured genetic risk are not redundant.20 Both can contribute to a better assessment of a patient's CHD risk because a GRS-based genetic risk measure is associated with CHD independent of self-reported family history of CHD, as well as established risk factors, that is, self-reported family history is not a substitute for genetic risk assessment. Since family history reflects both genetic and non-genetic factors, and since the accuracy of patient-reported family history is low, this non-redundancy of self-reported family history and genetic assessment is not surprising.
Since genetics is generally thought to play a more important role in CHD events that occur in younger individuals than in older individuals, we examined the 27-SNP and 50-SNP GRS risk estimates in different age groups and found that those risk estimates were higher in younger individuals than in older individuals. Moreover, younger individuals with no self-reported family history of CHD and with high GRS had greater risk than those with self-reported family history of CHD and a low GRS. This finding suggests that a GRS-based risk assessment could be particularly useful among younger individuals. The potential for a point-of-care GRS testing in routine clinical practice makes genetic testing among younger individuals—particularly those with borderline CHD risk by established risk factors—attractive since it could overcome barriers faced by other predictive tools, e.g. 24 h ambulatory blood pressure monitoring.
Our study has several limitations. This study was conducted in Swedish middle-aged individuals; hence, the generalizability to other ethnicities or age groups is uncertain. LDL cholesterol and HDL cholesterol levels were not available for our study population; therefore, we used the available apoA-I and apoB plasma levels as covariates in our established risk factors model. A different definition of self-reported family history might have produced different results. For example, the definition of self-reported family history of CHD in the Framingham Heart Study considers only CHD family member events that occurred before age 55 for men or 65 for women. However, in clinical practice, patients may not know the age at which their family member had a CHD event or may not be asked about the age. For these reasons, the family history question asked at baseline in MDC did not specify any limitations on the age of the family member at the time of event. The fraction of women in MDC is greater than that in other European-based population studies.11 However, since we found no interaction between GRS and sex, we believe that our results should be generalizable to populations with different proportion of women. Although our results suggest that genetic risk assessment could be useful in the young, this study population did not include sufficient number of individuals to provide risk estimates in those <45 years of age. Additional studies would be needed to address this question.
In conclusion, a GRS could improve risk assessment for future CHD when added to established risk factor models. We suggest that such genetic assessment could be considered for individuals whose established risk-based treatment decision is uncertain.
Supplementary material
Supplementary material is available at European Heart Journal online.
Funding
The Malmö Diet and Cancer study was made possible by grants from the Swedish Cancer Society, the Swedish Medical Research Council, the Swedish Dairy Association, and the Albert Påhlsson and Gunnar Nilsson Foundations and the Malmö city council. H.T. is supported by a grant from the Japanese Circulation Society to study in the USA. S.K. is supported by a Research Scholar award from the Massachusetts General Hospital (MGH), the Howard Goodman Fellowship from MGH, and the Donovan Family Foundation. O.M. is supported by the European Research Council (StG-282255), the Swedish Heart and Lung Foundation, Swedish Research Council; the Novo Nordisk Foundation, the Skåne University Hospital donation funds; the Medical Faculty, Lund University; the Governmental funding of clinical research within the national health services, the Albert Påhlsson Research Foundation, Region Skåne, the King Gustav V and Queen VictoriaFoundation, and the Marianne and Marcus Wallenberg Foundation. O.M. and S.K. are the recipients of an investigator-initiated grant from Quest Diagnostics. Funding to pay the Open Access publication charges for this article was provided by Quest Diagnostics.
Conflict of interest: D.S., J.Z.L., J.J.C., C.M.R., and J.J.D. are employees of Quest Diagnostics. S.K. is a member of a scientific advisory board at Quest Diagnostics.
Supplementary Material
References
- 1. Nichols M, Townsend N, Luengo-Fernandez R, Leal J, Gray A, Scarborough P, Rayner M. European Cardiovascular Disease Statistics 2012. Brussels/Sophia Antipolis: European Heart Network/European Society of Cardiology; 2012. [Google Scholar]
- 2. Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Judd SE, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Mackey RH, Magid DJ, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER III, Moy CS, Mussolino ME, Neumar RW, Nichol G, Pandey DK, Paynter NP, Reeves MJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Wong ND, Woo D, Turner MB; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2014 update: a report from the American Heart Association. Circulation 2014;129:e28–e292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Perk J, De Backer G, Gohlke H, Graham I, Reiner Z, Verschuren M, Albus C, Benlian P, Boysen G, Cifkova R, Deaton C, Ebrahim S, Fisher M, Germano G, Hobbs R, Hoes A, Karadeniz S, Mezzani A, Prescott E, Ryden L, Scherer M, Syvänne M, Scholte op Reimer WJ, Vrints C, Wood D, Zamorano JL, Zannad F; European Association for Cardiovascular Prevention & Rehabilitation (EACPR); ESC Committee for Practice Guidelines (CPG). European Guidelines on cardiovascular disease prevention in clinical practice (version 2012) The Fifth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of nine societies and by invited experts) Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur Heart J 2012;33:1635–1701. [DOI] [PubMed] [Google Scholar]
- 4. Goff DC Jr, Lloyd-Jones DM, Bennett G, Coady S, D'Agostino RB Sr, Gibbons R, Greenland P, Lackland DT, Levy D, O'Donnell CJ, Robinson J, Schwartz JS, Shero ST, Smith SC Jr, Sorlie P, Stone NJ, Wilson PW. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association task force on practice guidelines. J Am Coll Cardiol 2014;63:2935–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lloyd-Jones DM, Nam BH, D'Agostino RB Sr, Levy D, Murabito JM, Wang TJ, Wilson PW, O'Donnell CJ. Parental cardiovascular disease as a risk factor for cardiovascular disease in middle-aged adults: a prospective study of parents and offspring. JAMA 2004;291:2204–2211. [DOI] [PubMed] [Google Scholar]
- 6. Marenberg ME, Risch N, Berkman LF, Floderus B, de Faire U. Genetic susceptibility to death from coronary heart disease in a study of twins. N Engl J Med 1994;330:1041–1046. [DOI] [PubMed] [Google Scholar]
- 7. Murabito JM, Pencina MJ, Nam BH, D'Agostino RB Sr, Wang TJ, Lloyd-Jones D, Wilson PW, O'Donnell CJ. Sibling cardiovascular disease as a risk factor for cardiovascular disease in middle-aged adults. JAMA 2005;294:3117–3123. [DOI] [PubMed] [Google Scholar]
- 8. Joseph Y, McClelland RL, Polonsky TS, Burke GL, Sibley CT, O'Leary D, Carr JJ, Goff DC, Greenland P, Herrington DM. Comparison of novel risk markers for improvement in cardiovascular risk assessment in intermediate-risk individuals. JAMA 2012;308:788–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kashani M, Eliasson A, Vernalis M, Costa L, Terhaar M. Improving assessment of cardiovascular disease risk by using family history: an integrative literature review. J Cardiovasc Nurs 2013;28:E18–E27. [DOI] [PubMed] [Google Scholar]
- 10. Erdmann J, Grosshennig A, Braund PS, König IR, Hengstenberg C, Hall AS, Linsel-Nitschke P, Kathiresan S, Wright B, Trégouët DA, Cambien F, Bruse P, Aherrahrou Z, Wagner AK, Stark K, Schwartz SM, Salomaa V, Elosua R, Melander O, Voight BF, O'Donnell CJ, Peltonen L, Siscovick DS, Altshuler D, Merlini PA, Peyvandi F, Bernardinelli L, Ardissino D, Schillert A, Blankenberg S, Zeller T, Wild P, Schwarz DF, Tiret L, Perret C, Schreiber S, El Mokhtari NE, Schäfer A, März W, Renner W, Bugert P, Klüter H, Schrezenmeir J, Rubin D, Ball SG, Balmforth AJ, Wichmann HE, Meitinger T, Fischer M, Meisinger C, Baumert J, Peters A, Ouwehand WH; Italian Atherosclerosis, Thrombosis, and Vascular Biology Working Group; Myocardial Infarction Genetics Consortium; Wellcome Trust Case Control Consortium; Cardiogenics Consortium, Deloukas P, Thompson JR, Ziegler A, Samani NJ, Schunkert H. New susceptibility locus for coronary artery disease on chromosome 3q22.3. Nat Genet 2009;41:280–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. CARDIoGRAMplusC4D Consortium. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet 2013;45:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ripatti S, Tikkanen E, Orho-Melander M, Havulinna AS, Silander K, Sharma A, Guiducci C, Perola M, Jula A, Sinisalo J, Lokki ML, Nieminen MS, Melander O, Salomaa V, Peltonen L, Kathiresan S. A multilocus genetic risk score for coronary heart disease: case-control and prospective cohort analyses. Lancet 2010;376:1393–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Andrea G, Magnusson PKE, Pedersen NL, de Faire U, Reilly M, Ärnlöv J, Sundström J, Hamsten A, Ingelsson E. Multilocus genetic risk scores for coronary heart disease prediction. Arterioscler Thromb Vasc Biol 2013;33:2267–2272. [DOI] [PubMed] [Google Scholar]
- 14. Shah S, Casas JP, Gaunt TR, Cooper J, Drenos F, Zabaneh D, Swerdlow DI, Shah T, Sofat R, Palmen J, Kumari M, Kivimaki M, Ebrahim S, Smith GD, Lawlor DA, Talmud PJ, Whittaker J, Day INM, Hingorani AD, Humphries SE. Influence of common genetic variation on blood lipid levels, cardiovascular risk, and coronary events in two British prospective cohort studies. Eur Heart J 2013;34:972–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gui L, Wu F, Han X, Dai X, Qiu G, Li J, Wang J, Zhang X, Wu T, He M. A multilocus genetic risk score predicts coronary heart disease risk in a Chinese Han population. Atherosclerosis 2014;237:480–485. [DOI] [PubMed] [Google Scholar]
- 16. Yiannakouris N, Katsoulis M, Trichopoulou A, Ordovas JM, Trichopoulos D. Additive influence of genetic predisposition and conventional risk factors in the incidence of coronary heart disease: a population-based study in Greece. BMJ Open 2014;4:e004387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Berglund G, Elmstähl S, Janzon L, Larsson SA. The Malmo Diet and Cancer Study. Design and feasibility. J Intern Med 1993;233:45–51. [DOI] [PubMed] [Google Scholar]
- 18. Smith JG, Platonov PG, Hedblad B, Engström G, Melander O. Atrial fibrillation in the Malmö Diet and Cancer study: a study of occurrence, risk factors and diagnostic validity. Eur J Epidemiol 2010;25:95–102. [DOI] [PubMed] [Google Scholar]
- 19. Mega JL, Stitziel NO, Smith JG, Chasman DI, Caulfield M, Devlin JJ, Nordio F, Hyde C, Cannon CP, Sacks F, Poulter N, Sever P, Ridker PM, Braunwald E, Melander O, Kathiresan S, Sabatine MS. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy. Lancet 2015;385:2264–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kathiresan S, Melander O, Anevski D, Guiducci C, Burtt NP, Roos C, Hirschhorn JN, Berglund G, Hedblad B, Groop L, Altshuler DM, Newton-Cheh C, Orho-Melander M. Polymorphisms associated with cholesterol and risk of cardiovascular events. N Engl J Med 2008;358:1240–1249. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.