

Abstract
Study Objective
To investigate the pharmacodynamic and pharmacokinetic profiles of fluticasone furoate (FF)/vilanterol (VI) – a fixed‐dose combination of an inhaled corticosteroid (ICS) and a long‐acting β2‐agonist for the treatment of asthma and chronic obstructive pulmonary disease – after single and repeat administration in healthy Chinese subjects.
Design
Double‐blind, placebo‐controlled, single‐site, randomized, four‐way crossover study.
Setting
The Clinical Pharmacological Research Centre at Peking Union Medical College Hospital [PUMCH]) in Beijing, China.
Subjects
Sixteen healthy, nonsmoking Chinese adults.
Intervention
Subjects were randomized to receive FF/VI 50/25, 100/25, or 200/25 μg, or placebo once/daily in the morning, delivered by the Ellipta dry powder inhaler, for 7 consecutive days. The subjects then received the other three treatments, with each treatment period separated by a 7‐day washout period.
Measurements and Main Results
The co‐primary outcome measures reflected pharmacodynamic responses relating to recognized class effects of the two drug classes: reduced serum cortisol level (ICSs), and increased Fridericia's corrected QT interval (QTcF) and reduced serum potassium level (long‐acting β2‐agonists). Co‐primary pharmacodynamic endpoints were 0–24‐hour weighted mean serum cortisol level on day 7 (cortisol0–24 hr, Day 7), and 0–4‐hour weighted mean and maximum QTcF and weighted mean and minimum serum potassium level on days 1 and 7. Fluticasone furoate and VI plasma concentrations, derived pharmacokinetic parameters, and safety were also assessed. Of the 16 subjects randomized, 15 completed the study. Reductions in cortisol0–24 hour, Day 7 of 15% and 25% were observed with FF/VI 100/25 and 200/25 μg, respectively, versus placebo. Minor increases (< 10 msec) in maximum QTcF on day 7 were seen with FF/VI 50/25 and 100/25 μg but not with 200/25 μg. Slight decreases in serum potassium level were only observed in subjects receiving FF/VI 50/25 μg on day 1 and FF/VI 50/25 and 200/25 μg on day 7. Fluticasone furoate accumulation (day 7 vs day 1) for FF/VI 50/25–200/25 μg ranged from 38 to 54% for maximum observed concentration and 63–71% for area under the concentration‐time curve from 0 to 4 hours. Fluticasone furoate pharmacokinetics were less than dose proportional. The VI pharmacokinetic profiles were similar for all three FF/VI doses. Adverse events were all mild in intensity and were reported by 13 (81%) of the 16 subjects.
Conclusion
In healthy Chinese subjects, minimal and non–clinically relevant β‐adrenergic pharmacodynamic effects were observed with FF/VI doses ranging from 50/25 to 200/25 μg. FF dose‐dependent reductions in serum cortisol levels of 15–25% were seen after administration of FF/VI 100/25 and 200/25 μg. FF/VI was safe and well tolerated in these subjects at doses ranging from 50/25 to 200/25 μg.
Keywords: China; cortisol; fluticasone furoate, inhaled corticosteroids; long‐acting beta2 agonists; pharmacokinetics; pharmacodynamics; systemic exposure; vilanterol
Fluticasone furoate (FF)/vilanterol (VI) is a fixed‐dose, once‐daily inhaled corticosteroid (ICS)/long‐acting β2‐agonist (LABA) combination therapy approved in 25 countries and regions worldwide, including the United States, Canada, and European Union, for the treatment of chronic obstructive pulmonary disease (COPD) at doses of FF/VI 100/25 μg and asthma at doses of FF/VI 100/25 and 200/25 μg). The 100/25 and 200/25 μg nominal doses represent emitted doses from the ELLIPTA TA dry powder inhaler (DPI) (GSK, Brentford, UK) of 92/22 and 184/22 μg, respectively. In several large‐scale, multiregion, randomized, controlled studies, once‐daily FF/VI has demonstrated similar efficacy to twice‐daily fluticasone propionate (FP)/salmeterol, an established ICS/LABA for the treatment of asthma and COPD.1, 2, 3, 4
Fluticasone furoate and VI have been well tolerated at doses up to 800 μg and 100 μg/day, respectively5, 6, 7; healthy subjects receiving these doses separately or in combination experienced adverse events (AEs) at a similar frequency to those receiving placebo.7 In a placebo‐controlled study of therapeutic doses of FF/VI 100/25 and 200/25 μg in patients with persistent asthma, no evidence of an effect on hypothalamic–pituitary–adrenal (HPA) axis function was noted.8 A 7‐day repeat‐dose thorough QT study did not find any association between FF/VI 200/25 μg and any QTc changes of concern in mostly (74%) Caucasian healthy subjects.6
Genetic and physiological variation may result in pharmacokinetic (PK) differences across ethnic populations.9 Findings from a PK study with inhaled FF 200 μg in healthy East Asian subjects demonstrated modestly higher (< 2‐fold) systemic exposure to FF compared with Caucasians.10 Despite these FF systemic exposure differences, a 22% reduction in serum cortisol level relative to Caucasians was only observed in Japanese but not in Chinese or Korean subjects. In a separate study with FF monotherapy (200, 400, and 800 μg) in healthy Japanese subjects, significant and dose‐dependent serum cortisol level decreases were observed, which clearly correlated with FF systemic exposure (area under the concentration‐time curve from 0 to 24 hours [AUC0–24 hr]).11 A population PK analysis from global studies has suggested that the maximum observed concentration (C max) of VI may be higher in East Asian subjects, compared with non‐Asians, whereas AUC0–24 hour is consistent across ethnicities.12 There is no evidence to suggest an effect of increased VI C max on heart rate within 20 minutes of dosing.
This study aimed to assess the PK and pharmacodynamics (PD) of FF and VI after single and repeat administration of FF/VI combination therapy in healthy Chinese subjects. To our knowledge, PK and PD data for VI or FF/VI combination have not previously been reported in mainland Chinese subjects. In particular, we sought to establish whether FF and VI systemic exposure in healthy Chinese subjects is associated with clinically relevant changes in PD parameters. The co‐primary study endpoints reflect PD responses relating to recognized class effects of the two drug classes: reduced serum cortisol level (ICSs), and increased Fridericia's corrected QT interval (QTcF) and reduced serum potassium level (LABAs). Secondary endpoints were FF and VI plasma concentrations and derived PK parameters, and safety and tolerability.
Methods
Study Design and Setting
This was a randomized, double‐blind, placebo‐controlled, four‐way crossover, phase I, single‐ and repeat‐dose study conducted at a single center (Peking Union Medical College Hospital [PUMCH], Beijing, China). The study was conducted in accordance with International Conference on Harmonization Good Clinical Practice guidelines, all applicable subject privacy requirements, and the ethical principles outlined in the Declaration of Helsinki. The study design was approved by the ethics committee of PUMCH.
Subjects
Healthy, nonsmoking male or female adults, aged 18–45 years, with a body weight of 50 kg or greater and a body mass index of 19–24 kg/m2, were eligible for inclusion if they lived in mainland China and had Chinese nationality. Individuals who had used tobacco products within 12 months prior to study start or had a smoking history of more than 5 pack‐years were excluded, as were those with significant abnormalities in clinical laboratory, hematology, urinalysis, or 12‐lead electrocardiogram (ECG) assessments at screening or randomization. Other exclusion criteria were the use of oral corticosteroids within 8 weeks, or inhaled, intranasal, or topical steroids within 4 weeks of the screening visit. Written informed consent was obtained from each subject prior to any study‐specific procedures.
Study Treatments
A schematic of the study design is provided in Figure 1. All subjects attended the clinical research center for screening not more than 21 days before first dose. Subjects were randomized to receive FF/VI 50/25, 100/25, or 200/25 μg, or placebo once/daily in the morning, delivered by the Ellipta DPI, for 7 consecutive days. The subjects then received the other three treatments, with each treatment period separated by a 7‐day washout period. During the screening visit, all subjects were trained for correct use of the DPI and were required to demonstrate satisfactory competence in its use prior to randomization.
Figure 1.
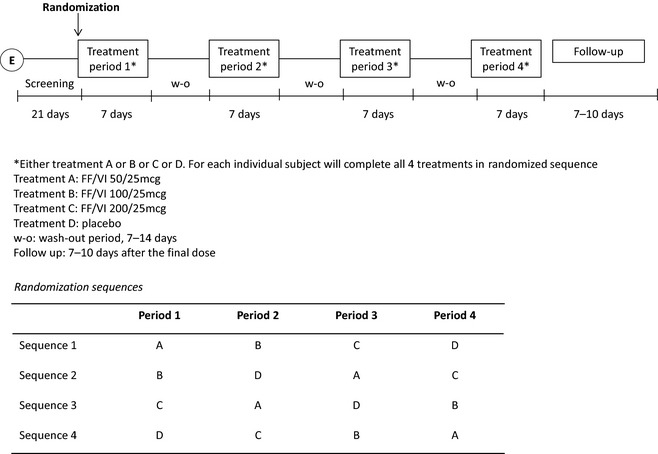
Study design schematic and randomization schedule. FF/VI = fluticasone furoate/vilanterol.
The order in which the treatments were administered was randomized based on a Williams Square design (Figure 1) and was generated by the GSK Statistics Group (R&D, Shanghai, China) prior to study start. Treatments were administered under double‐blind conditions.
Pharmacodynamic Analysis
Blood samples (3 ml/sample) for the measurement of serum cortisol levels were collected before dosing on day 1, and at 1, 2, 4, 6, 8, 10, 12, 16, and 24 hours after dosing on day 7. Twelve‐lead ECG was performed before dosing, and at 15 and 30 minutes, and at 1, 2, and 4 hours after dosing on days 1 and 7. Blood samples (3 ml/sample) for the measurement of potassium levels were collected before dosing, and at 30 minutes and 1, 2, and 4 hours after dosing on days 1 and 7.
Serum samples were assayed for cortisol levels by using a validated analytical method based on protein precipitation, followed by high‐performance liquid chromatography mass spectrometry (HPLC‐MS/MS) analysis. Calibration standards were prepared by using cortisol‐d4 based on the endogenous cortisol level in human serum once the equivalent response ratio of cortisol:cortisol‐d4 was confirmed. This ensured reliable quantitation of total cortisol concentrations in human serum. The lower limit of quantification (LLQ) was 5 ng/ml and the higher limit of quantification (HLQ) was 1000 ng/ml (WuXi AppTec, Shanghai, China). Quality control (QC) samples were prepared at four different concentration levels using either cortisol‐d4 (two lower concentrations) or cortisol (two higher concentrations). Quality control samples were analyzed with each batch of samples against the separately prepared calibration standards. The pre‐set guidelines for acceptable analysis stated that no more than one‐third of the QC sample results should deviate by > 15% from the nominal concentration, and ≥50% of results from each QC sample concentration should be within 15% of the nominal concentration. All predefined run acceptance criteria were met by applicable analytical runs.
Serum potassium concentration was measured by ion‐selective electrode analysis, using an AU5800 system (Beckman Coulter, Brea, CA, USA) in the PUMCH central laboratory (quantitation range 1.0–10.0 mmol/l).
Pharmacokinetic Analysis
Blood PK sampling (3 ml/sample) was performed before dosing on days 1 and 7, and at 5, 15, and 30 minutes, and at 1, 1.5, 2, 4, and 24 hours after dosing. On day 7, additional PK sampling was performed at 6, 8, and 12 hours after dosing.
Fluticasone furoate and VI plasma concentrations were analyzed by using two separately validated analytical methods based on solid‐phase extraction, followed by HPLC‐MS/MS analysis with a LLQ 10 pg/ml for both analytes (PUMCH). For both FF and VI, the LLQ was 10.0 pg/ml and the HLQ was 1000 pg/ml. Calibration standards and QC samples were prepared at three different analyte concentrations and analyzed with each batch of samples. The criteria for acceptable analysis were identical to those for serum cortisol level analysis. The applicable analytical runs met all predefined run acceptance criteria.
Plasma concentrations were listed by subject, period, treatment, and planned relative time, then summarized by treatment and planned sampling time relative to dosing. Noncompartmental PK analysis was conducted (ICON Plc, Marlow, UK) using Phoenix WinNonlin software, version 6.2.1 (Pharsight Corp., St Louis, MO, USA) to estimate PK parameters. Nonquantifiable values were imputed for C max (half of the LLQ) and AUC (half of the lowest observed AUC).
Outcome Measures
Co‐Primary Endpoints
The co‐primary study endpoints were evaluations of systemic PD effects commonly associated with either corticosteroid (reduction in 0–24‐hr weighted mean serum cortisol level on day 7)13 or β‐adrenergic drug classes (increase in 0–4‐hr maximum QTcF, assessed by 12‐lead ECG on days 1 and 7; reduction in 0–4‐hr weighted mean and minimum serum potassium level on days 1 and 7).14
Secondary Endpoints
The secondary study endpoints were FF/VI PK parameters following single and repeat dosing, derived from FF and VI plasma concentrations (Data S1).
Safety and Tolerability
Adverse events, vital signs, 12‐lead ECG measures, and clinical laboratory assessments were monitored from the start of study treatment until end of follow‐up. Changes in safety measures were evaluated as shifts relative to predefined reference ranges.
Statistical Analysis
A total of 16 subjects were randomized with the aim of achieving 8–10 evaluable subjects to meet the regulatory requirement for PK studies in China. No formal sample size calculations were performed.
Three analysis populations were predefined. The “all subjects” population, used in all listings other than those for PK and PD data, comprised all subjects who received at least one dose of study drug. The PK and PD populations, used for the PK and PD analyses, respectively, comprised all subjects in the “all‐subjects” population for whom PK or PD data were obtained and analyzed.
The co‐primary endpoints were analyzed for the PD population using mixed effects models. Logarithmically transformed 0–24‐hour weighted mean serum cortisol level (day 7) data were analyzed with period and treatment as fixed effects and subject as a random effect. Adjusted geometric mean (95% confidence interval [CI]) data were derived from this analysis; subsequently, ratios of adjusted means for each treatment group versus placebo were obtained. For QTcF and serum potassium level, a mixed model with period, treatment, day, and day by treatment as fixed effects and subject as a random effect was used to generate point estimates for each treatment group with associated 95% CIs and to obtain point estimates (90% CI) for differences between each treatment group and placebo. All analyses were performed by using SAS system software, version 9.1 (SAS Institute Inc., Cary, NC, USA). All co‐primary endpoints were summarized by using descriptive statistics and by treatment group and nominal time.
For the mean or median of concentration data, leading nonquantifiables (NQs), trailing NQs, and > 1 consecutive NQs between measurable concentrations were imputed as 0. Single NQ between measurable concentrations, measurable concentrations after > 1 consecutive mid‐profile NQ (defined as any NQ where measurable concentrations exist both before and after that NQ in the profile), and not reported values were set as missing.
Fluticasone furoate accumulation and dose proportionality were analyzed using the estimated PK parameters. The accumulation of log‐transformed AUC(0–4 hr) and C max (expressed as point estimates and 90% CI for the difference between days 7 and 1) was evaluated using a mixed effects model with day as a fixed effect and subject as a random effect, the results of which were subsequently back‐transformed to provide the ratios. Assessment of dose proportionality was performed using a mixed effects model on log‐transformed AUC(0–4 hr) and C max in which loge(dose) was a fixed effect and individual subject intercept was a random effect. An analysis of variance (ANOVA) model, in which dose was fitted as a categorical fixed effect and subject as a random effect, was also used to assess dose proportionality.
Results
Subject Disposition and Demographic Characteristics
The study was completed between November 21, 2012, and June 4, 2013; of the 16 subjects randomized, 15 completed the study. The demographics and disposition of the subjects are summarized in Table 1. All subjects were of Chinese heritage, with a mean age 30.4 years. The PD population included all 16 randomized subjects, including one who withdrew after receiving only placebo treatment; the PK population included the 15 subjects who completed the study.
Table 1.
Demographic Characteristics and Disposition of the 16 Study Subjects at Screening and Baseline
| All Subjects (n=16) | |
|---|---|
| Disposition of subjects | |
| Subjects randomized | 16 (100) |
| All subjects population | 16 (100) |
| PK population | 15 (94) |
| PD population | 16 (100) |
| Subjects completing the study | 15 (94) |
| Subjects withdrawn from the study | 1 (6) |
| Due to an adverse event | 1 (6) |
| Due to a serious adverse event | 0 |
| Demographics | |
| Age (yrs) | 30.4 ± 6.15 |
| Male sex | 15 (94) |
| BMI (kg/m2) | 22.04 ± 1.481 |
| Height (cm) | 169.8 ± 7.40 |
| Weight (kg) | 63.65 ± 7.238 |
| Asian or east Asian heritage | 16 (100) |
BMI = body mass index; PD = pharmacodynamics; PK = pharmacokinetics. Data are no. (%) of patients or mean ± SD values.
Two subjects used concomitant medication during the study. One, who was withdrawn due to experiencing an AE before receiving active treatment, used norfloxacin ear drops. A second subject, who completed the study, used a topical patch containing strong bone paste once on the fourth day of the third treatment period and was subsequently treated with paracetamol for a joint injury. Subjects were administered the study drug dose once/daily at the same subject‐specific time (from 8:00 to 8:42 A.M.).
Pharmacodynamics
0–24‐Hour Weighted Mean Serum Cortisol Level on Day 7
A concentration‐time profile of 0–24 hours geometric mean serum cortisol level on day 7 is provided in Figure 2. In subjects receiving FF/VI 100/25 and 200/25 μg, statistically significant decreases in weighted mean serum cortisol of 15% and 25% versus placebo, respectively, were observed (Table 2 and Figure 3). For subjects receiving FF/VI 50/25 μg for 7 days, there was no evidence of a statistically significant difference compared with placebo (Table 2). The observed circadian rhythm of cortisol levels was consistent with previously reported data.15
Figure 2.
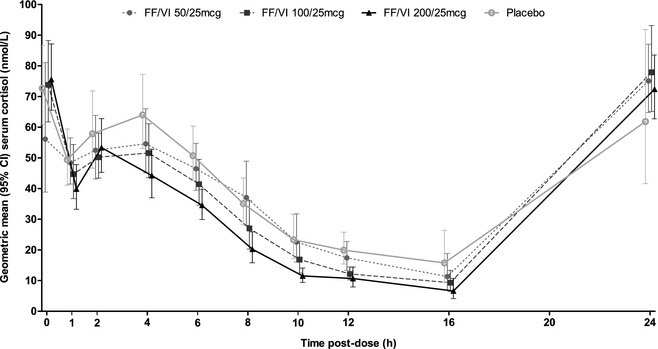
Mean (95% confidence interval [CI]) serum cortisol concentration from 0 to 24 hours on day 7 after administration of fluticasone furoate/vilanterol (FF/VI) 50/25, 100/25 and 200/25 μg (pharmacodynamics population).
Table 2.
Outcomes of Pharmacodynamics Analysis of Co‐Primary Endpoints for all Doses of Fluticasone Furoate/Vilanterol versus Placebo
| Treatment Comparison Specified Active Treatment vs Placebo | Day | Adjusted Mean Treatment | Adjusted Mean Placebo | Differences in Adjusted Means (90% CI) |
|---|---|---|---|---|
| Analysis of serum cortisol weighted mean 0–24 hrs (ng/ml) on day 7 | ||||
| FF/VI 50/25 vs placebo | 7 | 39.455 | 41.260 | 0.956 (0.890, 1.028) |
| FF/VI 100/25 vs placebo | 7 | 34.970 | 41.260 | 0.848 (0.789, 0.911) |
| FF/VI 200/25 vs placebo | 7 | 30.989 | 41.260 | 0.751 (0.699, 0.807) |
| Analysis of maximum QTcF 0–4 hrs (msec) | ||||
| FF/VI 50/25 vs placebo | 1 | 411.0 | 409.6 | 1.42 (‐1.88, 4.72) |
| 7 | 411.9 | 408.2 | 3.69 (0.60, 6.78) | |
| FF/VI 100/25 vs placebo | 1 | 412.8 | 409.6 | 3.25 (‐0.05, 6.55) |
| 7 | 413.5 | 408.2 | 5.25 (2.16, 8.35) | |
| FF/VI 200/25 vs placebo | 1 | 410.0 | 409.6 | 0.46 (‐2.84, 3.76) |
| 7 | 409.8 | 408.2 | 1.53 (‐1.56, 4.62) | |
| Analysis of serum potassium weighted mean 0–4 hrs (mmol/L) | ||||
| FF/VI 50/25 vs placebo | 1 | 3.845 | 3.963 | ‐0.118 (‐0.192, ‐0.043) |
| 7 | 3.888 | 3.975 | ‐0.087 (‐0.159, ‐0.015) | |
| FF/VI 100/25 vs placebo | 1 | 3.939 | 3.963 | ‐0.024 (‐0.099, 0.050) |
| 7 | 3.934 | 3.975 | ‐0.041 (‐0.114, 0.031) | |
| FF/VI 200/25 vs placebo | 1 | 3.905 | 3.963 | ‐0.058 (‐0.133, 0.017) |
| 7 | 3.887 | 3.975 | ‐0.088 (‐0.160, ‐0.015) | |
| Analysis of minimum serum potassium 0–4 h (mmol/L) | ||||
| FF/VI 50/25 vs placebo | 1 | 3.65 | 3.74 | ‐0.087 (‐0.188, 0.014) |
| 7 | 3.75 | 3.83 | ‐0.084 (‐0.164, ‐0.003) | |
| FF/VI 100/25 vs placebo | 1 | 3.79 | 3.74 | 0.049 (‐0.052, 0.150) |
| 7 | 3.82 | 3.83 | ‐0.014 (‐0.094, 0.066) | |
| FF/VI 200/25 vs placebo | 1 | 3.74 | 3.74 | ‐0.001 (‐0.102, 0.100) |
| 7 | 3.76 | 3.83 | ‐0.078 (‐0.158, 0.003) | |
CI = confidence interval; FF/VI = fluticasone furoate/vilanterol; QTcF = QT interval using Fridericia's correction.
Figure 3.
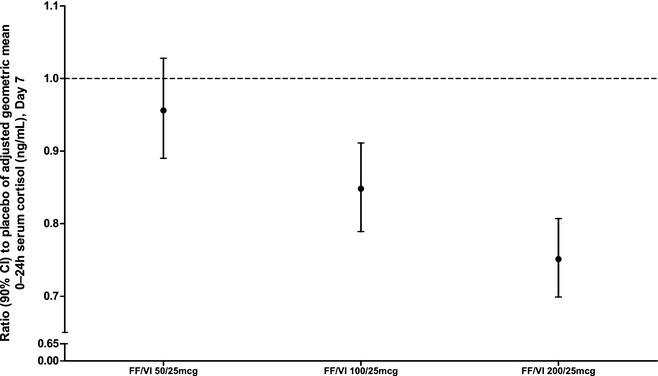
Ratio (90% confidence interval [CI]) to placebo of adjusted geometric mean 0–24‐hour serum cortisol level on day 7 in subjects receiving fluticasone furoate/vilanterol (FF/VI) 50/25, 100/25 and 200/25 μg (pharmacodynamics population).
Maximum 0–4‐Hour QTcF on Days 1 and 7
No statistically significant effects of FF/VI were observed compared with placebo on day 1. On day 7, small but statistically significant increases of 3.69 and 5.25 milliseconds in maximum 0–4‐hour QTcF were observed with FF/VI 50/25 and 100/25 μg, respectively. No effect was seen with FF/VI 200/25 μg. None of the effects on QTcF observed on days 1 or 7 were judged as clinically meaningful.
Weighted Mean and Minimum 0–4‐Hour Serum Potassium Level on Days 1 and 7
On day 1, a small but statistically significant decrease in weighted mean 0–4‐hour serum potassium level of 0.118 mmol/l was seen in subjects receiving the FF/VI 50/25‐μg dose only. After 7 days of dosing, similarly small but statistically significant decreases relative to placebo were observed in subjects receiving FF/VI 50/25 μg (0.087 mmol/l) and 200/25 μg (0.088 mmol/l); no effect was seen in subjects receiving FF/VI 100/25 μg.
For minimum 0–4‐hour serum potassium level, a statistically significant decrease was observed only in subjects receiving FF/VI 50/25 μg on day 7 (0.084 mmol/l).
None of the observed decreases in weighted mean or minimum serum potassium level were considered to be clinically meaningful.
Pharmacokinetics
Plasma concentration–time profiles are provided for FF (Figure 4) and VI (Figure 5), and derived PK parameters, following single‐ and repeat‐dose administration, are summarized in Table 3 (FF) and Table 4 (VI). Following 7 days' repeat‐dose administration, FF plasma concentrations were not quantifiable up to 24 hours after dosing after FF/VI 50/25 μg and were quantifiable in 10 (67%) of 15 subjects after FF/VI 100/25 μg and in all 15 subjects after FF/VI 200/25 μg. VI plasma concentrations were quantifiable up to 2 hours after dosing on day 7 in 13 (87%) of 15 subjects receiving FF/VI 100/25 μg and in 15 (100%) subjects receiving FF/VI 50/25 and 200/25 μg.
Figure 4.
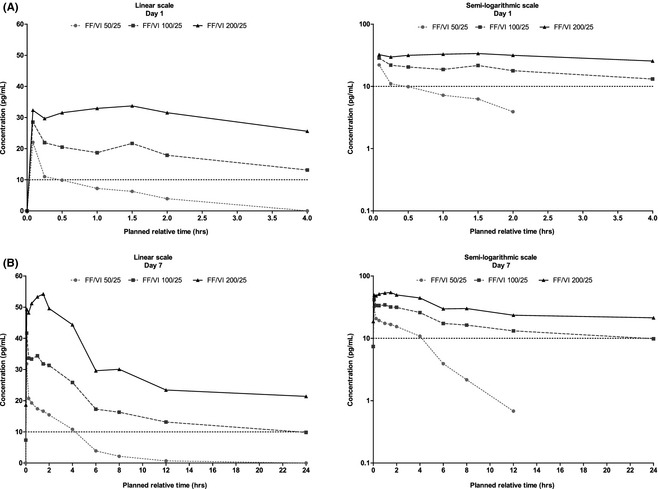
Mean fluticasone furoate plasma concentration–time profiles over 0–4 hours on day 1 (A) and over 0–24 hours on day 7 (B). FF/VI = fluticasone furoate/vilanterol.
Figure 5.
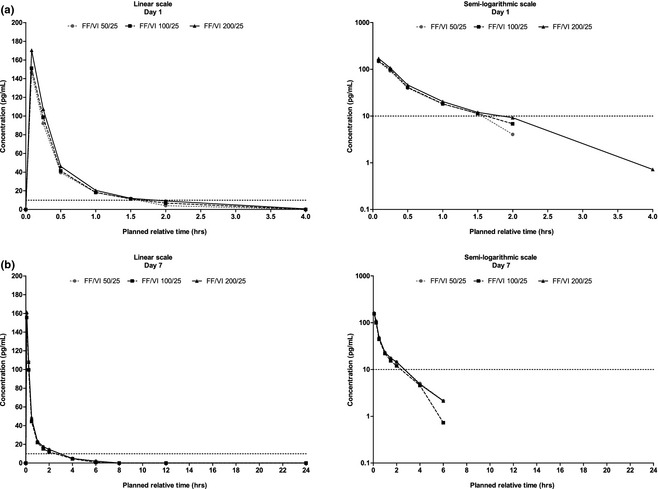
Mean vilanterol plasma concentration‐time profiles over 0–4 hours on day 1 (A) and over 0–24 hours on day 7 (B). FF/VI = fluticasone furoate/vilanterol.
Table 3.
Summary of Evaluated Pharmacokinetic Parameters for Fluticasone Furoate Following Single (Day 1) and Repeat‐Dose (Day 7) Administration of Fluticasone Furoate/Vilanterol (Pharmacokinetics Population)
| Parameter | FF/VI dose | Days | n | n | n* | Geometric Mean (CVb%) | 95% CI of Geometric Mean |
|---|---|---|---|---|---|---|---|
| AUCss (pg× hr/ml) | 50/25 | 7 | 15 | 15 | 15 | NA | NA |
| 100/25 | 7 | 15 | 14 | 4 | 364 (43.0) | (287, 461) | |
| 200/25 | 7 | 15 | 15 | 0 | 691 (19.8) | (620, 770) | |
| AUC(0–4 hr) (pg× hr/ml) | 50/25 | 1 | 15 | 14 | 14 | NA | NA |
| 50/25 | 7 | 15 | 15 | 3 | 55.8 (45.3) | (43.9, 70.9) | |
| 100/25 | 1 | 15 | 15 | 3 | 61.7 (50.4) | (47.4, 80.3) | |
| 100/25 | 7 | 15 | 14 | 0 | 129 (20.4) | (115, 145) | |
| 200/25 | 1 | 15 | 15 | 0 | 118 (19.6) | (106, 131) | |
| 200/25 | 7 | 15 | 15 | 0 | 193 (19.2) | (173, 214) | |
| C max (pg/ml) | 50/25 | 1 | 15 | 14 | 0 | 22.6 (32.5) | (18.8, 27.1) |
| 50/25 | 7 | 15 | 15 | 0 | 30.6 (29.0) | (26.2, 35.8) | |
| 100/25 | 1 | 15 | 15 | 0 | 26.8 (42.1) | (21.4, 33.5) | |
| 100/25 | 7 | 15 | 14 | 0 | 43.6 (22.9) | (38.2, 49.6) | |
| 200/25 | 1 | 15 | 15 | 0 | 36.4 (19.6) | (32.7, 40.5) | |
| 200/25 | 7 | 15 | 15 | 0 | 55.5 (21.2) | (49.4, 62.3) | |
| Css‐min (pg/ml) | 50/25 | 7 | 15 | 15 | 0 | 11.3 (9.5) | (10.8, 11.9) |
| 100/25 | 7 | 15 | 14 | 0 | 13.0 (15.4) | (11.9, 14.2) | |
| 200/25 | 7 | 15 | 15 | 0 | 20.4 (21.7) | (18.1, 23.0) | |
| RCmax | 50/25 | 7 | 15 | 14 | 0 | 1.41 (30.4) | (1.18, 1.67) |
| 100/25 | 7 | 15 | 14 | 0 | 1.52 (19.1) | (1.37, 1.70) | |
| 200/25 | 7 | 15 | 15 | 0 | 1.53 (15.0) | (1.40, 1.66) | |
| Ro | 50/25 | 7 | 15 | 0 | 0 | NA | NA |
| 100/25 | 7 | 15 | 12 | 0 | 1.72 (14.7) | (1.57, 1.89) | |
| 200/25 | 7 | 15 | 15 | 0 | 1.63 (15.5) | (1.50, 1.78) | |
| T max (hrs)a | 50/25 | 1 | 15 | 14 | 0 | 0.08 (0.08–0.08) | – |
| 50/25 | 7 | 15 | 15 | 0 | 0.08 (0.08–0.08) | – | |
| 100/25 | 1 | 15 | 15 | 0 | 0.08 (0.08–1.50) | – | |
| 100/25 | 7 | 15 | 14 | 0 | 0.08 (0.08–0.08) | – | |
| 200/25 | 1 | 15 | 15 | 0 | 1.00 (0.08–1.50) | – | |
| 200/25 | 7 | 15 | 15 | 0 | 1.00 (0.08–1.50) | – |
AUCss = area under the concentration‐time curve at steady state; AUC(0–4 hr) = AUC from 0 to 4 hrs; CI = confidence interval; C max = maximum observed concentration; Css‐min = minimum observed concentration at steady state; CVb%, between‐subject coefficient of variation; FF/VI = fluticasone furoate/vilanterol; NA = not applicable; NC = not calculated; NQ = not quantifiable; NR = not reported; RCmax = maximum observed concentration ratio; Ro = observed accumulation ratio; T max = time of occurrence of C max.
n = number of subjects with non‐missing observations (including imputed NC values).
n* = number of subjects for whom parameter cannot be derived because of NQ concentrations.
CVb% = 100 × sqrt(exp(logSD2)−1.
If all concentration values for one subject were NQ or NR, the AUC(0–4 hr) for this subject was set as missing. If some concentration values were NQ or NR, the AUC(0–4 hr) was calculated when the last quantifiable time point was > 4; otherwise, the AUC(0–4 hr) for this subject was reported as NC.
For C max and AUC, NCs were imputed prior to derivation of summary statistics when there were a small proportion (< 33%) of subjects with NCs. AUC imputed with ½ lowest observed AUC, C max imputed with ½ LLQ (5 pg/ml). When there was a large proportion (100% in this study) of subjects with NCs, the summary of statistics were set as NA.
Data are median (range).
Table 4.
Summary of Evaluated Pharmacokinetic Parameters for Vilanterol Following Single (Day 1) and Repeat‐Dose (Day 7) Administration of Fluticasone Furoate/Vilanterol (Pharmacokinetics Population)
| Parameter | FF/VI dose | Days | n | n | n* | Geometric Mean (CVb%) | 95% CI of Geometric Mean |
|---|---|---|---|---|---|---|---|
| AUC(0–t) (pg× hr/ml) | 50/25 | 1 | 15 | 15 | 0 | 61.7 (25.0) | (53.8, 70.7) |
| 50/25 | 7 | 15 | 15 | 0 | 94.5 (36.5) | (77.7, 115) | |
| 100/25 | 1 | 15 | 15 | 0 | 66.0 (23.1) | (58.2, 74.9) | |
| 100/25 | 7 | 15 | 15 | 0 | 82.9 (39.1) | (67.3, 102) | |
| 200/25 | 1 | 15 | 15 | 0 | 71.8 (42.6) | (57.2, 90.0) | |
| 200/25 | 7 | 15 | 15 | 0 | 91.9 (39.0) | (74.6, 113) | |
| AUC(0–1.5 hr) (pg× hr/ml) | 50/25 | 1 | 15 | 15 | 3 | 53.0 (44.3) | (41.9, 67.0) |
| 50/25 | 7 | 15 | 15 | 0 | 73.0 (20.0) | (65.4, 81.4) | |
| 100/25 | 1 | 15 | 15 | 3 | 55.1 (45.7) | (43.3, 70.1) | |
| 100/25 | 7 | 15 | 15 | 1 | 66.4 (31.6) | (56.0, 78.8) | |
| 200/25 | 1 | 15 | 15 | 3 | 61.0 (52.5) | (46.5, 80.2) | |
| 200/25 | 7 | 15 | 15 | 0 | 72.6 (23.5) | (63.9, 82.6) | |
| C max (pg/ml) | 50/25 | 1 | 15 | 15 | 0 | 144 (19.8) | (129, 160) |
| 50/25 | 7 | 15 | 15 | 0 | 155 (24.7) | (136, 178) | |
| 100/25 | 1 | 15 | 15 | 0 | 147 (26.3) | (127, 169) | |
| 100/25 | 7 | 15 | 15 | 0 | 151 (25.3) | (132, 174) | |
| 200/25 | 1 | 15 | 15 | 0 | 155 (36.7) | (127, 188) | |
| 200/25 | 7 | 15 | 15 | 0 | 157 (25.7) | (136, 180) | |
| T max(hrs)a | 50/25 | 1 | 15 | 15 | 0 | 0.08 (0.08–0.08) | – |
| 50/25 | 7 | 15 | 15 | 0 | 0.08 (0.08–0.08) | – | |
| 100/25 | 1 | 15 | 15 | 0 | 0.08 (0.08–0.08) | – | |
| 100/25 | 7 | 15 | 15 | 0 | 0.08 (0.08–0.08) | – | |
| 200/25 | 1 | 15 | 15 | 0 | 0.08 (0.08–0.25) | – | |
| 200/25 | 7 | 15 | 15 | 0 | 0.08 (0.08–0.08) | – |
AUC(0‐t) = area under the concentration‐time curve from time zero to last quantifiable time point; AUC(0–1.5 hr) = AUC from 0 to 1.5 hrs; CI = confidence interval; C max = maximum observed concentration; FF = fluticasone furoate; NA = not applicable; NC = not calculated; NQ = not quantifiable; NR = not reported; T max = time of occurrence of C max; VI = vilanterol.
CVb% = 100 × sqrt(exp(logSD2)−1.
n = number of subjects with non‐missing observations (including imputed NC values).
n* = number of subjects for whom parameter cannot be derived because of NQ concentrations.
If all concentration values for one subject were NQ or NR, the AUC(0–1.5 hr) for this subject was set as missing. If some concentration values were NQ or NR, the AUC(0–1.5 hr) was calculated when the last quantifiable time point was > 1.5, otherwise, the AUC(0–1.5 hr) for this subject was reported as NC.
For C max and AUC, NCs were imputed prior to derivation of summary statistics when there were a small proportion (< 33%) of subjects with NCs. AUC imputed with ½ lowest observed AUC, C max imputed with ½ LLQ (5 pg/ml). When there was a large proportion (100% in this study) of subjects with NCs, the summary of statistics were set as NA.
Data are median (range).
Accumulation of FF following repeat‐dose administration of FF/VI 100/25 μg on day 7 versus day 1 was estimated at 54% (90% CI 40–68%) based on C max and 71% (59–85%) based on AUC(0–4 hr). Fluticasone furoate dose proportionality assessments suggested less than dose proportional PK accumulation of FF (Table S1). The VI PK profiles were similar for all three FF/VI doses (Figure 5) with no evidence of any differences in VI exposure (AUC(0–t), AUC(0–1.5 hr) and C max; Table 4).
Six day 1 PK samples from five subjects were mislabelled because of operational error and were therefore excluded from the PK analyses. As these samples represented only 0.7% of the total 903 PK samples collected on days 1 and 7, this was not considered to meaningfully affect the overall outcome of the PK analysis.
Safety and Tolerability
In total, 13 (81%) of the 16 subjects experienced ≥1 AE during the study (Table 5). The overall number of AEs in the FF/VI 100/25 and 200/25‐μg groups (5 and 3, respectively) was similar to that experienced in the placebo group (n=5). Slightly more AEs (8) were reported in the FF/VI 50/25‐μg group. The most frequently reported AEs were decreased serum potassium level (4, all in the 50/25‐μg group) and oral ulcers (4, one in each treatment group). All reported AEs were of mild intensity and were deemed to be treatment related by the investigator, with the exception of one occurrence of joint injury. One subject withdrew on day 3 of period 1 due to otitis media and tympanic membrane perforation occurring 2 days before first dose administration and worsening after two doses of placebo. This was the only female to be randomized into the study.
Table 5.
Summary of All Adverse Events Occurring During the Study
| FF/VI 50/25 μg (n=15) | FF/VI 100/25 μg (n=15) | FF/VI 200/25 μg (n=15) | Placebo (n=16) | Total (n=16) | |
|---|---|---|---|---|---|
| Adverse events by subject | |||||
| Subjects with any adverse event | 8 (53) | 5 (33) | 3 (20) | 5 (31) | 13 (81) |
| All adverse events | |||||
| Laboratory results and vital signs | 4 (27) | 1 (7) | 1 (7) | 2 (13) | 7 (44) |
| Blood potassium level decreased | 4 (27) | 0 | 0 | 0 | 4 (25) |
| White blood cell count decreased | 0 | 1 (7) | 1 (7) | 0 | 2 (13) |
| Blood glucose level increased | 0 | 0 | 0 | 1 (6) | 1 (6) |
| Hemoglobin level decreased | 0 | 0 | 1 (7) | 0 | 1 (6) |
| Heart rate decreased | 0 | 0 | 0 | 1 (6) | 1 (6) |
| Infections | 2 (13) | 1 (7) | 1 (7) | 2 (13) | 5 (31) |
| Upper respiratory tract infection | 1 (7) | 0 | 1 (7) | 1 (6) | 3 (19) |
| Pharyngitis | 1 (7) | 1 (7) | 0 | 0 | 2 (13) |
| Otitis media | 0 | 0 | 0 | 1 (6) | 1 (6) |
| Gastrointestinal disorders | 1 (7) | 1 (7) | 1 (7) | 1 (6) | 4 (25) |
| Mouth ulceration | 1 (7) | 1 (7) | 1 (7) | 1 (6) | 4 (25) |
| Injury, poisoning, and procedural complications | 0 | 0 | 0 | 1 (6) | 1 (6) |
| Joint injury | 0 | 0 | 0 | 1 (6) | 1 (6) |
| Musculoskeletal and connective tissue disorders | 0 | 1 (7) | 0 | 1 (6) | 1 (6) |
| Musculoskeletal pain | 0 | 1 (7) | 0 | 0 | 1 (6) |
| Neck pain | 0 | 0 | 0 | 1 (6) | 1 (6) |
| Psychiatric disorders | 1 (7) | 1 (7) | 0 | 0 | 1 (6) |
| Tic | 1 (7) | 1 (7) | 0 | 0 | 1 (6) |
FF/VI = fluticasone furoate/vilanterol.
Data are no. (%) of patients.
All adverse events were of mild intensity. All adverse events except for joint injury were considered by the investigators to be treatment related.
No clinically significant or dose‐related changes were found in blood pressure records, hematology, or clinical chemistry assessments. Changes in ECG values from baseline to each time point were similar across treatment groups. Seven subjects had ECG values of potential clinical importance during the study, defined as QRS interval values of < 75 and > 110 milliseconds and PR interval values of < 110 and > 220 milliseconds, although none were reported as AEs or considered to be clinically significant.
Discussion
This study investigated the PD and PK of FF/VI combination therapy in mainland Chinese healthy subjects. The co‐primary PD endpoints were selected on the basis of established class effects of ICS (reduction in serum cortisol level) and LABAs (increase in QTcF and reduced serum potassium level). The analysis of 0–24‐hour weighted mean serum cortisol level following 7‐day repeat dosing of FF/VI showed that although there was no statistically significant effect of FF/VI 50/25 μg relative to placebo, FF/VI 100/25 μg and 200/25 μg were associated with statistically significant, dose‐related decreases in healthy Chinese subjects. As for QTcF and serum potassium levels, some statistically significant effects were seen with the active treatments relative to placebo on day 1 and/or day 7. However, these small effects were not considered clinically significant. In addition, other β‐agonist–associated systemic effects, such as increases in QTcF and decreases in serum potassium level, were not particularly marked and not seen consistently in all FF/VI treatment groups, despite containing the same VI 25‐μg dose.
The FF systemic exposure observed in this study was around 50% greater than that in healthy Caucasian subjects, consistent with previous findings in East Asian healthy subjects.10 Higher systemic exposure in Chinese subjects may be attributed to the differences in lung FF absorption across populations. An increased mean absorption time (estimated as the difference between mean residence time following inhaled and intravenous dosing) relative to Caucasians has been observed in East Asian subjects, potentially providing an explanation for the 20–50% higher FF bioavailability.10 The extent of FF accumulation and the less than dose‐proportional increase of exposures reported in this study are consistent with previously published data in healthy East Asian and Caucasian subjects.5, 10, 11 VI exposure was similar across the range of coadministered FF/VI doses. This was consistent with previously reported findings in Caucasian subjects, where similar VI exposure equivalence was seen when administered in combination with FF doses of 200–800 μg.5
Similar to other ICS,16 FF at supratherapeutic doses is associated with class‐related effects on the HPA axis.7, 11 The relationship between FF systemic exposure and cortisol suppression has been characterized: an FF AUC0–24 hour of 1000 pg× hour/ml is predicted to reduce 24‐hour serum cortisol level by 20% in a population comprising mostly Caucasian healthy subjects.17 Based on these findings, therapeutic FF doses are not expected to produce clinically meaningful suppression of the HPA axis. Furthermore, a 6‐week study of the effect of FF/VI 100/25 and 200/25 μg on the HPA axis in patients with persistent asthma found no evidence of significant cortisol level suppression.8
In a previous phase I study in healthy Japanese subjects, systemic exposure of FF 200 μg after 7 days of repeat dosing (geometric mean AUC0–τ 744 pg/ml; C max 62 pg/ml) was similar to that seen in the present study, and statistically significant serum cortisol level suppression of 32% compared with placebo was observed after 7 days of repeat FF 200 μg dosing.11 Taken together, our observations and those of another study11 suggest that the observed serum cortisol level suppression may be explained by a relatively higher sensitivity to FF's corticosteroid effect in healthy East Asian subjects versus patients with asthma or COPD.
East Asian patients with COPD or asthma receiving therapeutic FF doses have not displayed cortisol level suppression in previous studies. A 24‐week study in patients with COPD from China, Taiwan, South Korea, and the Philippines receiving FF/VI 50/25, 100/25, or 200/25 μg did not show statistically significant differences in serum cortisol levels,18 and a pooled data analysis from global studies in patients with asthma receiving FF/VI found no difference between patients from Japan and Korea and those from the rest of the world in change from baseline urinary cortisol level measurements.19 Also, in global phase III studies of up to 52 weeks' duration, once‐daily FF/VI administered at therapeutic doses did not result in clinically meaningful reductions in serum or urinary cortisol levels in patients with asthma or COPD, including those from East Asia.2, 20, 21, 22 All these findings suggest that FF does not affect the serum cortisol level when administered to patients at therapeutic doses, including patients of Asian origin with asthma or COPD.
Ethnic differences in FF (intravenous and inhaled) PK have been investigated in an open‐label, randomized, 2‐way crossover study of healthy Caucasian, Chinese, Japanese, and Korean subjects.10 No difference in FF (intravenous) PK parameters was observed between Asian and Caucasian subjects. Fluticasone furoate is primarily metabolized by cytochrome P450 (CYP) 3A4, and, indeed, CYP3A4 activity was reported to be similar across all ethnic groups.
A possible explanation for the PD difference between healthy volunteers and patients with asthma or COPD is differential PK exposure. Plasma exposure to ICS has previously been observed to be markedly greater in healthy subjects versus patients with asthma23 or COPD.24 Factors that affect the systemic bioavailability of ICS in patients with chronic respiratory disorders are yet to be identified. It has been hypothesized that airflow obstruction associated with asthma and COPD may contribute to increased proximal airway deposition of FP and therefore lower systemic FP level by increasing mucociliary clearance and elimination.25 PK data from patients with asthma or COPD and healthy subjects who received FF and VI support such an assumption, where markedly lower FF C max and AUC(0–24 hr) were observed in patients with asthma or COPD compared with healthy subjects.26
No cortisol‐related safety concerns associated with FF/VI were identified in this study, nor have any been reported in longer‐term phase III studies of the same FF/VI doses given to patients with asthma or COPD of East Asian (including Japanese) heritage.18, 27 In the aforementioned pooled analysis,19 no differences in drug‐ or class‐related AEs were observed among ethnicities. Our study did not show any clinically meaningful impairment on QTcF and other ECG measures, and no clinically relevant AEs were observed in serum potassium level, other clinical chemistry measures, hematology outcomes, or vital signs.
We employed a balanced crossover design, including a placebo arm, in which all subjects received all treatments, to enable the PD effects of three strengths of FF plus VI to be directly compared with placebo, with minimal confounding due to individual variation. The study was completed by 94% of subjects. The main limitation of the study was that we were unable to directly test the hypothesis of differential systemic exposure in healthy subjects versus patients with asthma or COPD in the ethnic population of interest. Further limitations were the lack of an ethnic comparator group and the relatively short‐term treatment period.
Conclusion
Minimal and non–clinically relevant β‐adrenergic PD effects were observed in healthy Chinese subjects receiving FF/VI 50/25–200/25 μg. Fluticasone furoate dose‐dependent reductions in serum cortisol level of up to 25% were observed after administration of FF/VI 100/25 and 200/25 μg. However, no evidence of such PD responses has been reported in previous studies of the same therapeutic FF/VI doses in patients with asthma or COPD. The FF/VI safety profile in healthy Chinese subjects demonstrated that this treatment was well tolerated at doses ranging from 50/25 to 200/25 μg.
Supporting information
Data S1. Assessment of Pharmacokinetic (PK) Parameters.
Table S1. Summary of Findings of Assessment of Dose Proportionality of FF PK Parameters Using a Power Model and an ANOVA Model (PK Population)
Acknowledgments
We thank Peking Union Medical College Hospital for their PK and PD bioanalytic services and WuXi AppTech Co., Ltd. (Shanghai, China) for their serum cortisol bioanalytic services. Editorial support in the form of development of the draft outline and manuscript first draft in consultation with the authors, editorial suggestions to draft versions of this paper, assembling tables and figures, collating author comments, copyediting, fact checking, referencing, and graphic services was provided by Ian Grieve, Ph.D., at Gardiner‐Caldwell Communications (Macclesfield, UK) and was funded by GSK.
This study was funded by GSK study no. HZA115199.
ClinicalTrials.gov identifier: NCT01711463.
References
- 1. Agusti A, de Teresa L, De Backer W, et al. A comparison of the efficacy and safety of once‐daily fluticasone furoate/vilanterol with fluticasone propionate/salmeterol in COPD patients. Eur Respir J 2014;43:763–72. [DOI] [PubMed] [Google Scholar]
- 2. Dransfield MT, Feldman G, Korenblat P, et al. Efficacy and safety of once‐daily fluticasone furoate/vilanterol (100/25 μg) versus twice‐daily fluticasone propionate/salmeterol (250/50 μg) in COPD patients. Respir Med 2014;108:1171–9. [DOI] [PubMed] [Google Scholar]
- 3. Woodcock A, Bleecker ER, Lötvall J, et al. Efficacy and safety of fluticasone furoate/vilanterol compared with fluticasone propionate/salmeterol combination in adult and adolescent patients with persistent asthma: a randomized trial. Chest 2013;144:1222–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Global Initiative for Asthma (GINA) . Global strategy for asthma management and prevention. 2014. Available from http://www.ginasthma.org/. Accessed March 16, 2015.
- 5. Allen A, Apoux L, Bal J, et al. The pharmacokinetics of fluticasone furoate (FF) and vilanterol (VI) trifenatate following single inhaled administration in combination and intravenous administration of individual components in healthy subjects. J Bioequiv Availab 2013;5:165–73. [Google Scholar]
- 6. Kempsford R, Allen A, Kelly K, Saggu P, Crim C. A repeat‐dose thorough QT study of inhaled fluticasone furoate (FF)/vilanterol (VI) combination in healthy subjects. Br J Clin Pharmacol 2014;77:466–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kempsford R, Allen A, Bareille P, Hamilton M, Cheesbrough A. The pharmacodynamics, pharmacokinetics, safety and tolerability of inhaled fluticasone furoate (FF) and vilanterol (VI) administered alone or simultaneously as FF/VI. Clin Pharmacol Drug Dev 2014;4:2–11. [DOI] [PubMed] [Google Scholar]
- 8. Allen A, Schenkenberger I, Trivedi R, et al. Inhaled fluticasone furoate/vilanterol does not affect hypothalamic‐pituitary‐adrenal axis function in adolescent and adult asthma: randomised, double‐blind, placebo‐controlled study. Clin Respir J 2013;7:397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huang S‐M, Temple R. Is this the drug or dose for you? Impact and consideration of ethnic factors in global drug development, regulatory review and clinical practice. Clin Pharmacol Ther 2008;84:287–94. [DOI] [PubMed] [Google Scholar]
- 10. Allen A, Bal J, Cheesbrough A, Hamilton M, Kempsford R. Pharmacokinetics and pharmacodynamics of intravenous and inhaled fluticasone furoate in healthy Caucasian and East Asian subjects. Br J Clin Pharmacol 2014;77:808–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nakahara N, Wakamatsu A, Kempsford R, et al. The safety, pharmacokinetics and pharmacodynamics of a combination of fluticasone furoate and vilanterol in healthy Japanese subjects. Int J Clin Pharmacol Ther 2013;51:660–72. [DOI] [PubMed] [Google Scholar]
- 12. GlaxoSmithKline . Relvar Ellipta 184/22mcg Summary of Product Characteristics (SPC). Available from http://public.gsk.co.uk/content/dam/Health/en_GB/DAM%20Content/Documents/Respiratory/Relvar/relvar_ellipta_spc_184mg.pdf. Accessed November 14, 2014.
- 13. Derendorf H, Hochhaus G, Meibohm B, Möllmann H, Barth J. Pharmacokinetics and pharmacodynamics of inhaled corticosteroids. J Allergy Clin Immunol 1998;101:S440–6. [DOI] [PubMed] [Google Scholar]
- 14. Taylor DR, Wilkins GT, Herbison GP, Flannery EM. Interaction between corticosteroid and beta‐agonist drugs: biochemical and cardiovascular effects in normal subjects. Chest 1992;102:519–24. [DOI] [PubMed] [Google Scholar]
- 15. Zhao Z, Xiea Y, Fua Y‐R, et al. Circadian rhythm characteristics of serum cortisol and dehydroepiandrosterone sulphate in healthy Chinese men aged 30 to 60 years. A cross‐sectional study. Steroids 2003;68:133–8. [DOI] [PubMed] [Google Scholar]
- 16. Dluhy RG. Clinical relevance of inhaled corticosteroids and HPA axis suppression. J Allergy Clin Immunol 1998;101:S447–50. [DOI] [PubMed] [Google Scholar]
- 17. Allen A. The relationship between fluticasone furoate systemic exposure and cortisol suppression. Clin Pharmacokinet 2013;52:885–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zheng JP, de Guia T, Wang‐Jairaj J, et al. The efficacy and safety of inhaled fluticasone furoate (FF)/vilanterol (VI) in Asian patients with COPD (abstract). Respirology 2013;18(Suppl 4):31. [Google Scholar]
- 19. Gross A, Goldfrad C, Hozawa S, et al. Ethnic sensitivity assessment of fluticasone furoate (FF)/vilanterol (VI) in asthma patients in Japan and Korea: a pre‐specified subgroup analysis (abstract). Respirology 2013;18(Suppl 4):157. [Google Scholar]
- 20. Busse WW, O'Byrne PM, Bleecker ER, et al. Safety and tolerability of the novel inhaled corticosteroid fluticasone furoate in combination with the beta2 agonist vilanterol administered once daily for 52 weeks in patients >=12 years old with asthma: a randomised trial. Thorax 2013;68:513–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kerwin EM, Scott‐Wilson C, Sanford L, et al. A randomised trial of fluticasone furoate/vilanterol (50/25 μg; 100/25 μg) on lung function in COPD. Respir Med 2013;107:560–9. [DOI] [PubMed] [Google Scholar]
- 22. Martinez FJ, Boscia J, Feldman G, et al. Fluticasone furoate/vilanterol (100/25; 200/25 μg) improves lung function in COPD: a randomised trial. Respir Med 2013;107:550–9. [DOI] [PubMed] [Google Scholar]
- 23. Brutsche MH, Carlen Brutsche I, Munawer M, et al. Comparison of pharmacokinetics and systemic effects of inhaled fluticasone propionate in patients with asthma and healthy volunteers: a randomised crossover study. Lancet 2000;356:556–61. [DOI] [PubMed] [Google Scholar]
- 24. Singh SD, Whale C, Houghton N, Daley‐Yates P, Kirby SM, Woodcock AA. Pharmacokinetics and systemic effects of inhaled fluticasone propionate in chronic obstructive pulmonary disease. Br J Clin Pharmacol 2003;55:375–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Harrison TW, Tattersfield AE. Plasma concentrations of fluticasone propionate and budesonide following inhalation from dry powder inhalers by healthy and asthmatic subjects. Thorax 2003;58:258–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. GSK . Data on file. Brentford, UK; 2014. [Google Scholar]
- 27. Muraki M, Soutome T, Hashimoto K, Tohda Y. Long‐term study of fluticasone furoate/vilanterol combination (FF/VI) and FF alone in Japanese adult patients with bronchial asthma. Allergol Immunol 2013;20:110–25. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Assessment of Pharmacokinetic (PK) Parameters.
Table S1. Summary of Findings of Assessment of Dose Proportionality of FF PK Parameters Using a Power Model and an ANOVA Model (PK Population)