

Abstract
A method of platinum quantification in whole blood samples after microwave digestion using sector field inductively coupled plasma mass spectrometry has been developed. The following analytical figures of merit have been established: limit of detection 1.1 µg/L for blood samples, dynamic range 3.6–200 µg/L, intra‐day precision (relative standard deviation, n = 9) did not exceed 5%. Spiked samples were analyzed for method validation. The method was used for pharmacokinetics studies of a novel anti‐cancer drug BP‐С1, a complex of cis‐configured platinum and benzene‐poly‐carboxylic acids. Main pharmacokinetic parameters (area under curve, maximum concentration, clearance, half‐life times for α‐ and β‐phase) were estimated for two dosage forms of BP‐C1 0.05 and 0.125 mass %. Pharmacokinetic curves were assessed for single and course administration. Studies were performed using rabbits (n = 6) as a model. BP‐C1 was injected intramuscularly. The study established dose proportionality of the tested dosage forms and suggested clinical dosing schedule: 5 days of injections followed by 2 days’ break. Platinum tissue distribution was studied in tissue samples collected 20 days after the last injection. Predominant platinum accumulation was observed in kidneys, liver, and muscles near injection site. ‘Slow’ phase of platinum excretion kinetics may be related to the muscles at the injection site. © 2015 The Authors. Drug Testing and Analysis published by John Wiley & Sons Ltd.
Keywords: platinum, breast cancer, pharmacokinetics, benzene‐poly‐carboxylic acids, inductively coupled plasma mass spectrometry
Introduction
Platinum (Pt) complexes, cytotoxic action of which is related to DNA binding1 and replication suppression,2 belong to the class of chemotherapeutic drugs. The first clinically approved3 and the most studied of the group is cis‐dichlorodiammineplatinum (II), cisplatin. High toxicity and severe side effects of cisplatin (nephro‐ and ototoxicity, peripheral nervous system disorders)4, 5, 6 inspired research into new platinum‐containing cytostatics. The second approved and less toxic compared to cisplatin was cis‐diammine(1,1‐cyclobutanedicarboxylato)platinum (II), carboplatin.7 In total, since the late 1960s, a considerable number of platinum and other d‐elements, e.g. palladium, ruthenium, osmium, iridium, etc., complexes were tested for antitumor activity.8, 9, 10 Although numerous studies related to new platinum compounds synthesis and their efficacy evaluation have been undertaken, insufficient selectivity for malignant cells, severe side effects and drug resistance developed by some tumours, are still a characteristic feature of these drugs.11, 12
Platinum‐containing chemotherapy agents with physiologically active ligands seem to be a perspective direction in anti‐cancer drug design. In any platinum‐containing drug a ligand should, first of all provide a stable therapeutic concentration of platinum in the system bloodstream. Consequently, pharmacokinetics studies are one of the key aspects of drug design for this group of compounds. The subject of the present study was pharmacokinetics of the novel anti‐cancer agent BP‐C1 (international patent WO2013/143549 A1), developed for therapy of metastatic breast and pancreas cancers. BP‐C1 is a complex of cis‐configured platinum (II) with benzene‐poly‐carboxylic acids. The novel ligand is assumed to possess highly functionalized poly‐phenolic backbone, which may contribute to additional tumour suppressing effect via immune modulation and inducing cytokine production (e.g. TNF‐α, IFN‐γ, IL‐1β, IL6, IL25).13 BP‐C1 was shown to activate pro‐apoptotic gene expression and inhibit oncogenes.14 The agent is undergoing controlled Phase II clinical trials.15
Chemical analysis plays an important role in the acquisition of pharmacokinetic data.16, 17 Rather strict requirements for analytical methods have stimulated a new method development for the analysis of drug dosage forms and raw materials as well as for the detection of pharmaceutical agents and their metabolites in biological media.18 Nowadays, one of the techniques available for platinum quantification in biological fluids and tissues is double‐focusing sector‐field inductively coupled plasma mass spectrometry (ICP‐MS). Providing high selectivity and low limit of detection (LOD),19 it may be called a technique of choice for elemental pharmaceutical analysis. The aim of the present study was method development for platinum determination in the blood and evaluation of pharmacokinetics of BP‐C1.
Materials and methods
Instrumentation
For platinum quantification, a double‐focusing sector‐field inductively coupled plasma mass spectrometer Thermo Scientific™ Element 2 (Thermo Fisher Scientific, Bremen, Germany) was used. Meinhard‐type concentric nebuliser and +4°C Peltier cooled glass spray chamber were employed for sample introduction. Microwave digestion system Start D (Milestone, Sorisole, Italy) with high‐pressure vials of polytetrafluoroethylene was used for sample pre‐treatment. Milli‐Q® Advantage A10 (Millipore, Molsheim, France) system was a source of ultra pure water.
Standards and chemicals
Calibration and spiking standard was CertiPUR® Platinum ICP standard 1000 mg/L Pt (Merck, Darmstadt, Germany). Rhodium CGRHN‐1 1001 ± 5 µg/mL (Inorganic Ventures, Christiansberg, VA, USA) was employed as an internal standard. For interference studies Multi‐Element Calibration Standards of Atomic Spectroscopy Standard series (PerkinElmer, Shelton, CT, USA) were analyzed. For blood and tissue sample digestion and calibration solution stabilization Suprapur® nitric acid (65%, Merck, Darmstadt, Germany) was used.
BP‐C1 was provided by Meabco A/S (Copenhagen, Denmark). Throughout this study two dosage forms of BP‐C1 were investigated: 0.125% injection solution in physiologic saline containing 110.6 µg/mL platinum (confirmed by sector field ICP‐MS) and 0.05% injection solution in physiologic saline containing 51.4 µg/mL platinum (confirmed by sector field ICP‐MS).
Animal keeping and study groups
The study was conducted in 6 adult male chinchilla rabbits (body weight 2900±192 g). Animals were kept in individual cages and provided with water and standard rabbit chow ad libitum. BP‐C1 (both 0.05 and 0.125% forms) was injected to rabbits intramuscularly in the volume of 1 mL per 3 kg of body weight in accordance with the study design. At the end of the study animals were euthanized with intravenous propofol injection and organs were excised for platinum tissue distribution studies. All animal experiments were conducted in accordance with ethical regulation and were approved by the ethics committee of the N.N. Petrov Research Institute of Oncology.
Pre‐clinical study design and sample collection
The pharmacokinetics study was performed in three stages within two series of experiments: kinetics studies of the single injection (the first administration), kinetics studies of the course treatment (multiple injections, stationary platinum level) and excretion kinetics (the last injection). In the first set of experiments rabbits 1, 2, and 3 received 1 mL of the 0.125% BP‐C1 dosage form (group 0.125%). Animals 4, 5 and 6 were given 1 mL of the 0.05% BP‐C1 dosage form (group 0.05%). In the second series, animals were crossed‐over, so that those previously given the higher dosage were switched to the 0.05% dosage and vice versa. The break between the series constituted 19 days.
Design of each series envisaged collection of blood samples each working day following the first test‐drug injection. The last blood sampling was performed 144 h after the initial injection. Afterwards, the same animals were used for studying the kinetics of multiple injections, which were given 22 injections (5 injections per week for 4 weeks). In the last week, animals were injected only twice. To study the single injection kinetics and excretion, samples were collected before BP‐C1 injection as well as 0.25, 0.5, 1, 2, 4, 8, 24, 48, and 144 h after the injection. For the multiple injection studies (stationary level) blood was sampled directly before the next injection (once per day). After the last injection (excretion kinetics) samples were collected similar to the single dose studies and also on days 8, 10, and 15. For details on injection and sampling design, see Table 1.
Table 1.
Groups, animals, injection and blood sampling schedule
| Group | Animals | Sampling after the first single injection | Injections and sampling for course treatment | Sampling for final excretion kinetics |
|---|---|---|---|---|
| 0.125% | 1, 2, 3 (series 1) | 0.25, 0.5, 1, 2, 4, 8, 24, 48 and 144 h after the first injection | Days 0, 1, 2, 4, 7, 8, 9, 10, 11, 14, 15, 16, 17, 18, 21, 22, 23, 24, 25, 28, 29, 30 | 0.25, 0.5, 1, 2, 4, 8, 24, 48, 144, 192, 240 and 360 h after the last injection |
| 4, 5, 6 (series 2) | ||||
| 0.05% | 4, 5, 6 (series 1) | |||
| 1, 2, 3 (series 2) |
Whole blood samples were used previously for platinum containing drugs pharmacokinetic studies, for example in the case of oxaliplatin, [(1R,2R)‐cyclohexane‐1,2‐diamine] (ethanedioato‐O,O') platinum(II).20, 21 In the current study, whole blood samples were taken from marginal ear vein. Lithium‐heparin (1300–1500 U/tube) 6 mL Vacutest® vacuum systems (Vacutest KIMA, Arzergrande, Italy) were used for blood sampling. Samples were frozen at –20°C and sent to analytical facility for analysis. Organs were sampled during autopsy, 20 days after the last BP‐C1 injection, using surgical instruments, tightly packed in sterile polyethylene bags, frozen at −20°C and transported for analysis. The following tissues were examined: brain, heart, left kidney, right kidney, liver, lungs, pancreas, urinary bladder, spleen, muscular tissue (near injection site), muscular tissue (intact), skin (near injection site) and skin (intact). Bones were not analyzed since this tissue was demonstrated to accumulate only minor levels of platinum.22
Sample preparation
Samples were thawed at +4°C. After that, 0.5 mL of whole blood or 100 mg of biological tissue were digested in microwave system Start D with 2.5 or 3 mL 65% Suprapure® nitric acid, for blood and tissue respectively, and 5 mL Milli‐Q® water. Mineralization was performed for 30 min (maximum autoclave temperature 185°C, power 1000 W). Solutions, acquired after digestion, were diluted to the final volume of 25 mL in PFA volumetric flasks. At this stage, internal standard (rhodium) was added to the final concentration of 1 µg/L. Samples were handled with all necessary precautions to avoid possible contamination.
Statistics
Method validation was performed in accordance with the United States Food and Drug Administration Guidelines on bioanalytical methods.23 Spiked samples were analyzed to estimate LODs, precision, accuracy, repeatability, linearity, and dynamic range. Blood and tissue samples were spiked with standard solution of platinum prior to digestion. Pharmacokinetics data was evaluated using both model‐independent calculation and three‐compartment model.24 Three‐compartment modelling was chosen since pilot study with two animals demonstrated prolonged ‘slow’ platinum excretion kinetics, which cannot be adequately described with two‐exponential representation. Standard Microsoft Excel® 2007 software (Microsoft, Redmond, WA, USA) was used for statistical calculations.
Results
Blood and tissue platinum assessment method development
The most abundant (33.832%) platinum isotope 195Pt was used for quantification with peak area registration. This isotope is not hampered with spectral interferences except for overlapping with 179Hf16O, 183W12C, 182W13C and 180Hf15N. However, according to the published data,25, 26 these interferences are insignificant for biological samples. Our own interference modelling supported this notion. However, for medium resolution mode (R = 4000) better intra‐day precision was observed. The following optimized parameters were employed: medium resolution (R = 4000), high frequency generator power 1170 W, plasma argon flow 16 L/min, auxiliary argon flow 0.72 L/min, nebulizer argon flow 0.93 L/min, spray chamber temperature +4°C.
Analytical procedure
The whole blood or biological tissues, treated as described in the ‘Sample preparation’ section, were analyzed using mass spectrometer Thermo Scientific Element 2. Analysis result was calculated as mean of 9 replicate measurements of the corresponding mass to charge ratio unless relative standard deviation (RSD) exceeded 5%. Otherwise, measurements were repeated after elimination of the cause of unsatisfactory precision. External calibration was made using aqueous standards of 0.6, 0.8, 1.0, 2.0, 4.0 and 5.0 µg/L platinum and 1 µg/L of internal standard (rhodium). Calibration was performed directly before analyzing real samples and its stability was controlled after each 10 samples. Control and correction of mass analyzer calibration, nebulizer performance and inductively coupled plasma parameters was performed daily prior to analysis.
Method validation
Samples containing standard additions of platinum were digested in accordance with the sample preparation procedure. Concentration of standard additions was 0, 50, 100, 200 µg/L Pt (adjusted for the final sample dilution to 1/50 by volume). Samples were prepared as follows: in digestion autoclaves 0, 250, 500, 750 and 1000 μL of 100 µg/L platinum standard, diluted from CertiPUR® Platinum ICP standard, were spiked into 0.5 mL of whole blood and 5.00, 4.75, 4.50, 4.25, and 4.00 mL of Milli‐Q® water was added. Microwave digested samples were diluted in PFA volumetric flasks of 25 mL with Milli‐Q® water. For validation studies each spiked sample was analyzed five times (n = 5).
LODs, linearity, and dynamic range
Detection limit corresponding to signal‐to‐noise ratio of 3.3 was found to be 1.10 µg/L. Linearity was evaluated using Fisher's F‐test. Dispersions between calculated and experimental values were compared to dispersion between average and individual values (F = 1.2 < F crit. = 2.1). Statistical significance of free parameter of linear regression (y = a + bx) was also estimated. According to Student's t‐test, free parameter a was found to be statistically equal to zero (tcalc. = 0.62 < t95%, n−2 = 23 = 2.07). Lower limit of quantification (LLOQ) corresponding to signal‐to‐noise ratio of 10 was 3.6 µg/L. Upper limit was 200 µg/L. Correlation coefficient for linear calibration was 1.0000.
Accuracy and repeatability
Method accuracy was evaluated using confidence half‐interval (P = 0.95; n = 5) for spiked/recovered ratios. Acquired value (4.5%) did not exceed established value for bioanalytical quantification methods (5%).23
Repeatability was estimated as relative standard deviation for 5 replicates within the dynamic range. It was found to be 4.4% which is statistically insignificant (β = 0.05) compared to the established value for method uncertainty (5%).23
Pharmacokinetics studies of BP‐C1
Pharmacokinetics curve for single and multiple BP‐C1 injections
Pharmacokinetic and dose proportionality studies of BP‐C1 in dosage forms of 0.05% and 0.125% were started from evaluation of kinetics of the single intramuscular injection. Experimental curves are shown in Figure 1. In Figure 2 stationary concentration studies for the course treatment are presented.
Figure 1.
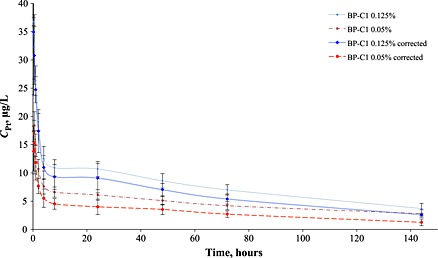
Animal averaged (n = 6) pharmacokinetics curves after the single BP‐C1 administration, corrected curves account previously administrated ‘background’ platinum in cross series and calculated using exponential modelling for background.
Figure 2.
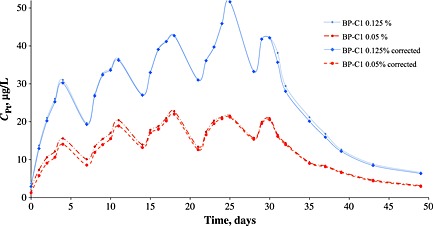
Animal averaged (n = 6) pharmacokinetics curves for the 30 days course treatment (22 injections, 5 days with 2 days’ break, 50 days of observation total), including after course platinum excretion, corrected curves account previously administrated ‘background’ platinum in cross series and calculated using exponential modelling for background.
Platinum tissue distribution after BP‐C1 course treatment (multiple injections)
Mean platinum concentrations in animal tissues and standard deviations (SD) are presented in Table 2. To examine platinum distribution, relative concentrations of platinum in respective tissue to platinum level for blood collected prior to euthanasia were calculated. Ratios for higher and lower dosage were also studied to eradicate primary accumulation organs.
Table 2.
Tissue mean platinum concentration (n = 3) 20 days after the end of the cross‐over course for each group of animals. Blood platinum concentration before euthanasia (Mean ± SD) was 6.60 ± 1.43 and 3.17 ± 0.07 µg/L for dosages of 0.125% and 0.05% (ratio 2.08), respectively
| Tissue | Dose 0.125%, µg/kg | Dose 0.05%, µg/kg | 0.125%/0.05% ratio | ||||
|---|---|---|---|---|---|---|---|
| Mean | SD | Tissue / blood | Mean | SD | Tissue / blood | ||
| Left kidney | 1101 | 75.9 | 167 | 579 | 79.2 | 183 | 1.90 |
| Right kidney | 1099 | 125 | 167 | 576 | 62.9 | 182 | 1.91 |
| Liver | 816 | 24.9 | 124 | 574 | 63.6 | 181 | 1.42 |
| Lung | 29.7 | 2.19 | 4.49 | 18.7 | 1.86 | 5.89 | 1.59 |
| Pancreas | 17.7 | 1.86 | 2.68 | 13.7 | 2.19 | 4.32 | 1.29 |
| Prostate | 41.0 | 5.29 | 6.21 | 24.3 | 4.67 | 7.68 | 1.68 |
| Urinary bladder | 57.7 | 5.24 | 8.74 | 32.0 | 6.11 | 10.1 | 1.80 |
| Spleen | 757 | 197 | 115 | 335 | 109 | 106 | 2.26 |
| Brain | 8.67 | 0.88 | 1.31 | 4.00 | 1.00 | 1.26 | 2.17 |
| Heart | 39.0 | 4.58 | 5.91 | 23.7 | 1.67 | 7.47 | 1.65 |
| Muscles, injection site | 1198 | 701 | 181 | 3455 | 1155 | 1091 | 0.35 |
| Muscles, intact | 12.0 | 3.51 | 1.82 | 14.3 | 3.53 | 4.53 | 0.84 |
| Skin, injection site | 220 | 135 | 33.4 | 401 | 208 | 127 | 0.55 |
| Skin, intact | 44.3 | 3.48 | 6.72 | 32.3 | 2.03 | 10.2 | 1.37 |
Discussion
Developed method
Comparing the dynamic range (3.6–200 µg/L) of the developed method to the results acquired for digested blood samples (Figures 1 and 2) and biological tissues (Table 2) as well as evaluation of the method accuracy (4.5%) confirmed compliance of the method with bioanalytical method requirements.23 Recently, Kristiansen et al. 27 reported BP‐C1 efficacy in veterinary treatment of canine mammary tumours; they also presented pharmacokinetics data in dogs for single subcutaneous injection of BP‐C1 within 48 h of observation and for 16 days course treatment consisting of 8 subcutaneous injections. However, pharmacokinetics data from this study and our own are difficult to compare since different biological species (dogs vs. rabbits) and different dosages (3.7 µg/kg Pt vs. 38.1 and 17.7 µg/kg Pt for the current study) were employed. Nevertheless, Kristiansen et al. 27 also showed quite slow excretion of platinum. To the best of our knowledge (Web of Science Core Collection and Scopus databases), there is no published pharmacokinetics data on platinum‐benzene‐poly‐carboxylic acid complexes after intramuscular injection in rabbits.
In general, major pharmacokinetics data of platinum‐containing chemotherapy drugs pertain to both pre‐clinical28, 29 and clinical trials.30, 31 The most frequently used techniques are ICP‐MS31, 32, 33, 34 and atomic absorption spectrometry (AAS) with flameless atomization.35, 36 Chromatographic methods and molecular mass spectrometry are also applied quite frequently.2, 37, 38, 39 Advantages of AAS are good instrumental availability and established methodical approaches to biological sample analysis.40 Nevertheless, relatively high limit of detection in the range of < 0.3− ~ 30 µg/L (depending on sample analyzed and sample pretreatment procedure41, 42, 43, 44, 45), considerably confines application of AAS, especially for analysis of samples from ‘slow’ excretion phases of studies. ICP‐MS makes possible lowering detection limit to the level of 0.0005 µg/L for blood plasma ultrafiltrate.31 Moreover, use of double focusing sector field ICP‐MS enables both detection limit and selectivity improvement leading to reliable assessment of platinum concentration especially at ‘slow’ excretion phases.46, 47 In the current study, in order to achieve higher sample output rather high sample dilution (1/50 per volume) was used. That resulted in elevated lower limit of quantification for sector field ICP‐MS. However, it should be noted that dynamic range corresponded to measured platinum concentration in the analyzed samples.
BP‐C1 pharmacokinetics
For the single injection kinetics (Figure 1) experimental curve could be rather easily divided into tissue distribution stage (α‐phase) and platinum excretion stage (β‐phase). The β‐phase could be further subdivided into two semi‐phases β1 and β2. Distribution phase took less than 2 h; after that, distribution to elimination transition stage (ca 2–24 h post injection) and per se β‐phase followed.
Based on acquired results major pharmacokinetics parameters were estimated: area under curve (AUC), platinum maximum concentration (Cmax), clearance, and half‐life times corresponding to α, β1 and β2 phases (Table 3). Maximum concentration was achieved at ~ 0.5 h for the 0.125% concentration and only at ~ 0.25 h for the 0.05%. Notably, experimental pharmacokinetics curves (Figure 1) for BP‐C1 were in concordance with previously published results for other platinum‐containing drugs, having rather fast metal redistribution into organs and tissues from the bloodstream within several hours after administration as well as prolonged platinum excretion.27, 35
Table 3.
Pharmacokinetics parameters (Mean ± SD) calculated for the first single injection, averaged for all animals (n = 6) after both stages of the study. Three compartment modelling and model‐independent calculation were used
| Parameter | Mean ± SD | |
|---|---|---|
| Group 0.125% | Group 0.05% | |
| Сmax *, µg/L | 35.2 | 15.3 |
| Сmax, µg/L | 31.1 ± 1.2 | 16.0 ± 0.2 |
| AUC0‐144 *, µg.h/L | 1023 | 462 |
| AUC0‐∞, (µg.h)/L | 1682 ± 145 | 1387 ± 83 |
| Cl, L/(h.kg) | 0.022 ± 0.002 | 0.012 ± 0.0007 |
| T½α, h | 0.810 ± 0.016 | 0.797 ± 0.016 |
| T½β1, h | 63.0 ± 1.4 | 64.2 ± 0.5 |
| T½β2, h | 142 ± 30 | 231 ± 49 |
– direct model‐independent calculation.
Direct model‐independent calculation of maximum concentration (Сmax *) and area under curve (AUC0‐144 * ) showed dose proportionality of the two studied forms of BP‐C1, since both Cmax and AUC increased proportionally for the higher dose. Noteworthy, for three‐compartment modelling average area under curve (AUC0‐∞) for the lower dosage of 0.05% was found somewhat overestimated. That may be related to residual excretion of platinum remained after the first set of BP‐C1 injections as it was typical for animals, which received the 0.125% dosage form within the first experimental series. Presence of ‘residual platinum’ from the previous BP‐C1 administration is also supported with unexpectedly high value of T½β2 for the 0.05% dosage form.
Under the course treatment with previously described design stationary blood platinum concentration was established 10 days after the first injection for the 0.05% dosage form and not earlier than in 15 days in case of the 0.125% form (Figure 2). Pharmacokinetics studies of multiple injections support the following dosing schedule: 5 days intramuscular injections of BP‐C1 (from Monday to Friday) with two days break for a weekend. Such dosing schedule could be called ‘patient‐oriented’ as it makes hospitalization non‐mandatory and allows ambulatory treatment or even treatment at home. This is very important from the patient psychological state standpoint and also lowers the costs of the treatment. Finally, BP‐C1 post‐course kinetics revealed a tendency for delayed excretion of lower dosages of the agent and prolonged elimination of the metal from the bloodstream. The ‘slow’ excretion established platinum deposition in biological tissues. Thus, further analyses were devoted to the investigation of platinum distribution in biological tissues.
Platinum tissue distribution after BP‐C1 course treatment (multiple injections)
Organs of primary platinum accumulation were kidneys, liver, spleen and muscular tissue near the injection site (Table 2). Low platinum concentration in the brain indicated slight transport of BP‐C1 through the blood‐brain‐barrier, which is in concordance with the data established for other platinum‐containing agents, for example cisplatin, carboplatin and oxaliplatin28, 48 as well as for platinum metal complexes in general.49 Although animal sample was small (three animals per dosage form), some tendencies for platinum elimination from the organs could be demonstrated. Muscular tissue near the injection site may be responsible for the ‘slow’ excretion stage. Even 20 days after the last injection, this tissue indicated high platinum concentration as well as the smallest higher‐to‐lower dose ratio that could be explained by platinum accumulation during the first experimental series, when sacrificed animals of the 0.05% dosage subgroup received higher dose of 0.125%. Notably, intact muscles (taken from forelegs with no BP‐C1 injections) also showed somewhat elevated platinum accumulation along with pancreas and skin.
Tissue distribution of platinum‐containing agents was not studied thoroughly enough, especially in case of the course treatment. For Saliplatin, SM‐54111 [cis‐3,5‐diisopropylsalylic cyclohexano‐diaminoplatinum (II)] primary accumulation was demonstrated to take place in spleen, liver, and kidneys (0.5–3 h of observation),50 but data for course treatment are absent. Shimakura et al. 51 studied platinum tissue distribution in canine tissues after single intra‐arterial administration of SM‐11355 [cis[((1R,2R)‐1,2‐cyclohexanediamine‐N,N’)bis(myristato)]platinum (II)] simultaneously with lipiodol; main accumulation sites were liver and gall bladder. Egger et al. 10 investigated spatial platinum distribution in murine kidney and spleen after single intraperitoneal injection of cisplatin; main platinum accumulation was detected in kidneys and, secondly, in liver. For orally administered [(OC‐6‐43)‐bis (acetato) (1‐adamantylamine) amminedichloroplatinum (IV)] main accumulation organs in rats 24 and 48 h after injection were also liver, kidneys and spleen.52 Similar results were obtained for [cis‐diammine‐1,1‐(3‐cyclobutanone) dicarboxylate platinum (II)] after intravenous injection in rats.53 Esteban‐Fernandez et al. 54 studied distribution of platinum, originating from cisplatin, carboplatin, and oxaliplatin, in cellular compartments of side effect affected organs – inner ear, brain, kidney and liver; primary accumulation was found for kidney and liver for all compounds, whereas for carboplatin was found to be much less distributed into brain and inner ear. Wang et al.55 reported blood plasma pharmacokinetics and tissue distribution of cisplatin, carboplatin and 5 experimental anti‐cancer agents; main deposition was found for kidney for investigated agents. Noteworthy, in most distribution studies, which concern platinum‐containing drugs, mainly intravenous (for human‐based designs) or also intra‐peritoneal (for non‐clinical studies) routes of administration are employed.54, 56, 57, 58, 59 That is why media such as skin and muscle tissue are not investigated as usual. However, in case of BP‐C1, we observed some platinum retention even for intact skin and muscles, so this is also possible for other agents. That was previously observed only for carboplatin injected intravenously to rats.60 Considerable platinum content was observed for skin and muscle comparable to spleen, intestine, blood and even liver but still much lover than that of kidney.60 Interestingly, for our results kidney also showed quite high platinum retention, however, if compared to other studies,54, 55, 60 lower ratio of kidney to other platinum accumulating tissues (liver, spleen and muscle) was observed. Thus, lower nephrotoxicity may be assumed for BP‐C1 than that of existing platinum‐containing anticancer drugs. Finally, preliminary protein binding studies for BP‐C1 conducted in our group (unpublished results) showed higher proportion of protein‐unbound platinum compared to existing platinum‐containing agents, first of all cisplatin, and their slower elimination from the system bloodstream. Anyway, truly prolonged elimination from the organism is very typical for platinum. For instance, for mature teratoma platinum is detected in the tissue more than 7 years after cisplatin administration.22
Conclusion
A method of platinum quantification in whole blood and biological tissues after microwave digestion using sector field mass spectrometry was developed. Main analytical figures of merit – detection limit, dynamic range, accuracy, and repeatability – were estimated. Pharmacokinetics of the novel antitumor agent BP‐C1 (dosage forms 0.125% and 0.05%) was studied in rabbits after intramuscular injection using the developed method. Two dosage forms of BP‐C1 were shown to be dose proportional. The possibility of BP‐C1 administration for 5 days followed by 2 days breaks was shown. Stationary concentration was observed after ca. 2 weeks of course treatment. Platinum tissue distribution in rabbit tissue was studied 20 days after the end of the course treatment. Primary metal accumulation was shown in kidneys, liver and muscles near the injection site. Observed ‘slow’ platinum elimination may also be related to the injection site muscles. Next stage of BP‐C1 pharmacokinetics testing should be devoted to examination of ultrafilterable platinum originating from the agent.
Conflict of interest
None of the authors are employed or own stocks in Meabco A/S. Meabco A/S had no role in the study design, data collection, analysis and interpretation, drafting, writing and editing of the current paper.
Acknowledgements
The authors wish to thank Valerian Trefilov (Institute of Toxicology of Federal medico‐biological agency, St Petersburg, Russia) for assistance in statistical data processing and pharmacokinetics modelling, Professor Stig Larsen (Norwegian University of Life Sciences, Oslo, Norway) for his valuable suggestions concerning study cross‐design and Sergey Pigarev (Nobel LTD, St Petersburg, Russia) for reading the manuscript. The authors are also grateful to the collaborative research group at the N.N. Petrov Research Institute of Oncology and to Meabco A/S for providing the test product and financial support of the study.
Navolotskii, D. V. , Ivanenko, N. B. , Solovyev, N. D. , Fedoros, E. I. , and Panchenko, A. V. (2015) Pharmacokinetics and tissue distribution of novel platinum containing anticancer agent BP‐C1 studied in rabbits using sector field inductively coupled plasma mass spectrometry. Drug Test. Analysis, 7: 737–744. doi: 10.1002/dta.1824.
References
- 1. Takahara P. M., Frederick C. A., Lippard S. J.. Crystal structure of the anticancer drug cisplatin bound to duplex DNA. J. Am. Chem. Soc. 1996, 118, 12309. [Google Scholar]
- 2. Zayed A., Jones G. D. D., Reid H. J., Shoeib T., Taylor S. E., Thomas A. L., Wood J. P., Sharp B. L.. Speciation of oxaliplatin adducts with DNA nucleotides. Metallomics. 2011, 3, 991. [DOI] [PubMed] [Google Scholar]
- 3. Rosenberg B., Vancamp L., Trosko J. E., Mansour V. H.. Platinum compounds ‐ a new class of potent antitumour agents. Nature. 1969, 222, 385. [DOI] [PubMed] [Google Scholar]
- 4. Greaves E. D., Angeli‐Greaves M., Jaehde U., Drescher A., von Bohlen A.. Rapid determination of platinum plasma concentrations of chemotherapy patients using total reflection X‐ray fluorescence. Spectrochim. Acta B. 2006, 61, 1194. [Google Scholar]
- 5. Shimada M., Itamochi H., Kigawa J.. Nedaplatin: a cisplatin derivative in cancer chemotherapy. Cancer Manage. Res. 2013, 5, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Piccolini V. M., Bottone M. G., Bottiroli G., De Pascali S. A., Fanizzi F. P., Bernocchi G.. Platinum drugs and neurotoxicity: effects on intracellular calcium homeostasis. Cell Biol. Toxicol. 2013, 29, 339. [DOI] [PubMed] [Google Scholar]
- 7. Rozencweig M., Nicaise C., Beer M., Crespeigne N., Vanrijmenant M., Lenaz L., Kenis Y.. Phase‐I study of carboplatin given on a 5‐day intravenous schedule. J. Clin. Oncol. 1983, 1, 621. [DOI] [PubMed] [Google Scholar]
- 8. Laine A. L., Passirani C.. Novel metal‐based anticancer drugs: a new challenge in drug delivery. Curr. Op. Pharmacol. 2012, 12, 420. [DOI] [PubMed] [Google Scholar]
- 9. Matczuk M., Przadka M., Aleksenko S. S., Czarnocki Z., Pawlak K., Timerbaev A. R., Jarosz M.. Metallomics for drug development: a further insight into intracellular activation chemistry of a ruthenium(III)‐based anticancer drug gained using a multidimensional analytical approach. Metallomics. 2014, 6, 147. [DOI] [PubMed] [Google Scholar]
- 10. Egger A. E., Theiner S., Kornauth C., Heffeter P., Berger W., Keppler B. K., Hartinger C. G.. Quantitative bioimaging by LA‐ICP‐MS: a methodological study on the distribution of Pt and Ru in viscera originating from cisplatin‐ and KP1339‐treated mice. Metallomics. 2014, 6, 1616. [DOI] [PubMed] [Google Scholar]
- 11. Galluzzi L., Marsili S., Vitale I., Senovilla L., Michels J., Garcia P., Vacchelli E., Chatelut E., Castedo M., Kroemer G.. Vitamin B6 metabolism influences the intracellular accumulation of cisplatin. Cell Cycle. 2013, 12, 417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marrache S., Pathak R. K., Dhar S.. Detouring of cisplatin to access mitochondrial genome for overcoming resistance. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 10444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kirkin A., Dzhandzhugazyan K., Fang J., Lindkair‐Jensen S., Guldberg P.. A novel anticancer agent, benzene‐poly‐carboxylic acids complex with cis‐diammineplatinum (II) dichloride, activates multiple immunologic mechanisms of an antitumor response. J. Clin. Oncol. 2013, 31, e12024. [Google Scholar]
- 14. Fares F., Azzam N., Fares B., Larsen S., Lindkaer‐Jensen S.. Benzene‐Poly‐Carboxylic Acid Complex, a Novel Anti‐Cancer Agent Induces Apoptosis in Human Breast Cancer Cells. Plos One. 2014, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Larsen S. E., Dewi S., Srimuninnimit V., Lu Y. S., Manuaba T., Jensen S. L.. Estimation of maximum tolerated dose and minimum efficient dose of BP‐C1 in the treatment of stage IV breast cancer patients: A phase I response surface pathway designed study. J. Clin. Oncol. 2012, 30, 1.22105825 [Google Scholar]
- 16. Zufia L., Aldaz A., Castellanos C., Giraldez J.. Simple and rapid determination of carboplatin in plasma by highperformance liquid chromatography. Error pattern and application to clinical pharmacokinetic studies. J. Chromatog. B. 2001, 764, 457. [DOI] [PubMed] [Google Scholar]
- 17. Bosch M. E., Sanchez A. J. R., Rojas F. S., Ojeda C. B.. Analytical methodologies for the determination of cisplatin. J. Pharmaceut. Biomed Anal. 2008, 47, 451. [DOI] [PubMed] [Google Scholar]
- 18. Michalke B.. Platinum speciation used for elucidating activation or inhibition of Pt‐containing anti‐cancer drugs. J. Trace Elem. Med. Biol. 2010, 24, 69. [DOI] [PubMed] [Google Scholar]
- 19. Becker S.. Inorganic Mass Spectrometry: Principles and Applications, John Wiley & Sons Ltd., Chichester, UK, 2007. [Google Scholar]
- 20. Ehrsson H., Wallin I., Yachnin J.. Pharmacokinetics of oxaliplatin in humans. Med. Oncol. 2002, 19, 261. [DOI] [PubMed] [Google Scholar]
- 21. Baur M., Drescher A., Gneist M., Dittrich C., Jaehde U.. Pharmacokinetics of oxaliplatin in patients with severe hepatic dysfunction. Cancer Chemother. Pharmacol. 2008, 61, 97. [DOI] [PubMed] [Google Scholar]
- 22. Dikhoff T., De Goeij J. J. M., McVie J. G.. Long‐term body retention and tissue distribution of platinum in cisplatin treated cancer patients. J. Radioanalyt. Nuclear Chem. 1998, 236, 81. [Google Scholar]
- 23. Guidance for Industry, Bioanalytical Method Validation. FDA, US, 2001, p. 22.
- 24. Bonate P. L.. Pharmacokinetic‐Pharmacodynamic Modeling and Simulation, Second Edition, Springer US, Deerfiels, IL, USA, 2011. [Google Scholar]
- 25. Iavicoli I., Bocca B., Petrucci F., Senofonte O., Carelli G., Alimonti A., Caroli S.. Biomonitoring of traffic police officers exposed to airborne platinum. Occup. Environ. Med. 2004, 61, 636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Frazzoli C., Cammarone R., Caroli S.. Investigation of palladium and platinum levels in food by sector field inductively coupled plasma mass spectrometry. Food Add. Contam. 2007, 24, 546. [DOI] [PubMed] [Google Scholar]
- 27. Kristiansen V. M., Dewi S., Horsberg T. E., Jonasdottir T. J., Moe L., Berlinger B., Lindkær‐Jensen S., Larsen S.. Tolerability and pharmacokinetic profile of a novel benzene‐poly‐carboxylic acids complex with cis‐diammineplatinum (II) dichloride in dogs with malignant mammary tumours. Vet. Compar. Oncol. 2015. In Press, DOI: 10.1111/vco.12144 [DOI] [PubMed] [Google Scholar]
- 28. Jacobs S. S., Fox E., Dennie C., Morgan L. B., McCully C. L., Balis F. M.. Plasma and cerebrospinal fluid pharmacokineties of intravenous oxaliplatin, cisplatin, and carboplatin in nonhuman primates. Clin. Cancer Res. 2005, 11, 1669. [DOI] [PubMed] [Google Scholar]
- 29. Levy‐Nissenbaum E., Khan W., Pawar R. P., Tabakman R., Naftali E., Winkler I., Kaufman O., Klapper L., Domb A. J.. Pharmacokinetic and efficacy study of cisplatin and paclitaxel formulated in a new injectable poly(sebacic‐co‐ricinoleic acid) polymer. Eur. J. Pharm. Biopharm. 2012, 82, 85. [DOI] [PubMed] [Google Scholar]
- 30. Peng B., English M. W., Boddy A. V., Price L., Wyllie R., Pearson A. D. J., Tilby M. J., Newell D. R.. Cisplatin pharmacokinetics in children with cancer. Eur. J. Cancer. 1997, 33, 1823. [DOI] [PubMed] [Google Scholar]
- 31. Clerico A., Cappelli C., Ragni G., Caroli S., De Ioris M. A., Sordi A., Petrucci F., Bocca B., Alimonti A.. Evaluation of carboplatin pharmacokinetics in pediatric oncology by means of inductively coupled plasma mass spectrometry. Ann. Ist. Super. Sanita. 2006, 42, 461. [PubMed] [Google Scholar]
- 32. Zoriy M., Matusch A., Spruss T., Becker J. S.. Laser ablation inductively coupled plasma mass spectrometry for imaging of copper, zinc, and platinum in thin sections of a kidney from a mouse treated with cis‐platin. Int. J. Mass Spectrom. 2007, 260, 102. [Google Scholar]
- 33. Janssens T., Brouwers E. E. M., de Vos J. P., Schellens J. H. M., Beijnen J. H.. Determination of platinum originating from cartoplatin in canine sebum and cerumen by inductively coupled plasma mass spectrometry. J. Pharm. Biomed. Anal. 2011, 54, 395. [DOI] [PubMed] [Google Scholar]
- 34. Zhao D., Zhang Y., Xu C., Dong C., Lin H., Zhang L., Li C., Ren S., Wang X., Yang S., Han D., Chen X.. Pharmacokinetics, Tissue Distribution, and Plasma Protein Binding Study of Platinum Originating from Dicycloplatin, a Novel Antitumor Supramolecule, in Rats and Dogs by ICP‐MS. Biol. Trace Elem. Res. 2012, 148, 203. [DOI] [PubMed] [Google Scholar]
- 35. Yasumasu T., Ueda T., Uozumi J., Mihara Y., Kumazawa J.. Comparative‐study of cisplatin and carboplatin on pharmacokinetics, nephrotoxicity and effect on renal nuclear‐dna synthesis in rats. Pharmacol. Toxicol. 1992, 70, 143. [DOI] [PubMed] [Google Scholar]
- 36. Najafi N. M., Shahparvizi S., Rafati H., Ghasemi E., Alizadeh R.. Preconcentration and determination of ultra‐traces of platinum in human serum using the combined electrodeposition‐electrothermal atomic absorption spectroscopy (ED‐ETAAS) and chemometric method. J. Pharm. Biomed. Anal. 2010, 53, 58. [DOI] [PubMed] [Google Scholar]
- 37. Hanada K., Nagai N., Ogata H.. Quantitative‐determination of unchanged cisplatin in rat‐kidney and liver by high‐performance liquid‐chromatography. J. Chromatog. B. 1995, 663, 181. [DOI] [PubMed] [Google Scholar]
- 38. Peleg‐Shulman T., Gibson D.. Cisplatin‐protein adducts are efficiently removed by glutathione but not by 5 '‐guanosine monophosphate. J. Am. Chem. Soc. 2001, 123, 3171. [DOI] [PubMed] [Google Scholar]
- 39. Laghari A. J., Khuhawar M. Y., Zardari L. A., Bhatti A. G.. GC determination of cisplatin in serum and urine of cancer patients after chemotherapy as platinum (II) pyrrolidinedithiocarbamate chelate. Chromatographia. 2008, 67, 748. [Google Scholar]
- 40. Ivanenko N. B., Ganeev A. A., Solovyev N. D., Moskvin L. N.. Determination of trace elements in biological fluids. J. Anal. Chem. 2011, 66, 784. [Google Scholar]
- 41. Cantarero A., Gomez M. M., Camara C., Palacios M. A.. Online preconcentration and determination of trace platinum by flow‐injection atomic‐absorption spectrometry. Anal. Chim. Acta. 1994, 296, 205. [Google Scholar]
- 42. Milacic R., Cemazar M., Sersa G.. Determination of platinum in tumour tissues after cisplatin therapy by electrothermal atomic absorption spectrometry. J. Pharm. Biomed. Anal. 1997, 16, 343. [DOI] [PubMed] [Google Scholar]
- 43. Raghavan R., Mulligan J. A.. Low‐level (PPB) determination of cisplatin in cleaning validation (rinse water) samples. I. An atomic absorption spectrophotometric method. Drug Dev. Indus. Pharmacy. 2000, 26, 423. [DOI] [PubMed] [Google Scholar]
- 44. da Costa A. C., Vieira M. A., Luna A. S., de Campos R. C.. Determination of platinum originated from antitumoral drugs in human urine by atomic absorption spectrometric methods. Talanta. 2010, 82, 1647. [DOI] [PubMed] [Google Scholar]
- 45. Zalba S., Navarro‐Blasco I., Moreno D., Garrido M. J.. Application of non‐aggressive sample preparation and electrothermal atomic absorption spectrometry to quantify platinum in biological matrices after cisplatin nanoparticle administration. Microchem. J. 2010, 96, 415. [Google Scholar]
- 46. Parsons P. J., Barbosa F.. Atomic spectrometry and trends in clinical laboratory medicine. Spectrochim. Acta B. 2007, 62, 992. [Google Scholar]
- 47. Ivanenko N. B., Ivanenko A. A., Solovyev N. D., Zeimal' A. E., Navolotskii D. V., Drobyshev E. J.. Biomonitoring of 20 trace elements in blood and urine of occupationally exposed workers by sector field inductively coupled plasma mass spectrometry. Talanta. 2013, 116, 764. [DOI] [PubMed] [Google Scholar]
- 48. Eiseman J. L., Beumer J. H., Rigatti L. H., Strychor S., Meyers K., Dienel S., Horn C. C.. Plasma pharmacokinetics and tissue and brain distribution of cisplatin in musk shrews. Cancer Chemother. Pharmacol. 2015, 75, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sabbioni E., Fortaner S., Manenti S., Groppi F., Bonardi M., Bosisio S., Di Gioacchino M.. The metallobiochemistry of ultratrace levels of platinum group elements in the rat. Metallomics. 2015, 7, 267. [DOI] [PubMed] [Google Scholar]
- 50. Liu Q. F., Li X., Su Q., Luo G. A., Wang Y. M., Liu W. P.. Investigation on pharmacokinetics, tissue distribution and excretion of a novel anticancer platinum compound by inductively coupled plasma mass spectrometry after intravenous administration to rats. Arch. Pharm. Res. 2009, 32, 1621. [DOI] [PubMed] [Google Scholar]
- 51. Shimakura J., Fujimoto K., Komuro S., Nakano M., Kanamaru H.. Long‐term disposition of a novel lipophilic platinum complex SM‐11355 in dog after intrahepatic arterial administration: highly sensitive detection of platinum and radioactivity. Xenobiotica. 2002, 32, 399. [DOI] [PubMed] [Google Scholar]
- 52. Sova P., Chladek J., Zak F., Mistr A., Kroutil A., Semerad M., Slovak Z.. Pharmacokinetics and tissue distribution of platinum in rats following single and multiple oral doses of LA‐12 [(OC‐6‐43)‐bis(acetato)(1‐adamantylamine)amminedichloroplatinum(IV)]. Int. J. Pharm. 2005, 288, 123. [DOI] [PubMed] [Google Scholar]
- 53. Zhao J., Wen Y., Zhang W., Zhao D., Fan A., Zhang Y., Deng S., Wang X., Liu Q., Lu Y., Wang Z., Gou S., Chen X.. Investigation on pharmacokinetics, tissue distribution and excretion of a novel platinum anticancer agent in rats by inductively coupled plasma mass spectrometry (ICP‐MS). Xenobiotica. 2014, 44, 757. [DOI] [PubMed] [Google Scholar]
- 54. Esteban‐Fernandez D., Verdaguer J. M., Ramirez‐Camacho R., Palacios M. A., Gomez‐Gomez M. M.. Accumulation, fractionation, and analysis of platinum in toxicologically affected tissues after cisplatin, oxaliplatin, and carboplatin administration. J.Anal. Toxicol. 2008, 32, 140. [DOI] [PubMed] [Google Scholar]
- 55. Wang X., Au‐Yeung S. C., Ho Y. P.. Pharmacokinetics and tissue distribution of novel traditional Chinese medicine‐platinum anticancer agents in rats. J. Inorg. Biochem. 2007, 101, 909. [DOI] [PubMed] [Google Scholar]
- 56. Hanada K., Suda M., Kanai N., Ogata H.. Pharmacokinetics and toxicodynamics of oxaliplatin in rats: application of a toxicity factor to explain differences in the nephrotoxicity and myelosuppression induced by oxaliplatin and the other platinum antitumor derivatives. Pharm. Res. 2010, 27, 1893. [DOI] [PubMed] [Google Scholar]
- 57. Gholap D., Verhulst J., Ceelen W., Vanhaecke F.. Use of pneumatic nebulization and laser ablation‐inductively coupled plasma‐mass spectrometry to study the distribution and bioavailability of an intraperitoneally administered Pt‐containing chemotherapeutic drug. Anal. Bioanal. Chem. 2012, 402, 2121. [DOI] [PubMed] [Google Scholar]
- 58. Minami T., Tohno Y., Tohno S., Utsumi M., Yamada M., Hashii K., Tateyama I., Kadota E., Okazaki Y.. Tissue platinum after clinical treatment with cisplatin or carboplatin in tumor‐bearing patients. Biol. Trace Elem. Res. 1997, 58, 77. [DOI] [PubMed] [Google Scholar]
- 59. da Silva Faria M. C., Santos N. A., Carvalho Rodrigues M. A., Rodrigues J. L., Barbosa Junior F., Santos A. C.. Effect of diabetes on biodistribution, nephrotoxicity and antitumor activity of cisplatin in mice. Chem. Biol. Interact. 2015, 229, 119. [DOI] [PubMed] [Google Scholar]
- 60. Siddik Z. H., Boxall F. E., Harrap K. R.. Flameless atomic‐absorbtion spectrophotometric determination of platinum in tissues solubilized in hyamine hydroxide. Anal. Biochem. 1987, 163, 21. [DOI] [PubMed] [Google Scholar]