

Abstract
Molecular crosstalk between cancer cells and fibroblasts has been an emerging hot issue in understanding carcinogenesis. As oral submucous fibrosis (OSF) is an inflammatory fibrotic disease that can potentially transform into squamous cell carcinoma, OSF has been considered to be an appropriate model for studying the role of fibroblasts during early stage carcinogenesis. In this sense, this study aims at investigating whether areca nut (AN)‐exposed fibroblasts cause DNA damage of epithelial cells. For this study, immortalized hNOF (hTERT‐hNOF) was used. We found that the levels of GRO‐α, IL‐6 and IL‐8 increased in AN‐exposed fibroblasts. Cytokine secretion was reduced by antioxidants in AN‐exposed fibroblasts. Increase in DNA double strand breaks (DSB) and 8‐oxoG FITC‐conjugate was observed in immortalized human oral keratinocytes (IHOK) after the treatment of cytokines or a conditioned medium derived from AN‐exposed fibroblasts. Cytokine expression and DNA damage were also detected in OSF tissues. The DNA damage was reduced by neutralizing cytokines or antioxidant treatment. Generation of reactive oxygen species (ROS) and DNA damage response, triggered by cytokines, were abolished when NADPH oxidase (NOX) 1 and 4 were silenced in IHOK, indicating that cytokine‐triggered DNA damage was caused by ROS generation through NOX1 and NOX4. Taken together, this study provided strong evidence that blocking ROS generation might be a rewarding approach for cancer prevention and intervention in OSF.
Keywords: fibroblasts, oral submucous fibrosis, carcinogenesis, cytokines, DNA damage
Short abstract
What's new?
Fibroblasts in the tumor microenvironment influence tumor initiation and growth and are of particular interest in oral submucous fibrosis (OSF), a progressive fibrotic disease of malignant potential. This study shows that the release of tumor‐promoting cytokines by fibroblasts exposed to areca nut, the primary cause of OSF, induce DNA damage in oral keratinocytes. The findings suggest that fibroblasts indirectly promote epithelial transformation in OSF by secreting cytokines, whereby DNA damage of epithelial cells is inflicted by reactive oxygen species generated via NADPH oxidases. These insights could inform the development of new therapeutic approaches for OSF.
Abbreviations
- AN
areca nut
- CAF
cancer associated fibroblasts
- DSB
double strand break
- hNOF
normal human gingival fibroblasts
- hTERT‐hNOF
immortalized normal human gingival fibroblasts
- IHOK
immortalized human oral keratinocytes
- NOM
Normal oral mucosa
- NOX
NADPH oxidase
- OSCC
oral squamous cell carcinoma
- OSF
oral submucous fibrosis
- ROS
reactive oxygen species
Tissue microenvironments have been focused on playing crucial roles in carcinogenesis. In particular, cancer associated fibroblasts (CAF) have emerged as a prominent modifier of cancer initiation and progression.1, 2 CAF produces inflammatory cytokines and chemokines, and these secretory products facilitate communication between CAFs and cancer cells, resulting in cancer progression.1, 2, 3 Hence, inflammatory reaction has emerged as one of the main causes of cancer development.4, 5 Even though the causal relationship between cancer and surrounding inflammation has not been well elucidated, inflammation has been accepted as an indispensable causal factor for development of various tumors.6, 7 A large number of genes related to inflammations are associated with cancer development,8 accumulating evidence suggests that various inflammatory cytokines such as TNF‐α, IL‐6, IL‐8 and GRO‐α may play a critical role in carcinogenesis.9, 10, 11
Oral submucous fibrosis (OSF) is an inflammatory fibrotic disease that mainly occurs in Southeast Asian people.12 OSF has received great attention because it can potentially transform into oral squamous cell carcinoma (OSCC); OSCC occurs in 7–13% of OSF.13 The main cause of OSF is the consumption of Areca Nut (AN).13 AN was categorized as a Grade 1 carcinogen according to the International Agency for Research on Cancer (IARC).14 Considering that OSF in its initial stage is characterized by chronic inflammation and epithelial atrophy accompanied by fibrosis, we assumed that OSF deserved special consideration as a unique model for studying the role of fibroblasts at the early stage of carcinogenesis. Moreover, the localization of cytokines such as IL‐6, TGF‐β and bFGF in OSF supported our assumptions.15
Thus, this study aimed to search for evidence that fibroblasts are actively involved in early‐stage carcinogenesis by secreting cytokines and subsequently modulating DNA damage in epithelial cells. To achieve this goal, we first investigated whether cytokines are released from gingival fibroblasts upon AN exposure and then evaluated whether these cytokines induce DNA damage in oral keratinocytes using in vitro analyses and human OSF samples. In this study, we found that gingival fibroblasts exposed to AN produced several cytokines, eventuating in DNA damage through generation of reactive oxygen species (ROS) in immortalized oral keratinocytes. This study provided an insight into understanding the role of fibroblasts in early‐stage carcinogenesis and will likely contribute to development of novel therapeutic modalities to prevent malignant transformation of oral potentially malignant lesions.
Material and Methods
Human tissue samples
OSF tissues were obtained from the SriLankan patients who had AN‐chewing habit. Normal oral mucosa (NOM) tissues for immunohistochemical staining were also obtained from SriLankan outpatients who had no AN‐chewing habit. Information about tobacco or cigarette smoking was not investigated. The tissues were ethically approved by the IRB of the Faculty of Dental Sciences, University of Peradeniya, Sri Lanka (FDS‐RERC/2009/03).
Cell culture
For conducting this study, we used immortalized human gingival fibroblasts obtained by hTERT transfection (hTERT‐hNOF), which were previously described.16 For evaluating whether hTERT‐hNOF were appropriate for this study, we obtained human gingival fibroblasts (hNOF) by explant culture taken from a healthy individual. Normal human epidermal keratinocytes (HEK) were also obtained for this study. Generation of hNOF and HEK was ethically approved by the Institutional Review Board (IRB) of the Yonsei Dental Hospital, Yonsei University Health System, Seoul, Republic of Korea (IRB‐2‐2009‐0002). Both hNOF and HEK were used below the 10th passage. For evaluating epithelial cell damage, immortalized human oral keratinocytes (IHOK) obtained by human papilloma virus (HPV) 16 E6/E7 transfection were used, which has been previously described.17 YD10B OSCC cell line18 was also used to compare cytotoxicity with IHOK. The culture media used for each cell line were shown in Table 1, Supporting Information. All cell lines were maintained in a humidified incubator at 37°C, in an atmosphere containing 5% CO2 and culture medium was changed every 3 days for serial subculture.
Preparation of AN‐extract
One kg of dried, ripened AN (without the husks) were powdered using a mortar and pestle before being boiled for 1 hrs in 2 L of distilled water. After filtration, the extract was lyophilized and stored at −20°C until use.19 The lyophilized AN‐extract was weighed and dissolved in sterilized ice cold distilled water with vortexing at 4°C overnight and then centrifuged at 5000 rpm for 15 min. The supernatant was then filtered using sterilized filter paper (Whatman®) with 0.45 µm pore size syringe filters (Sartorius Stedim Biotech) and stored at −80°C until use.
The AN‐extract contents were analyzed by Liquid Chromatography and Gas Chromatography Mass Spectrometer (LC‐MS/GC‐MS) in the Yonsei Center for Research Facilities. LC was performed using a 1260 Infinity LC (Agilent Technologies). The detail was described in Table 2, Supporting Information. The GC‐MS analysis was carried out in Agilent 7890A‐GC/5975‐MS/Combi‐Pal‐Headspace. A 1 μl portion of the derivative extract was injected in splitless mode onto the HP‐5 ms column; the injection temperature at 290°C, at a flow rate of 1 ml/min.
Cytotoxicity assessment
To identify suitable AN‐extract concentrations, the cytotoxic effect of fibroblasts was assessed by MTT assay. In brief, 2 ×104 cells were seeded in to 96 well plates and concentration gradients of AN‐extract (10 −160 µg/ml) were applied. Mitochondrial dehydrogenase activity was assessed by MTT assay at 24 hr intervals for 96 hr. We also observed cytotoxicity of IHOK by the treatment of recombinant human GRO‐α, IL‐6 and IL‐8 (R&D Systems). IHOK cells (5 × 103) were exposed to a 10 ng/ml concentration of cytokines. The control was treated with media alone. The optical density was measured at a wavelength of 570 nm using a microplate reader (Bio‐Rad).
Assessment of cytokines released from fibroblasts
On the basis of the cytotoxicity assay results, 30 µg/ml of AN‐extract was selected as suitable dose for the stimulation of fibroblasts. First, the Human Cytokine Antibody Array 3 kit (Ray Biotech) was used to screen cytokine secretion in the conditioned media (CM) of AN‐exposed fibroblasts, following the manufacturer's instructions (Fig. S1a, Supporting Information). Controls were maintained in the absence of AN‐extract treatment (culture media alone). The relative expression level of the cytokines was determined by comparing signal intensities. The “Quantity one program” (Bio‐Rad) was used for densitometric analysis of the results. Sandwich Enzyme‐Linked Immunosorbent Assay (ELISA) was employed to measure cytokine levels in the CM according to AN treatment. Both cells were stimulated by AN‐extract for 24 hr. The supernatant was collected, centrifuged and filtered before storing at −80°C. To evaluate antioxidant effect, the levels of cytokines were also measured after a treatment of Epigallocatechin‐3‐gallate (EGCG; 12.5 μM; DSM Nutritional Products), l‐Glutathione‐reduced (Glu; 5 mM; Sigma‐aldrich), N‐acetyl‐cysteine (NAC; 10 mM; Enzo Life Sciences). All reagents for ELISA are described in Table 3, Supporting Information. Cytokine secretions were normalized according to the number of cells at the time of harvesting CM.
For morphological detection of cytokine secretions in tissue samples, 10 OSF and 5 NOM tissues were used for immunohistochemical staining. Mouse immunoglobulin fraction served as negative controls.
Measurement of oxidative stress
Reactive oxygen species (ROS) were measured with the fluorescent probe 2′7′‐dichlorofluorescin diacetate (H2DCFDA) dye (Molecular Probes) according to the instructions. The cells were exposed to the 10 µM H2DCFDA in PBS in the dark at 37°C for 20 min. For detection of ROS by AN exposure (30 µg/ml) in hTERT‐hNOF cells, 4 × 105 cells were seeded into 6‐well plates and incubated for 24 and 48 hr. To investigate the pathway of ROS generation in hTERT‐hNOF cells, several pharmacologic inhibitors were pre‐incubated for 1 hr before AN treatment; the inhibitors were used as follows: PD98059 (Sigma‐aldrich), Erk inhibitor; SP600125 (Sigma‐aldrich), JNK inhibitor; SB203580 (Enzo Life Sciences), p38 inhibitor; BAY‐11‐7082 (Sigma‐aldrich), NF‐κB inhibitor. All of the inhibitors were used at 10 μM.
For detection of ROS by AN exposure (30 µg/ml) in IHOK cells, 2.5 × 105 cells were seeded into 6‐well plates and incubated for 24 and 48 hr. For detection of ROS in IHOK by cytokine treatment, 2.5 × 105 cells were seeded into 6‐well plates and cytokines (10 ng/ml) were added individually (or in combination) into the wells and incubated for the desired time periods in dark. Serum‐free media alone served as a negative control and 10 µM H2O2 served as a positive control. For the analysis of a flow cytometry (Becton Dickinson, Beckman coulter), the cells (1 × 104) were then analyzed by at an excitation and emission wavelength of 485 and 535 nm, respectively. Cell‐Quest software (BD Biosciences) was utilized for data analysis. For confocal microscopic examination of ROS, IHOK cells (1 × 104) grown in chamber slide were stained with H2DCFDA, as previously described.20 The nuclei were stained with DAPI. ROS‐producing cells were counted and calculated into percentages.
Detection of oxidative DNA damage
To detect oxidative DNA damage caused by cytokines or CM, the OxyDNA assay kit (Caibiochem) consisted of FITC‐conjugated probe specific for 8‐oxoguanine (8‐oxoG) (1:10 dilution) was utilized and assessed by both flow cytometry and confocal microscopy, according to the manufacturer's protocol.21 IHOK cells (1 × 106) treated with cytokines or CM from AN‐exposed hTERT‐hNOF cells or VAS2870 (10 μg/ml) (Sigma‐aldrich) were stained with the 8‐oxoG FITC‐conjugate and fluorescence intensity was read using a flow cytometry at an excitation wavelength of 495 nm (Becton Dickinson). Serum‐free media, alone, served as a negative control while 10 µM H2O2 served as a positive control. For morphological examination, IHOK cells (1 × 104) grown per chamber were treated with cytokines or CM for the indicated time, and were stained using a 8‐oxoG FITC‐conjugate and stored at 4°C overnight. Slides were also stained with a 10 µg/ml DAPI and photomicrographed using confocal microscopy. For neutralizing cytokines in CM from AN‐exposed hTERT‐hNOFs, mouse monoclonal anti‐human GRO‐α (1 µg/ml), IL‐6 (8 µg/ml) and IL‐8 (1 µg/ml) antibodies (R&D Systems) were applied.
We observed oxidative DNA damage in human OSF and NOM samples. Staining with an 8‐oxoG FITC‐conjugate was carried out as previously reported.21 In brief, the tissue sections were autoclaved for 20 min in 50 mM Tris‐EDTA buffer (pH 9), after being deparaffinized and rehydrated. Next, the sections were blocked and incubated overnight with 8‐oxoG FITC‐conjugate at 4°C. Thereafter, the slides were stained with a 10 µg/ml DAPI and photomicrographed using confocal microscopy.
Detection of DNA double strand breaks (DSB) by histone H2A.X
To detect DNA damage caused by cytokines, the phospho‐Histone H2A.X rabbit monoclonal antibody (Cell Signaling Technology) was utilized22 and described in Table 3, Supporting Information. IHOK cells (1 × 104) were seeded in chamber slides and were treated with cytokines for 72 hr. The positive cells were counted in five randomly selected fields and calculated into percentages. To find out whether oxidative stress induced by cytokines causes DNA damage in IHOK, we compared DNA damaged cells after treatment with 5 mM of Glu.
To detect phospho‐Histone H2A.X in human OSF tissues, human OSF and NOM samples were immunohistochemically stained. Human colon cancer tissues were used as a positive control. Immunoperoxidase stained cells were visualized and photomicrographed. Rabbit IgG (DakoCytomation) was used instead of the primary antibody as a negative control.
Protein extract and western blot
hTERT‐hNOFs (1 × 106) were harvested in Cell Lysis Buffer (Cell Signaling Technology), containing PMSF (Sigma‐aldrich). The lysates were incubated for 30 min on ice with vortexing every 5 min and, samples were centrifuged at 15,000 rpm for 10 min at 4°C to remove insoluble debris. After centrifuging, the supernatant is transferred to new micro‐centrifuge tube and then mixed with 5× SDS sample buffer for 5 min at 100°C. Proteins (40 μg) were loaded and separated on a SDS–PAGE and transferred on polyvinylidene difluoride membranes. The information of antibodies is listed in Table 3, Supporting Information.
Reverse transcriptase (RT)‐PCR for detection of NAPDH oxidase 1 and 4 (NOX1/NOX4)
Total RNA was extracted from each cell lysate by using an RNeasy kit (Qiagen) and then cDNA was synthesized from 1 μg of the RNA by using a RT&GO‐MasterMix (MP Biomedicals) according to the manufacturer's protocols. The following primers were used for RT‐PCR: NADPH oxidase 1 (NOX1), 5′‐TTCCTGGTTCAACAACCTGT‐3′ (forward) and 5′‐GGG TGGGAGGTAGCTATTGT‐3′ (reverse); NADPH oxidase 4 (NOX4), 5′‐TCTGGAAAACCTTCTTGCTG‐3′ (forward) and 5′‐GGCTGCAGTTGAGGTTAAGA‐3′ (reverse); GAPDH, 5′‐GAAGGTGAAGGTCGGAGT‐3′ (forward) and 5′‐GAAGAT GGTGATGGGATTTC‐3′ (reverse). The reaction mixture was subjected to 35 PCR amplification cycles of 40 sec at 94°C, 60 sec at 48°C and 40 sec at 72°C. The PCR products were identified using ethidium bromide in 1.5% agarose gel.
NOX4 small interfering RNA transfection and NOX inhibition
NOX4 small interfering RNA(siRNA) had the following sense and antisense sequences: Sense 5′‐CUGUUG UGGACCCAAUUCA‐3′ and Antisense 5′‐UGAAUUGGGU CCACAACAG‐3′. A negative control siRNA was purchased from Bioneer. Transfection of siRNA was performed using the Lipofectamine RNAiMAX (Invitrogen) according to the manufacturers' instructions. In brief, IHOK cells (4 × 105) were seeded per well in a six‐well plate and cultured for 24 hr. NOX4 siRNA was diluted with 250 μl Opti‐MEM (Invitrogen). These two dilution mixtures were combined, mixed gently, and incubated for 20 min at room temperature to facilitate complex formation. The 500 μl siRNA‐Lipofectamine mixture was then added to cells. The transfected cells were cultured for 24 hr and then assayed. To inhibit the NOX proteins in IHOK, the cells (4 × 105) were seed per well in a six‐well plate and cultured for 24 hr. The cells were preincubated with VAS2870 10 μg/ml (Sigma‐aldrich) as a NOX inhibitor for 1 hr. Cytokines were then added to cells for indicated time.
Statistical analysis
All results shown are representative of multiple repeats. The Mann–Whitney U test was used for analyzing AN or cytokine treatment effects between groups. Two‐way analysis of variance (ANOVA) using Tukey's multiple test was used to assess the significance of proliferation rate between groups. All statistical analysis was processed in SPSS software version 20.0. p‐values of <0.05 was considered significant in all statistical methods.
Results
The selection of the optimal concentration of AN‐extract
Both hNOF and hTERT‐hNOF were exposed to a concentration gradient of AN‐extract and assessed for cell viability. Cytotoxic effects were not observed in both cell lines after 24 hr. Growth was retarded in both types of fibroblasts when they were exposed to more than 40 µg/ml concentration after 72 hr. We selected 30 µg/ml for this study, since this concentration showed no noticeable changes in cell viability, even after 96 hr (Fig. S2, Supporting Information).
Arecoline (1.97 ppm) was detected in 30 µg/ml of AN‐extract by LC‐MS (Table 2, Supporting Information). Cytotoxic effect by Arecoline treatment showed no growth change in both fibroblasts in less than 0.05 mM (11.8 ppm) (Fig. S3, Supporting Information). Taken together, the present study applied a 30 µg/ml concentration of AN‐extract. Arecaidine and phenolics were not detected by GC‐MS (data not shown).
Secretion level of GRO‐α, IL‐6 and IL‐8 increased in AN‐exposed fibroblasts and OSF tissue samples
To determine whether the AN‐extract induces cytokine secretion from gingival fibroblasts, the cytokine antibody array analysis was employed. Figure 1a shows sequential cytokine expression pattern in both hNOF and hTERT‐hNOF. The present study selected GRO‐α, IL‐6 and IL‐8, for these cytokines showed a greater increase in both cell lines than in nontreated control (Fig. 1a, Fig. S1b, Supporting Information) and were also known to have a role in crosstalk with cancer cells.23 Next, we carried out ELISA in AN‐exposed fibroblasts. As treated with AN‐extract for 24 hr, the levels of GRO‐α, IL‐6 and IL‐8 were 1.15‐/1.34‐, 1.2‐/1.26‐ and 1.97‐/1.73‐ fold higher in hNOF/hTERT‐hNOF, respectively, as compared with nontreated controls (Fig. 1b,c). We used hTERT‐hNOF for all in vitro study thereafter, since both hNOF and hTERT‐hNOF exhibited the same cytotoxicity concentration (Fig. S2, Supporting Information) and the same pattern of cytokine secretion after the treatment of AN‐extract (Figs. 1b and 1c).
Figure 1.
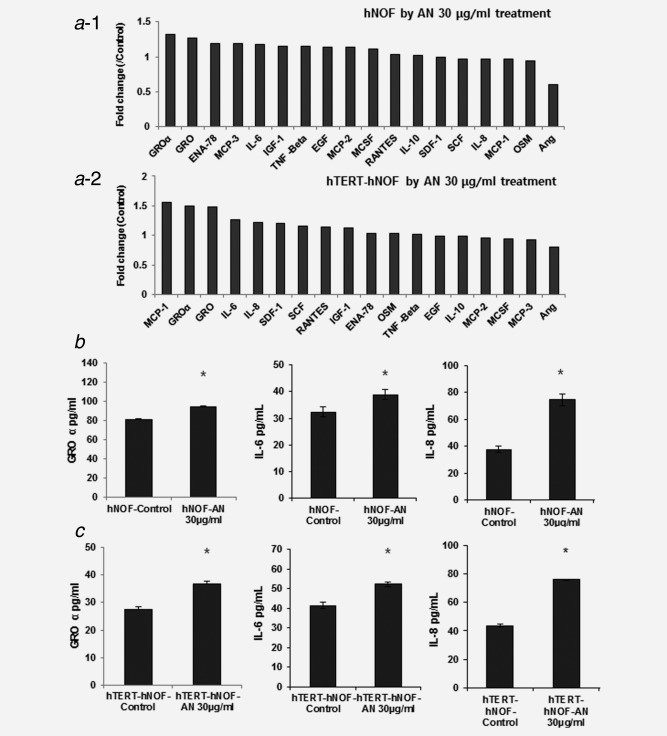
Cytokine secretion increased by AN‐extract treatment. (a) RayBio® Human Cytokine Antibody Array 3 kit (Ray Biotech) was used to screen cytokine secretion in the AN‐exposed CM. The relative expression level of the cytokines was determined by densitometric analysis using the ‘Quantity one program’. Quantification of the 18 cytokines and growth factors was available in both hNOF (a−1) and hTERT‐hNOF (a−2) by AN‐extract for 24 hr. (b,c) GRO‐α, IL‐6 and IL‐8 were measured by ELISA. As 30 μg/ml AN‐extract were treated for 24 hr in both hNOF (b) and hTERT‐hNOF (c), each cytokine was increased than in nontreated CM. Results shown are representative of multiple repeats (means ± SD of triplicates) (*p ≤ 0.05 by Mann–Whitney U‐test).
We observed cytokine expression morphologically in OSF tissue samples. GRO‐α, IL‐6 and IL‐8 were more highly expressed in the fibroblasts of OSF tissue samples, as compared with those of NOM (Fig. 2a).
Figure 2.
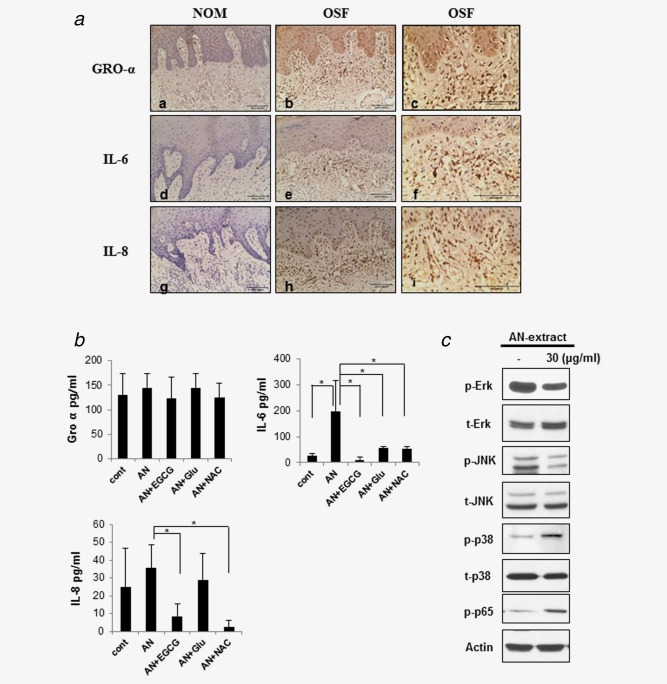
Cytokine expression in OSF tissues and AN‐exposed fibroblasts. (a) Immunohistochemical staining for GRO‐α, IL‐6 and IL‐8 in OSF and NOM. OSF (b, c, e, f, h, i) showed increased expressions of GRO‐α (a, b, c), IL‐6 (d, e, f) and IL‐8 (g, h, i) as compared to NOM (a, d, g). Representative photomicrographs, the figures of c, f, i: ×200 magnification of b, e, h (×100), respectively. (b) Antioxidants reduced cytokine secretion in AN‐exposed fibroblasts. GRO‐α, IL‐6 and IL‐8 were measured by ELISA. As 30 μg/ml AN‐extract were treated for 24 hr in hTERT‐hNOF, each cytokine was increased than in nontreated cell. The AN‐induced increase of IL‐6 and IL‐8 secretion was 3.7‐ to 19.5‐fold/1.2‐ to 4.3‐fold decreased by antioxidants (EGCG, Glu and NAC), while GRO‐α showed no noticeable change. Results shown are representative of multiple repeats (means ± SD of triplicates) (*p < 0.05 by Mann–Whitney U‐test). (c) NF‐κB and MAPKinase pathway were involved in cytokine secretion of AN‐exposed fibroblasts. hTERT‐hNOFs (1 × 106) were seeded in 100 mm dishes and then stabilized for 24 hr. hTERT‐hNOFs were harvested after exposure of 30 μg/ml AN‐extract for 24 hr, respectively. β‐actin was used as a loading control. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
AN‐exposed fibroblasts induce cytokine secretion via ROS generation
To determine whether AN‐extract induces ROS in cells, we first measured the level of ROS in hTERT‐hNOF. The level of ROS was significantly higher in AN‐exposed cells than in the controls (Fig. S4, Supporting Information).
To evaluate whether the cytokines released from AN‐exposed fibroblasts were induced by ROS, AN‐exposed hTERT‐hNOF was treated with antioxidants. As results, IL‐6 and IL‐8 induced by AN‐extract were significantly abolished by antioxidants (EGCG, Glu and NAC), while no noticeable change was observed in the level of GRO‐α (Fig. 2b). These results showed that ROS induces IL‐6 and IL‐8 secretion in AN‐exposed fibroblast. As a candidate of molecular pathway to release cytokines from AN‐exposed hTERT/hNOF, NF‐κB and MAPKinase pathway were examined. The p38 and p65 were phosphorylated by AN‐extract in hTERT‐hNOF, while the Erk and JNK were inactivated (Fig. 2c), suggesting that AN‐extract might induce ROS generation through p38 and NF‐κB pathways. To further verify whether AN‐induced ROS was modulated through p38 and NF‐κB pathway, we used a variety of the specific pharmacologic inhibitors for blocking their downstream pathways. Inhibiors of NF‐κB significantly reduced AN‐induced ROS generation. In addition, both the NF‐κB and p38 inhibitors blocked AN‐induced ROS generation more effectively than treatment NF‐κB inhibitor only (Fig. S5, Supporting Information). Thus, we demonstreated that AN‐exposed hTERT/hNOF induced cytokine secretion via ROS generated through NF‐κB pathway.
Cytokines induced by AN‐exposure promote oxidative DNA damage in IHOK
To identify whether the cytokines from AN‐exposed fibroblasts elicit DNA damage in epithelial cells, we examined oxidative DNA damage in IHOK after treatment of cytokine or CM from AN‐exposed hTERT‐hNOF. A 10 ng/ml concentration of each cytokine was applied because this concentration showed no impact on cell viability in HEK and IHOK (Fig. S6, Supporting Information). Flow cytometric analysis showed that the number of 8‐oxoG FITC‐conjugate labeled cells were more than 2‐fold increased when treated with AN‐exposed CM than in the unexposed‐CM or the negative control. The positive cells were reduced to the control level by the treatment of neutralizing antibodies against each cytokine (Fig. 3a‐1, Supporting Information). These results were also morphologically confirmed (Fig. 3a‐2). Supporting Information Fig. S7a−1 showed the increase of 8‐oxoG‐labelled cells morphologically in IHOK treated by respective and combined treatment of cytokines. The damaged cells could not be augmented by the combined treatment, compared with the respective treatment. Supporting Information Fig. S7a−2 demonstrated that 8‐oxoG‐ labeled cells were significantly increased by the combined treatment of cytokines compared with the controls. Additionally, we examined DNA DSB by staining phospho‐Histone H2A.X in the IHOK and counted the positive cells by confocal microscopy. The positive cells were more than 4‐ to 5‐fold higher in cytokines‐treated IHOK than in the controls (Fig. 3b‐1.2). Morphologically, no positive cell was found in HEK by cytokine treatment at this concentration (Supporting Information Fig. S8).
Figure 3.
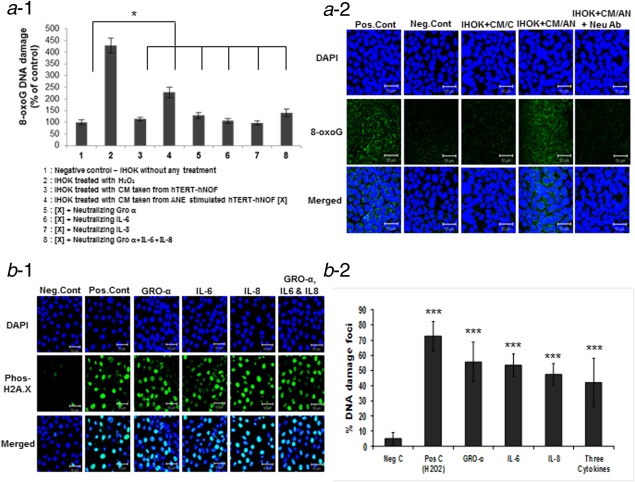
Oxidative DNA damage in IHOK was detected by OxyDNA assay. (a−1) The positive cells for 8‐oxoG FITC‐conjugate were 2‐fold higher in IHOK by treating CM of AN‐exposed hTERT‐hNOF, compared to nontreated CM or negative control. The positive cells were reduced by the treatment of neutralizing antibodies (*p < 0.05 by Mann–Whitney U‐test). (a−2) Confocal microscopy showed that the 8‐oxoG‐ labeled cells were reduced in IHOK by treating neutralizing antibodies (Neu Ab). Scale bar 50 µm. (b−1) DNA DSB was detected by staining phospho‐Histone H2A.X in cytokine‐treated IHOK. IHOK were treated with cytokines individually (GRO‐α, IL‐6 and IL‐8) and in combination for 72 hr and tested for DNA DSB. Representative photomicrographs are shown. Scale bar 50 µm. Cont: control. (b−2) Statistical photomicrographs are shown. Cytokine treatment caused 4‐ to 5‐fold increase of DNA DSB in IHOK. The results shown are representative of multiple repeats (***p < 0.001 by Mann–Whitney U‐test). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Oxidative DNA damage was observed in OSF tissue
To confirm in vitro results, we measured DNA DSB and 8‐oxoG in OSF tissues. OSF and NOM tissues were stained using anti‐phospho‐Histone H2A.X. Human colon cancer tissue served as a positive control. Immunohistochemical staining showed positively stained cells in OSF mucosa, especially in the basal cell layer of all OSF cases tested. NOM showed negative response for phospho‐Histone H2A.X staining (Fig. 4a). We carried out the OxyDNA assay by staining the 8‐oxoG FITC‐conjugate in OSF tissues. In all OSF tissues tested, fluorescent signaling was evident as a band in the basal and parabasal layers, while no signal was found in the NOM tissues (Fig. 4b).
Figure 4.
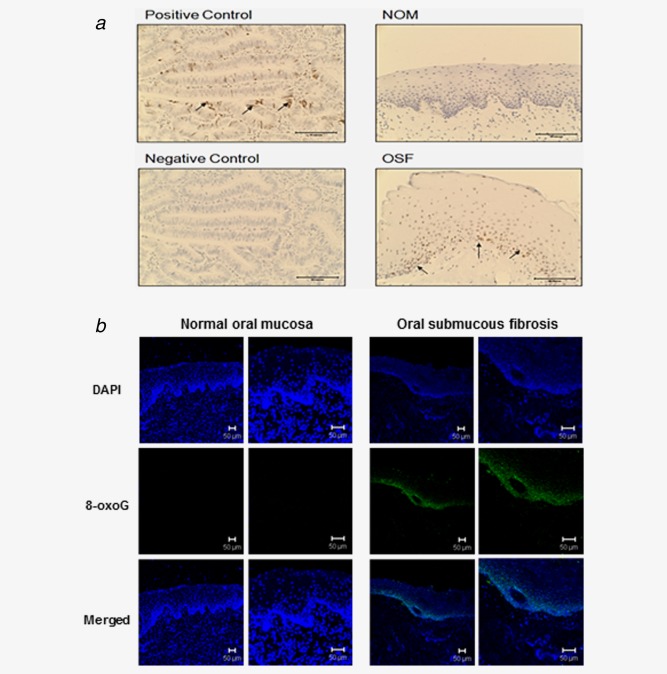
DNA DSB and oxidative DNA damage were detected in OSF tissues (a) Immunohistochemical staining for phospho‐Histone H2A.X in OSF tissue. OSF tissue showed DNA damage foci in basal cell layer as compared to NOM. Arrows indicate positive cells. Positive control: human colon cancer. Representative photomicrographs are shown. Scale bar 50 µm. C: control (b) OSF tissues showed positive staining for 8‐oxoG FITC‐conjugate in basal cell area seen by confocal microscopy. Scale bar 50 µm. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Cytokine‐triggered oxidative DNA damage was caused by ROS generation via NOX4
To evaluate whether the DNA damage of IHOK induced by cytokine treatment is also due to ROS generation, ROS generation of IHOK by cytokine treatment was quantified using a fluorescent probe. After exposure to each cytokine, a 7‐ and 8‐fold higher number of cells produced ROS by respective and combined treatments of cytokines, respectively, as compared to the negative control (Fig. 5a‐1,2). To confirm whether the cytokine‐induced DNA damage in IHOK is due to oxidative stress, we treated cytokine‐exposed IHOK with an antioxidant and stained the cells with phospho‐Histone H2A.X. As results, the positive cells were reduced in antioxidant‐treated IHOK, supporting that DNA DSB were induced by ROS following the cytokine (Fig. S9, Supporting Information).
Figure 5.
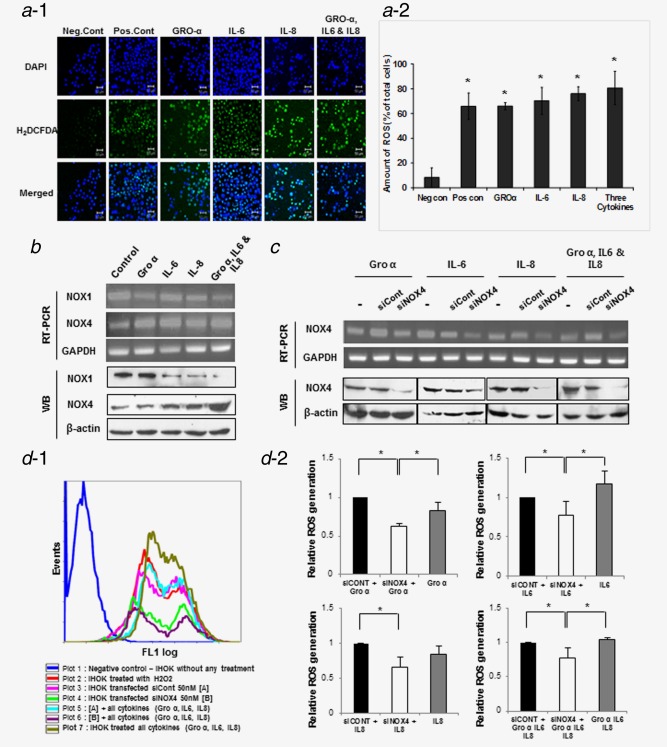
Cytokines induced ROS production via NOX4 in IHOK. (a−1) ROS generation by cytokine treatment in IHOK was detected using H2DCFDA dye. IHOK were treated with cytokines individually (GRO‐α, IL‐6 and IL‐8) and in combination for 72 hr. Green fluorescent staining detected using confocal microscopy. (a−2) ROS‐generating cells were 7‐ to 8‐fold higher in cytokine‐treated IHOK than in nontreated cells (*p < 0.05 by Mann–Whitney U test). Representative photomicrographs shown. Scale bar 50 µm. (b) The expression of NOX1 and NOX4 was analyzed by RT‐PCR and Western blot. NOX4 was increased after stimulation with each cytokine and combined cytokine treatment for 72 hr in IHOK. (c) IHOK cells were transfected with NOX4 siRNA (siNOX4) 50 nM or control siRNA (siCont) 50 nM and stabilized for 24 hr. After stabilization, the cells were cultured with cytokine for 1 hr. Knockdown of NOX4 was accomplished as above and confirmed by RT‐PCR and Western blot. (d−1) Flow cytometry analysis of positive cell staining H2DCFDA dye for detection of ROS generation by NOX4 knockdown. (d−2) Graphs shown were as a quantitative analysis. Each of IHOK cell transfected with siCont (Black stick), siNOX4 (White stick) and the cell without any transfection (Gray stick) treated with indicated cytokine and then analyzed. In spite of cytokine treatment, NOX4 knockdown‐IHOK decreased ROS generation compare to cell transfected with siCont and cell without any transfection. The results shown are representative of multiple repeats (*p ≤ 0.05 by Mann–Whitney U‐test). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
To determine whether NADPH oxidases (NOXs) participate at generating ROS in IHOKs, we measured the mRNA and protein levels of NOX1 and NOX4 in IHOK treated with respective or combined cytokines. NOX4 expression increased in IHOK treated with the cytokines as compared to the control, whereas NOX1 expression was reduced (Fig. 5b). siRNA targeting to NOX4 significantly reduced ROS generation compared with the control. Intracellular ROS levels were 0.63‐/0.78‐/0.65‐/0.77‐fold lower than siControl in NOX4‐knockdown cells (Fig. 5d‐1,2).
NOX1 and NOX4 contribute to DNA damage in IHOK
To identify whether NOX4 mediates DNA damage by cytokine, we measured the oxidative DNA damage by the use of 8‐oxoG FITC‐conjugates. Silencing NOX4 alone did not block cytokine‐induced oxidative DNA damage (Fig. S10, Supporting Information). To evaluate whether DNA damage response was mediated by NOX4 in combination with NOX1, we used an inhibitor of NOXs, VAS2870, for blocking both these proteins. In mRNA and protein levels, VAS2870 treatment induced a dose‐dependent decrease of both NOX1 and NOX4. To determine the influence of VAS2870 on cytokine‐induced ROS generation, the cells were pretreated with VAS2870 10 μg/ml. ROS levels showed the 2.3‐/1.8‐/1.9‐/2.0‐fold decrease in IHOK by treatment of GRO‐α, IL‐6, IL‐8 and the combination of three cytokines, respectively, as compared with control (Fig. 6b‐1,2). In addition, the NOX inhibitor reduced cytokine‐induced 8‐oxoG positive cells. As shown in Figure 6c, 15–27% of the positive cells were reduced by preincubation of VAS2870 in each cytokine‐treated IHOK, however, the reduction of 8‐oxoG positive cells was not found in combined treatment of three cytokines (Fig. 6c‐1,2). Together, these results suggest both NOX1 and NOX4 mediate cytokine‐induced oxidative DNA damage by regulating ROS production.
Figure 6.
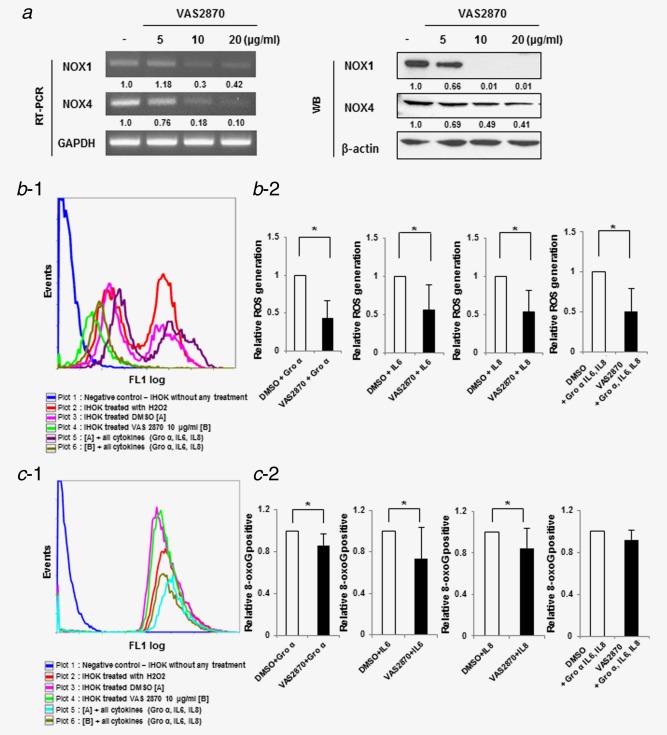
Cytokines‐induced DNA damage was caused by ROS production through NOX1 and NOX4 in IHOK. (a) Effect of NOX1 and NOX4 by VAS2870, NOX inhibitor. Knockdown of NOX1 and NOX4 were accomplished in dose‐dependent manner. (b−1) The cells were incubated with cytokine for 1 hr. Flow cytometry analysis of positive cell stained H2DCFDA dye for ROS generation. (b−2) Statistical graphs are shown. NOX inhibitor, VAS2870, inhibited cytokine‐induced ROS production (p < 0.05* by Mann–Whitney U‐test). (c−1) The cells were incubated with cytokine for 20 hr. Flow cytometry analysis of positive cell stained using FITC‐conjugated OxyDNA assay for detection of Oxidative DNA damage by knockdown both NOX1 and NOX4. (c−2) Cytokine‐triggered oxidative DNA damage was blocked by VAS2870 in IHOK, except for combined treatment. The results shown are representative of multiple repeats (p < 0.05* by Mann–Whitney U‐test). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Discussion
DNA damage in AN‐exposed oral epithelial cells has been well acknowledged. Safrole‐like DNA adducts24 or acrolein‐derived 1,N2‐propanodeoxyguanosine DNA adducts25 were formed in AN chewers. Moreover, DNA single‐strand breaks and DNA‐protein crosslinks were induced in AN‐exposed oral epithelial cells.19 On the basis of the fact that OSF caused by AN‐chewing habit is characterized by chronic inflammation with fibrosis, the aim of this study was to investigate whether DNA damage in epithelial cells occurs indirectly via secretory cytokines released from AN‐exposed fibroblasts. To achieve this goal, we attempted to evaluate whether AN‐exposed fibroblasts secrete cytokines and whether the secreted cytokines induced DNA damage in IHOK.
In that immortalized hTERT‐hNOF16 have the same biological feature with normal fibroblasts and are more stable for in vitro study than primary cultured fibroblasts, we first compared the cytokine secretion patterns of hTERT‐hNOF with hNOF after AN exposure, to determine whether hNOF can be replaced with hTERT‐hNOF in this study. Since the secretion pattern of cytokines after AN exposure was similar in both fibroblasts, we used hTERT‐hNOF for this study.
Accumulating evidence has indicated that GRO‐α, IL‐6 and IL‐8 promote carcinogenesis in various human cancers. For instance, GRO‐α is a potential mediator of cancer invasion in several human cancers such as gastric cancer26 and colon adenocarcinoma.27 IL‐8 is capable of promoting cancer progression by regulating angiogenesis through autocrine and paracrine mechanisms.28, 29 Autocrine effects of IL‐6 in tumorigenesis have been identified in human cancers such as prostatic,30 breast9 and head and neck cancer.31 Our finding which showed the proliferation of OSCC cells after cytokine treatment was in agreement with those studies (Fig. S6, Supporting Information).
The present study detected 8‐oxoG as evidence of oxidative DNA damage, based on the notion that an oxidative stress‐induced DNA damage pathway underlies its significance at the earliest stages of cancer development.32, 33, 34 The oxidized guanine bases were elevated in various human cancers and thus, oxidative DNA damage is expected to be an etiology of human cancers.35, 36 Our study showed that the level of 8‐oxoG‐labeled cells increased in IHOK treated with either cytokines or CM from AN‐exposed fibroblasts. Further, the 8‐oxoG‐positive cells were reduced when cytokines in the CM were neutralized, suggesting that DNA damage has been triggered by cytokines released from AN‐exposed fibroblasts. As DNA DSB is one of the most deleterious forms of DNA damage and is accumulated in premalignant lesions as well as in OSCC,22, 37 the continuous formation of DNA DSB may contribute to genomic instability and consequently to carcinogenesis.34 In our study, the DNA DSB‐positive cells increased in cytokine‐treated IHOK compared to the nontreated controls. Collectively, the present study showed that DNA damage in IHOK cells occurred by cytokines released from AN‐exposed fibroblasts.
AN‐exposed human keratinocytes are directly damaged via their own oxidative stress pathway.38 Our study attempted to determine indirect DNA damage of epithelial cells via AN‐exposed fibroblasts. Herein, we used immortalized HPV E6/E7 transfected keratinocytes to examine DNA damage. Considering that DNA damage in oral keratinocytes by AN‐exposed fibroblasts has been secondarily affected in the damaged epithelium in advance by AN exposure, we thought that IHOK could be substituted for oral keratinocytes irritated by AN exposure. Supporting our assumption, our data showed that IHOK produced ROS by AN‐exposure (Fig. S4, Supporting Information). Moreover, cytokine treatment to normal keratinocytes (HEK) failed to induce DNA damage (Fig. S8, Supporting Information), suggesting that DNA damage by cytokine treatment may be limited to the genetically transformed keratinocytes like IHOK.
A 10 ng/ml concentration of cytokine was 50‐ to 100‐fold higher than the concentration produced from AN‐exposed fibroblasts in our experiment. Nevertheless, we extrapolated that these cytokines induce oxidative DNA damage to epithelial keratinocytes transformed by carcinogens, because oxidative DNA damage occurred through CM obtained from AN‐exposed fibroblasts. These in vitro data were confirmed by the detection of DNA damage in OSF tissues. However, the causes of DNA damage observed in OSF tissues cannot be proven with the present study; whether this damage is due to direct injury by AN‐extract or indirect injury by cytokines released from AN‐exposed fibroblasts.
Oxidative stress is a well‐known mechanism of causing cell damage by AN‐extract.39 In our study, antioxidants reduced IL‐6 and IL‐8 levels in AN‐exposed fibroblasts, supporting that cytokine secretion by AN‐exposed fibroblasts was due to ROS generation.40 However, Gro‐α expression was not reduced by antioxidants, indicating that Gro‐α may be modulated by different pathway from IL‐6 and IL‐8. It has been well established that cytokines are regulated by the activation of NF‐κB linked with ROS generation.41, 42 In our study, phosphorylated p65 (subunit of NF‐κB family) and p38 were increased in AN‐exposed fibroblasts, compared with nontreated control (Fig. 2c). These results are consistent with previous reports.39, 43 Also, we confirmed that AN‐induced ROS generation was regulated through NF‐κB pathway, because the inhibitor of NF‐κB significantly attenuated ROS generation by AN‐extract (Fig. S5, Supporting Information). Thus, it can explain that AN‐extract induces ROS generation through NF‐κB and then triggers secretion of cytokines. In addition, Arecoline, a major alkaloid among the components in AN‐extract, can induce cytokine production.44 Since the present study could detect Arecoline from AN‐extract, we could extrapolate that Arecoline might induce secretion of cytokines in our study.
ROS generation could be a mechanism of causing DNA damage induced by cytokine.20 ROS generation is mediated through activation of NADPH oxidase.45 In our study, NOX4 expression increased in cytokine‐treated IHOK, whereas NOX1 expression was reduced. It has been reported that NOX1 is regulated by recruitment of cytosolic subunits to redoxosomes, whereas NOX4‐dependent ROS generation is confined to the nuclear envelope.46, 47 Considering that intracellular signaling by cytokine binding proceeds by endocytosis, NOX1 may be primarily affected by cytokine binding, resulting in a decrease of NOX1 activity. The overexpression of NOX4 may be caused by a compensatory mechanism between NOX1 and NOX4.48 In our study, the downregulation of both NOX1 and NOX4 decreased ROS generation and DNA damage, suggesting that GRO‐α, IL‐6 and IL‐8 induced ROS generation in IHOK through NOX4 in combination with NOX1.
We observed DNA damage by the combination treatment of three cytokines as well as by the respective treatment of each cytokine, presuming that several cytokines work together in vivo condition. Our study showed the increase of 8‐oxoG‐labelled cells in IHOK treated by three cytokines together (Fig. S7a−2, Supporting Information) and these results were also confirmed by the treatment of neutralizing antibodies (Fig. 3a‐2), accepting that the combination of three cytokines also induce DNA damage. However, the damaged cells could not be augmented by combined treatment, as compared with the respective treatment. Moreover, a NOX inhibitor had no impact on the reduction of 8‐oxoG positive cells in the combined treatment of three cytokines, whereas a NOX inhibitor reduced 8‐oxoG positive cells by respective treatment. Collectively, these in vitro data should be confirmed by further in vivo study for clinical application.
On the basis of these data, we could conclude that fibroblasts secrete cytokines in response to external stimuli such as AN, subsequently triggering oxidative DNA damage and DNA DSB in adjacent epithelia cells. Accumulation of DNA damage that ensues in the epithelial cells as a result of continuous insult by the cytokine‐mediated ROS generation might contribute to the malignant transformation of OSF. Thus, inhibition of ROS generation could be a promising mode of blocking the deleterious effects of cytokines secreted from fibroblasts, as it can prevent malignant transformation of potentially malignant lesions such as OSF tissues.
Supporting information
Supporting Information Figure 1
Supporting Information Figure 2
Supporting Information Figure 3
Supporting Information Figure 4
Supporting Information Figure 5
Supporting Information Figure 6
Supporting Information Figure 7
Supporting Information Figure 8
Supporting Information Figure 9
Supporting Information Figure 10
Supporting Information Table 1
Supporting Information Table 2
Supporting Information Table 3
Supporting Information
Acknowledgements
The authors thank Professor Young Ran Kim, Dongshin University, Republic of Korea, for preparing the AN extract. They would also like to extend sincere thanks to Professor Darren R. Williams, Gwangju Institute of Science and Technology, Republic of Korea, for critical reading of the manuscript.
References
- 1. Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature 2004;432:332–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.2. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer 2006;6:392–401. [DOI] [PubMed] [Google Scholar]
- 3.3. Orimo A, Tomioka Y, Shimizu Y, et al. Cancer‐associated myofibroblasts possess various factors to promote endometrial tumor progression. Clin Cancer Res 2001;7:3097–105. [PubMed] [Google Scholar]
- 4. Raman D, Baugher PJ, Thu YM, et al. Role of chemokines in tumor growth. Cancer Lett 2007;256:137–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Scheller J, Ohnesorge N, Rose‐John S. Interleukin‐6 trans‐signalling in chronic inflammation and cancer. Scand J Immunol 2006;63:321–9. [DOI] [PubMed] [Google Scholar]
- 6. Coussens LM, Werb Z. Inflammation and cancer. Nature 2002;420:860–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hussain SP, Harris CC. Inflammation and cancer: an ancient link with novel potentials. Int J Cancer 2007;121:2373–80. [DOI] [PubMed] [Google Scholar]
- 8. Atsumi T, Singh, R , Sabharwal L, et al. Inflammation amplifier, a new paradigm in cancer biology. Cancer Res 2014;74:8–14. [DOI] [PubMed] [Google Scholar]
- 9. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell 2010;140:883–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Grivennikov SI, Karin M. Dangerous liaisons: STAT3 and NF‐kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev 2010;21:11–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Aggarwal BB, Vijayalekshmi RV, Sung B. Targeting inflammatory pathways for prevention and therapy of cancer: short‐term friend, long‐term foe. Clin Cancer Res 2009;15:425–30. [DOI] [PubMed] [Google Scholar]
- 12. Pindborg JJ, Bhonsle RB, Murti PR, et al. Incidence and early forms of oral submucous fibrosis. Oral Surg Oral Med Oral Pathol 1980;50:40–4. [DOI] [PubMed] [Google Scholar]
- 13. Tilakaratne WM, Klinikowski MF, Saku T, et al. Oral submucous fibrosis: review on aetiology and pathogenesis. Oral Oncol 2006;42:561–8. [DOI] [PubMed] [Google Scholar]
- 14. Humans IACR Working Group on the Evaluation of Carcinogenic Risks to Humans. Betel‐quid and areca‐nut chewing and some areca‐nut derived nitrosamines. IARC Monogr Eval Carcinog Risks Hum 2004;85:1–334. [PMC free article] [PubMed] [Google Scholar]
- 15. Haque MF, Harris M, Meghji S, et al. Immunolocalization of cytokines and growth factors in oral submucous fibrosis. Cytokine 1998;10:713–9. [DOI] [PubMed] [Google Scholar]
- 16. Illeperuma RP, Park YJ, Kim JM, et al. Immortalized gingival fibroblasts as a cytotoxicity test model for dental materials. J Mater Sci Mater Med 2012;23:753–62. [DOI] [PubMed] [Google Scholar]
- 17. Lee HJ, Guo HY, Lee SK, et al. Effects of nicotine on proliferation, cell cycle, and differentiation in immortalized and malignant oral keratinocytes. J Oral Pathol Med 2005;34:436–43. [DOI] [PubMed] [Google Scholar]
- 18. Lee EJ, Kim J, Lee SA, et al. Characterization of newly established oral cancer cell lines derived from six squamous cell carcinoma and two mucoepidermoid carcinoma cells. Exp Mol Med 2005;37:379–90. [DOI] [PubMed] [Google Scholar]
- 19. Sundqvist K, Liu Y, Nair J, et al. Cytotoxic and genotoxic effects of areca nut‐related compounds in cultured human buccal epithelial cells. Cancer Res 1989;49:5294–8. [PubMed] [Google Scholar]
- 20. Seidelin JB, Nielsen OH. Continuous cytokine exposure of colonic epithelial cells induces DNA damage. Eur J Gastroenterol Hepatol 2005;17:363–9. [DOI] [PubMed] [Google Scholar]
- 21. Roper JM, Mazzatti DJ, Watkins RH, et al. In vivo exposure to hyperoxia induces DNA damage in a population of alveolar type II epithelial cells. Am J Physiol Lung Cell Mol Physiol 2004;286:L1045–54. [DOI] [PubMed] [Google Scholar]
- 22. Celeste A, Fernandez‐Capetillo O, Kruhlak MJ, et al. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol 2003;5:675–9. [DOI] [PubMed] [Google Scholar]
- 23. Bae JY, Kim EK, Yang DH, et al. Reciprocal interaction between carcinoma‐associated fibroblasts and squamous carcinoma cells through interleukin‐1alpha induces cancer progression. Neoplasia 2014;16:928–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen CL, Chi CW, Chang KW, et al. Safrole‐like DNA adducts in oral tissue from oral cancer patients with a betel quid chewing history. Carcinogenesis 1999;20:2331–4. [DOI] [PubMed] [Google Scholar]
- 25. Chung FL, Krzeminski J, Wang M, et al. Formation of the acrolein‐derived 1,N2‐propanodeoxyguanosine adducts in DNA upon reaction with 3‐(N‐carbethoxy‐N‐nitrosamino)propionaldehyde. Chem Res Toxicol 1994;7:62. [DOI] [PubMed] [Google Scholar]
- 26. Jung JJ, Noh S, Jeung HC, et al. Chemokine growth‐regulated oncogene 1 as a putative biomarker for gastric cancer progression. Cancer Sci 2010;101:2200–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wen Y, Giardina, SF , Hamming D, et al. GROalpha is highly expressed in adenocarcinoma of the colon and down‐regulates fibulin‐1. Clin Cancer Res 2006;12:5951–9. [DOI] [PubMed] [Google Scholar]
- 28. Yao PL, Lin YC, Wang CH, et al. Autocrine and paracrine regulation of interleukin‐8 expression in lung cancer cells. Am J Respir Cell Mol Biol 2005;32:540–7. [DOI] [PubMed] [Google Scholar]
- 29. Inoue K, Slaton JW, Eve BY, et al. Interleukin 8 expression regulates tumorigenicity and metastases in androgen‐independent prostate cancer. Clin Cancer Res 2000;6:2104–19. [PubMed] [Google Scholar]
- 30. Rojas A, Liu G, Coleman I, et al. IL‐6 promotes prostate tumorigenesis and progression through autocrine cross‐activation of IGF‐IR. Oncogene 2011;30:2345–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yadav A, Kumar B, Datta J, et al. IL‐6 promotes head and neck tumor metastasis by inducing epithelial‐mesenchymal transition via the JAK‐STAT3‐SNAIL signaling pathway. Mol Cancer Res 2011;9:1658–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gorgoulis VG, Vassiliou LV, Karakaidos P, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005;434:907–13. [DOI] [PubMed] [Google Scholar]
- 33. Bartkova J, Horejsi, Z , Koed K, et al. DNA damage response as a candidate anti‐cancer barrier in early human tumorigenesis. Nature 2005;434:864–70. [DOI] [PubMed] [Google Scholar]
- 34. Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene‐induced DNA damage model for cancer development. Science 2008;319:1352–5. [DOI] [PubMed] [Google Scholar]
- 35. Tanaka H, Fujita N, Sugimoto R, et al. Hepatic oxidative DNA damage is associated with increased risk for hepatocellular carcinoma in chronic hepatitis C. Br J Cancer 2008;98:580–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Valavanidis A, Vlachogianni T, Fiotakis C. 8hydroxy‐2' ‐deoxyguanosine (8‐OHdG): a critical biomarker of oxidative stress and carcinogenesis. J Environ Sci Health C: Environ Carcinog Ecotoxicol Rev 2009;27:120–39. [DOI] [PubMed] [Google Scholar]
- 37. Chou SJ, Alawi F. Expression of DNA damage response biomarkers during oral carcinogenesis. Oral Surg Oral Med: Oral Pathol Oral Radiol Endod 2011;111:346–53. [DOI] [PubMed] [Google Scholar]
- 38. Thangjam GS, Kondaiah P. Regulation of oxidative‐stress responsive genes by arecoline in human keratinocytes. J Periodontal Res 2009;44:673–82. [DOI] [PubMed] [Google Scholar]
- 39. Lu HH, Kao SY, Liu TY, et al. Areca nut extract induced oxidative stress and upregulated hypoxia inducing factor leading to autophagy in oral cancer cells. Autophagy 2010;6:725–37. [DOI] [PubMed] [Google Scholar]
- 40. Bulua AC, Simon A, Maddipati R, et al. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1‐associated periodic syndrome (TRAPS). J Exp Med 2011;208:519–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Patil CS, Kirkwood KL. p38 MAPK signaling in oral‐related diseases. J Dent Res 2007;86:812–25. [DOI] [PubMed] [Google Scholar]
- 42. Gloire G, Legrand‐Poels S, Piette J. NF‐kappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol 2006;72:1493–505. [DOI] [PubMed] [Google Scholar]
- 43. Lin SC, Lu SY, Lee SY, et al. Areca (betel) nut extract activates mitogen‐activated protein kinases and NF‐kappaB in oral keratinocytes. Int J Cancer 2005;116:526–35. [DOI] [PubMed] [Google Scholar]
- 44. Tsai CH, Yang SF, Chen YJ, et al. Regulation of interleukin‐6 expression by arecoline in human buccal mucosal fibroblasts is related to intracellular glutathione levels. Oral Dis 2004;10:360–4. [DOI] [PubMed] [Google Scholar]
- 45. Kodama R, Kato M, Furuta S, et al. ROS‐generating oxidases Nox1 and Nox4 contribute to oncogenic Ras‐induced premature senescence. Genes Cells 2013;18:32–41. [DOI] [PubMed] [Google Scholar]
- 46. Spencer NY, Engelhardt JF. The basic biology of redoxosomes in cytokine‐mediated signal transduction and implications for disease‐specific therapies. Biochemistry 2014;53:1551–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Spencer NY, Yan Z, Boudreau RL, et al. Control of hepatic nuclear superoxide production by glucose 6‐phosphate dehydrogenase and NADPH oxidase‐4. J Biol Chem 2011;286:8977–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pendyala S, Gorshkova IA, Usatyuk PV, et al. Role of Nox4 and Nox2 in hyperoxia‐induced reactive oxygen species generation and migration of human lung endothelial cells. Antioxid Redox Signal 2009;11:747–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure 1
Supporting Information Figure 2
Supporting Information Figure 3
Supporting Information Figure 4
Supporting Information Figure 5
Supporting Information Figure 6
Supporting Information Figure 7
Supporting Information Figure 8
Supporting Information Figure 9
Supporting Information Figure 10
Supporting Information Table 1
Supporting Information Table 2
Supporting Information Table 3
Supporting Information