

Abstract
Significant progress has been made in the understanding of the underlying cancer biology of castration‐resistant prostate cancer (CRPC) with the androgen receptor (AR) signalling pathway remaining implicated throughout the prostate cancer disease continuum. Reactivation of the AR signalling pathway is considered to be a key driver of CRPC progression and, as such, the AR is a logical target for therapy in CRPC. The objective of this review was to understand the importance of AR signalling in the treatment of patients with metastatic CRPC (mCRPC) and to discuss the clinical benefits associated with inhibition of the AR signalling pathway. A search was conducted to identify articles relating to the role of AR signalling in CRPC and therapies that inhibit the AR signalling pathway. Current understanding of prostate cancer has identified the AR signalling pathway as a logical target for the treatment of CRPC. Available therapies that inhibit the AR signalling pathway include AR blockers, androgen biosynthesis inhibitors, and AR signalling inhibitors. Enzalutamide, the first approved AR signalling inhibitor, has a novel mode of action targeting AR signalling at three key stages. The direct mode of action of enzalutamide has been shown to translate into clinical responses in patients with mCRPC. In conclusion, the targeting of the AR signalling pathway in patients with mCRPC results in numerous clinical benefits. As the number of treatment options increase, more trials evaluating the sequencing and combination of treatments are required. This review highlights the continued importance of targeting a key driver in the progression of CRPC, AR signalling, and the clinical benefits associated with inhibition of the AR signalling pathway in the treatment of patients with CRPC.
Keywords: androgen receptor signalling, enzalutamide, mechanism of action, prostate cancer, metastatic, castration‐resistant
Introduction
The development and function of the prostate gland is dependent on androgen regulation via the androgen receptor (AR) signalling pathway. The AR is a 110 kDa member of the steroid‐receptor family and contains four modular domains: the ligand‐binding domain (LBD), the hinge region, the DNA‐binding domain (DBD) and the N‐terminal domain (NTD) (Fig. 1 1, 2, 3) 1. The C‐terminal LBD mediates activation of transcription by the binding of ligand 1. The DBD facilitates binding to DNA and the hinge region is important for nuclear localisation 1. The NTD is responsible for regulation of transcription 1.
Figure 1.
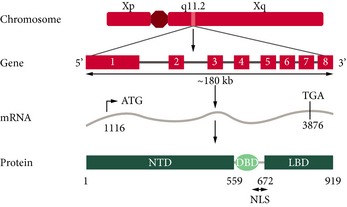
Schematic representation of the structure of the human AR 1, 2, 3. Adapted with permission from Hu et al. 2 and Quigley et al. 3. NLS, nuclear localisation signal.
Within a normal prostate, the AR [regulated by testosterone and dihydrotestosterone (DHT)], has a homeostatic function to balance the rate of cell proliferation with the rate of apoptosis. In prostate epithelial and stromal cells, testosterone is converted into the more active form, DHT, by 5α‐reductase 2, 4. Testosterone or DHT binds to the LBD of the AR, causing a conformational change and activation of the receptor. This leads to homodimer formation and translocation of the AR from the cytoplasm to the nucleus. Within the nucleus, the AR binds to androgen response elements of DNA, recruiting co‐factors (co‐activators or co‐repressors) that regulate transcription of target genes, e.g. PSA. In malignant prostate cells, the AR signalling pathway drives uncontrolled growth and the balance between the rate of cell proliferation and the rate of apoptosis is lost (Fig. 2) 4. The AR signalling pathway plays a key role in all phases of prostate cancer, from disease initiation to disease progression, including metastatic transformation and spread 4, 5.
Figure 2.
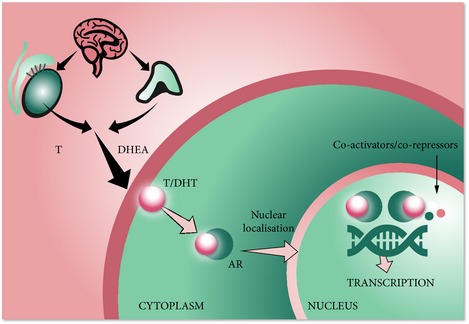
The androgen signalling pathway 4. DHEA, dehydroepiandesterone; T, testosterone.
Different stages within the prostate cancer disease continuum can be clinically defined by the presence or absence of detectable metastases and whether testosterone concentrations are in the castrate range 6. Treatment of prostate cancer, at any stage in the disease continuum, is determined by tumour characteristics, PSA level, extent of tumour spread, estimated life‐expectancy of the patient, whether or not symptoms are present, and progression on previous treatments 7.
For most patients with metastatic prostate cancer, androgen‐deprivation therapy (ADT) with LHRH agonists or antagonists, or orchidectomy, constitutes first‐line therapy 7, 8. Medical ADT with LHRH agonists or antagonists can involve monotherapy or can be combined with antiandrogens (e.g. bicalutamide) that block the effect of any residual testosterone. This is known as combined androgen blockade. ADT strategies can produce a dramatic improvement of symptoms, but invariably prostate cancer stops responding to androgen suppression and progresses. The average time to development of castration‐resistant prostate cancer (CRPC) after commencement of ADT is 2–3 years 9, 10. While this state has previously been referred to as ‘hormone refractory’ or ‘androgen independent’, advancements in the understanding of disease progression and the continued involvement of the AR have led to this state being termed ‘castration‐resistant prostate cancer’ 10. CRPC is defined by serial rises in PSA values despite castrate levels of testosterone, and/or evidence of disease progression on imaging studies 11. Compared with hormone‐sensitive prostate cancer, the prognosis for patients with CRPC is poor and survival is reduced 12. In clinical trials, the median survival of patients with CRPC varies between 9 and 30 months 12.
This review highlights the continued importance of AR signalling in the treatment of patients with CRPC and discusses the clinical benefits associated with inhibition of the AR signalling pathway maintained even in later stages of the disease.
Evidence Acquisition
A literature search was conducted, using PubMed and congress abstracts, to identify articles relating to the role of AR signalling in CRPC and therapies that inhibit the AR signalling pathway. Key words included AR signalling, mCRPC, abiraterone and enzalutamide.
Evidence Synthesis
Continued AR Signalling in CRPC
Significant progress has been made in the understanding of the underlying cancer biology of CRPC. The AR signalling pathway is reactivated in CRPC and is a key driver of tumour progression in this condition. This is demonstrated by levels of PSA, which is a protein known to be regulated by AR signalling, which is present in high levels in most patients with CRPC 13. In addition, many genes known to be regulated by the AR signalling pathway are also expressed in CRPC 13.
Various molecular mechanisms have been proposed to explain how prostate tumour cells continue to use the AR signalling pathway for growth despite castrate levels of testosterone (Fig. 3) 2, 4, 13, 14, 15, 16. The most commonly reported mechanisms of continued AR signalling are extragonadal steroidogenesis and AR gene amplification.
Figure 3.
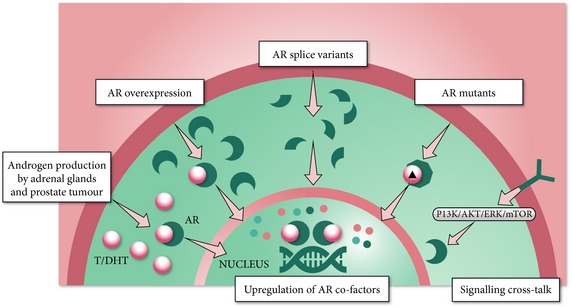
Potential mechanisms of continued AR signalling in CRPC 2, 4, 16, 33. T, testosterone.
Androgen synthesis by the adrenal glands is known to continue despite medical or surgical castration 2. Intratumoral testosterone/DHT production is also implicated in the pathogenesis of CRPC. In addition, the synthesis of androgen from circulating precursors occurs within prostate tumour cells, reactivating AR signalling in CRPC despite serum levels of testosterone in the castrate range 5.
The incidence of AR gene amplification in untreated primary prostate cancer is low 14. In comparison, studies have shown that 50–85% of tumours in CRPC have an increased AR gene copy number causing upregulation of AR gene expression. This leads to overexpression of AR in the cytoplasm of CRPC cells and consequent sensitisation of the tumour to low levels of androgens, which confers a survival and growth advantage upon tumours 14, 15, 17. This suggests that AR gene amplification and AR overexpression contribute to the progression of prostate cancer to a castration‐resistant state 14, 18. AR overexpression also appears to promote the conversion of first‐generation antiandrogens, e.g. bicalutamide, to full agonists 14.
Raised levels of AR splice variants have also been found in some CRPC tissue specimens 16. Transcriptional active splice variants have also been detected in normal prostate tissue 16. AR splice variants are a result of alternative splicing of the AR primary gene transcript. Up to seven different AR splice variants that lack the LBD have been identified in a range of human prostate tissues 16. In particular, expression levels of AR‐V7 are found to be elevated 20‐fold in CRPC when compared with hormone‐naïve prostate cancer 16, 19. Although AR splice variants are associated with the progression of prostate cancer, the exact role they play in CRPC is complex and not yet fully understood. Some AR splice variants that lack the LBD have been shown to be constitutively active, suggesting that AR signalling could occur in the complete absence of ligand binding 16, 19. This mechanism of continued AR signalling may drive resistance to therapies that rely on binding to the LBD for their activity. AR splice variants are more common when residual androgen levels are low, suggesting that AR splice variants may arise as a result of ADT 20, 21. Constitutively active AR splice variants are also associated with increased expression of N‐cadherin, a protein that plays an important role in cell adhesion, which in turn induces epithelial–mesenchymal transition (EMT) 22. EMT is a physiological process whereby epithelial cells are converted into mesenchymal cells during embryonic development as part of organ development 22. After embryogenesis, it was assumed that EMT is ‘switched off’; however, it is now thought that EMT is reactivated by malignant cells 22. This transformation may be responsible for the formation of metastatic tumours via dissemination of cells from the primary tumour site to tissue sites such as the liver, lungs or bone marrow 22.
AR gene mutations constitute another genetic alteration involved in the progression to CRPC. The incidence of AR gene mutations has been found to increase with cancer stage, with ≈10% of CRPC tumours found to have such mutations 2, 4. Most of the AR mutations characterised to date occur in the LBD and may alter ligand binding. This may allow activation of the AR signalling pathway by alternative ligands, such as some first‐generation antiandrogens and steroids 23. The AR LBD mutations may provide a mechanistic explanation for the development of resistance to antiandrogen therapy 4.
Another proposed biological mechanism for the reactivation of AR signalling in CRPC is cross‐talk between signal transduction pathways 2, 4. Two or more signal transduction pathways often cross‐talk via the activation or inhibition of downstream signalling molecules or target genes common to all pathways 2, 4. Cross‐talk between growth factor signalling pathways may influence the phosphorylation of AR and AR co‐regulators to stimulate AR activity 2, 4. Growth factor kinase signalling pathways, including phosphoinositide‐3‐kinase (PI3K), protein kinase B (AKT), extracellular signal‐regulated kinase (ERK) and mammalian target of rapamycin (mTOR), have been shown to stimulate AR target gene expression in the absence of an AR ligand 2, 4. Studies have shown that the PI3K/AKT signalling pathway is upregulated in 30–50% of prostate cancer cases, with the associated loss of phosphatase and tensin homolog (PTEN) function shown to enhance CRPC disease development 24. The loss of PTEN function increases the expression and signalling of the pro‐inflammatory chemokine ligand 8 (CXCL8) in prostate cancer cells. CXCL8 signalling is associated with the activation of additional signalling pathways, leading to upregulation of anti‐apoptotic proteins and, ultimately, the survival of cancer cells 25.
The AR Signalling Pathway and Current Treatment Options in mCRPC
As our understanding of the biological mechanisms underlying disease progression expands, the treatment landscape in CRPC has been evolving. Cytotoxic chemotherapy agents, such as docetaxel and cabazitaxel, have shown survival benefits in patients with mCRPC 26, 27, 28. Taxanes (docetaxel and cabazitaxel) cause apoptosis of prostate cancer cells by stabilising their microtubule network 28, 29. The stabilisation of microtubules inhibits their disassembly, preventing cell division and promoting cell‐cycle arrest 28, 29. An indirect consequence of the stabilisation of microtubules by taxanes is inhibition of AR nuclear translocation 30. Docetaxel was the first agent to show improvement in overall survival (OS), in addition to palliative benefits in patients with mCRPC who had progressed on ADT 28. Cabazitaxel has shown an OS benefit in patients with mCRPC whose disease has progressed during or after treatment with docetaxel 26.
Current understanding of prostate cancer has identified the AR signalling pathway as a logical target for the treatment of CRPC. Available therapies that inhibit the AR signalling pathway include AR blockers (bicalutamide, nilutamide and flutamide), androgen biosynthesis inhibitors (ketoconazole and abiraterone) and AR signalling inhibitors (enzalutamide). Other available treatments with different mechanisms of actions include sipuleucel‐T, an autologous cellular immunotherapy and radium‐223, a radiopharmaceutical that targets bone metastases. Agents that do not affect the AR signalling pathway are not covered in the present review.
AR blockers
Traditionally, antiandrogens (bicalutamide, nilutamide or flutamide) have been added to ADT at PSA progression, with the objective of achieving a more complete androgen blockade. Mainly used in advanced prostate cancer, these agents provide modest survival benefits of ≈3% improvement in survival at 5 years 31.
After long‐term use of antiandrogens combined with ADT, some patients will respond to the selective discontinuation of the antiandrogen. This is known as ‘antiandrogen withdrawal syndrome’, which is characterised by decreasing PSA levels and regression of the tumour on discontinuation of the antiandrogen 32, indicating that antiandrogens can serve as AR agonists under specific circumstances.
AR overexpression occurs in most cases of CRPC and is perhaps the most common mechanism whereby antiandrogens gain agonist activity 14, 17. In vitro studies of AR overexpressing cells indicate that first‐generation antiandrogens induce changes in the AR that continue to allow nuclear translocation, DNA binding and co‐activator recruitment at variable efficiencies 14, 33. Up‐regulation of co‐activators may also allow activation of wild‐type AR by some antiandrogens, thus demonstrating agonist activity 4, 34. In the presence of AR gene mutations, some antiandrogens may confer agonist activity 4.
Androgen biosynthesis inhibitors
The androgen biosynthesis inhibitor ketoconazole has been suggested to have limited clinical efficacy in CRPC 35. However, its widespread use in this condition is now restricted due to significant side‐effects and the need to co‐administer with steroids.
Abiraterone irreversibly and selectively blocks cytochrome P450 17A1 (CYP17A1), indirectly inhibiting production of androgens from the testes, adrenal glands and from the prostate tumour itself 36. Elevated mineralocorticoid levels due to CYP17 blockade by abiraterone require co‐administration with prednisone to suppress adrenocorticotrophic hormone and reduce the adverse events (AEs) associated with mineralocorticoid excess. Abiraterone has shown efficacy over placebo in both chemotherapy‐naïve and post‐docetaxel patients with mCRPC 37, 38. In the chemotherapy‐naïve setting, the AEs of fatigue, arthralgia, peripheral oedema, grade 3 or 4 mineralocorticoid‐related AEs, and abnormalities on liver‐function testing were reported more frequently in the abiraterone‐prednisone group than in the prednisone‐alone group 38. A similar tolerability profile was reported for abiraterone‐prednisone in the post‐docetaxel study 37.
AR signalling inhibitors in mCRPC
The AR signalling pathway remains implicated throughout the prostate cancer disease continuum and reactivation of the AR signalling pathway is thought to be a key driver of CRPC progression 5. The underlying molecular mechanisms of CRPC progression are considered to affect tumour growth and some have been shown to potentiate agonist activity of first‐generation antiandrogens, such as the AR blocker bicalutamide 14, 33. The AR signalling pathway is a logical target for novel therapies in CRPC. AR blockers, e.g. bicalutamide, have provided the starting point for development of AR signalling inhibitors. These therapies would need to be potent AR inhibitors capable of avoiding significant agonist activity.
Enzalutamide is the first approved AR signalling inhibitor (also described as an AR inhibitor), with a novel mechanism of action, that distinguishes it from both androgen biosynthesis inhibitors (e.g. abiraterone) and first‐generation antiandrogens (e.g. bicalutamide) 39, 40.
Enzalutamide was rationally designed by conducting a chemical synthesis programme to identify novel chemical structures that would be potent AR inhibitors in CRPC without significant agonist activity 41. Based on structure‐activity relationships and optimisation of half‐life and oral bioavailability, enzalutamide was selected for further preclinical and clinical studies 41, 42. This approach to drug discovery has provided a new model for the rational design and development of AR signalling inhibitors.
Other AR signalling inhibitors currently being investigated include ODM‐201 and ARN‐509. ODM‐201 has been shown to inhibit AR nuclear translocation in preclinical studies and 86% of patients had a ≥50% PSA level decrease with a 1 400‐mg dose of ODM‐201 in a Phase I/II study 43, 44. ARN‐509 targets the AR signalling pathway by binding to AR and a Phase I study has demonstrated a ≥50% PSA level decrease in 46.7% of patients 45, 46. The Phase III SPARTAN trial will evaluate the efficacy and safety of ARN‐509 in patients with non‐metastatic CRPC at high risk of progression 47.
Direct AR Inhibition with Enzalutamide
Extensive in vitro studies have shown that enzalutamide targets the AR signalling pathway at three key stages, exerting its effect by blocking binding of androgens to AR, by inhibiting nuclear translocation of activated AR, and by impairing binding of activated AR with DNA (Fig. 4) 16, 33. In preclinical CRPC models, enzalutamide has been shown to competitively inhibit androgen binding to the receptor by binding with five‐ to eight‐times higher affinity than bicalutamide, but without partial agonist activity 33. Molecular modelling suggests that enzalutamide sits in the ligand binding domain in a manner that is distinct from bicalutamide and offers mechanism for the partial agonist activity of bicalutamide 48. AR localisation studies have shown that enzalutamide inhibits nuclear translocation of the AR and impairs DNA binding and activation in preclinical CRPC models 33.
Figure 4.
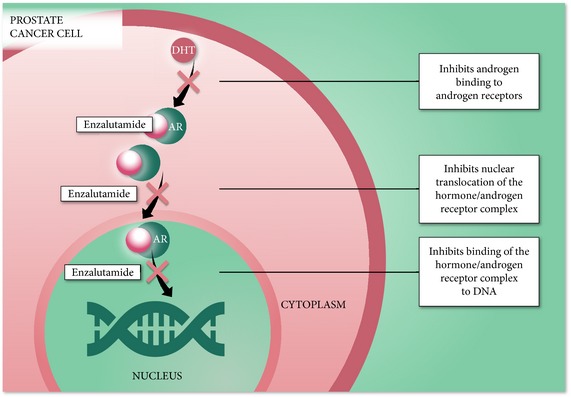
Enzalutamide Clinical Studies
The theoretical benefit of enzalutamide, designed to interrupt the androgen signalling mechanism in prostate cancer cells, has been shown to translate into clinical responses in patients with mCRPC 49, 50.
A Phase I/II, open‐label, uncontrolled, dose‐escalation study in patients with progressive CRPC with or without detectable metastases, demonstrated the antitumour activity of enzalutamide, both in patients who had previously received chemotherapy and those who had not 51. At the end of the study, maximal PSA level decrease did not differ significantly by prior chemotherapy status 51.
The clinical efficacy and safety of enzalutamide in patients with mCRPC has been shown in two Phase III randomised clinical trials, thus further supporting the AR signalling pathway as a therapeutic target in mCRPC 49, 50.
In the Phase III AFFIRM trial, 1 199 men with mCRPC who had received prior docetaxel‐based chemotherapy were randomly assigned to receive either enzalutamide (160 mg as a single oral dose, once‐daily) or placebo in a 2:1 ratio 50. Concomitant therapy with steroids was permitted but not required 50. At the time of the interim analysis (after 520 deaths), a significant benefit in the primary endpoint of OS was seen, with a 37% reduction in risk of death favouring enzalutamide compared with placebo (hazard ration [HR] = 0.63, 95% CI 0.53–0.75; P < 0.001, median OS 18.4 vs 13.6 months, respectively). This effect was consistent across all patient subgroups analysed. Enzalutamide was associated with significant improvements in all secondary efficacy endpoints compared with placebo, including radiographic progression‐free survival (rPFS), time to PSA progression, PSA response rate, and soft‐tissue response rate 50. Enzalutamide was also associated with significantly better patient‐related outcomes compared with placebo (i.e. health‐related quality of life, time to first skeletal‐related event and pain) 50, 52. Additional secondary analyses have shown consistent benefits in OS, rPFS and time to PSA progression with enzalutamide in elderly (≥75 years) and younger (<75 years) patients 53, in patients with different levels of disease severity assessed by baseline PSA levels, and in both North American‐ and European‐treated patients 54, 55.
In the Phase III PREVAIL trial, 1 717 chemotherapy‐naïve patients with progressive mCRPC who had failed on ADT were randomly assigned to receive either enzalutamide (160 mg as a single oral dose, once‐daily) or placebo in a 1:1 ratio 49. The patient population included ≈12% of patients with visceral metastases, typically not included in CRPC trials. At the time of the planned interim analysis (after 540 deaths) enzalutamide treatment resulted in significant benefits in both co‐primary endpoints vs placebo, significantly reducing the risk of radiographic progression by 81% (HR 0.19, 95% CI 0.15–0.23; P < 0.001) or death by 29% (HR 0.71, 95% CI 0.60–0.84; P < 0.001). The rPFS and OS benefits were seen across all subgroups including age, baseline pain intensity, geographical region, and type of disease progression at entry 49. In addition, enzalutamide was associated with significant benefits over placebo across all secondary outcome measures (i.e. time to chemotherapy initiation, time to PSA progression, reduction in PSA level, objective soft‐tissue response, and quality of life maintenance) 49.
In both the AFFIRM and PREVAIL studies, enzalutamide was generally well tolerated 49, 50. In AFFIRM, the most common AEs reported more frequently in the enzalutamide group compared with placebo included fatigue (34% vs 29%), diarrhoea (21% vs 18%), and hot flushes (20% vs 10%) 50. The rate of discontinuations due to AEs was low in both the enzalutamide and placebo groups (8% vs 10%) 50. Of 800 patients treated with enzalutamide, five (0.6%) had a seizure, whereas no seizures occurred in patients receiving placebo 50. One additional patient was identified with an event termed syncope with features suggestive of a seizure, and another patient was diagnosed with a seizure after the interim analysis cut‐off date 52. Enzalutamide was not associated with liver or cardiac toxicity 50.
In the PREVAIL trial in patients with chemotherapy‐naïve mCRPC, enzalutamide showed a generally favourable tolerability profile, despite the reporting period for enzalutamide being more than twice that for placebo 49. AEs that occurred in ≥20% of patients receiving enzalutamide at a rate that was at least 2% higher than that in the placebo group were fatigue (36% vs 26%), back pain (27% vs 22%), constipation (22% vs 17%), and arthralgia (20% vs 16%). Cardiac AEs were seen in 10% of patients receiving enzalutamide compared with 8% of placebo patients. Hypertension was more commonly seen in the enzalutamide group than in the placebo group (13% vs 4%). Two seizures were reported during the study, one in each treatment arm. The seizure in the enzalutamide treatment arm occurred after the data cut‐off date.
Sequencing of Hormonal Therapies
While the number of treatments available across the prostate cancer disease continuum that target or have an effect on the AR continues to grow, treatment decisions in the management of patients with mCRPC are becoming more complex.
There have been several retrospective reports in the literature, with some studies involving patients initially included in early access or compassionate use programmes prior to commercial availability of enzalutamide in various countries. Typically they were based on a few heavily pre‐treated patients, and none of the studies were designed to specifically investigate sequential treatment in patients with mCRPC in the post‐chemotherapy setting (Table 1) 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71. In this population of heavily pre‐treated patients with advanced mCRPC, the effectiveness of enzalutamide appeared to be attenuated by prior treatment with abiraterone, with a generally smaller proportion of patients experiencing ≥50% decline in PSA level with enzalutamide after prior treatment with abiraterone (Table 1) compared with patients treated with enzalutamide post‐docetaxel in the AFFIRM Phase III study (54%) 50. None of the reports identified any reliable predictors of response to enzalutamide. Similarly, subsequent treatment with abiraterone in patients who progressed on enzalutamide treatment also only experienced modest responses (Table 1). However, significant cabazitaxel activity was reported in patients with mCRPC progressing after abiraterone or enzalutamide 71.
Table 1.
Retrospective studies of hormonal sequencing with enzalutamide in patients with mCRPC
| Reference | Sequence of hormonal therapies | Type of study | Number of patients | ≥50% decline in PSA with second treatment, % | Key study notes |
|---|---|---|---|---|---|
| Docetaxel, abiraterone → enzalutamide | |||||
| Badrising et al. 56 | Docetaxel and abiraterone prior to enzalutamide | Retrospective | 61 | 21 | Enzalutamide had modest clinical activity in patients with mCRPC who previously received docetaxel and abiraterone; PSA response to docetaxel and abiraterone did not predict PSA response to enzalutamide |
| Bournakis et al. 57 | Abiraterone and/or orterenol (25 of 35 patients) prior to enzalutamide | Patient access programme | 35 | 40 (six out of 15 evaluable patients) | Enzalutamide benefited a subset of patients resistant to prior abiraterone treatment |
| Scholz et al. 58 | Docetaxel, abiraterone prior to enzalutamide | Retrospective | 66 | NR; 29% had >30% decline in PSA | Enzalutamide had activity in a heavily pre‐treated population of men resistant to abiraterone and docetaxel |
| Caffo et al. 59 | Docetaxel prior to second‐ and third‐line agent (abiraterone, cabazitaxel, or enzalutamide) | Retrospective | 260 | 20 (70 out of 260 patients received enzalutamide as third‐line agent) | No difference in clinical outcomes of abiraterone, cabazitaxel, or enzalutamide as third‐line treatment was seen, regardless of the previous treatment |
| Schrader et al. 60 | Docetaxel and abiraterone prior to enzalutamide | Retrospective | 35 | 29 | Patients progressing after abiraterone achieved only a modest response rate with enzalutamide; a small but significant number of patients showed significant benefit from sequential treatment |
| Bianchini et al. 61 | Docetaxel and abiraterone prior to enzalutamide | Retrospective | 39 | 13 | Limited activity of enzalutamide was reported in the post‐docetaxel and post‐abiraterone patient population |
| Thomsen et al. 62 | Docetaxel and abiraterone prior to enzalutamide | Retrospective | 24 | 17 | Previous abiraterone therapy was associated with a less marked reduction in PSA level following enzalutamide treatment, compared with reported results in randomised studies |
| Brasso et al. 63 | Docetaxel and abiraterone prior to enzalutamide | Compassionate use programme | 137 | 18 | Modest PSA level responses and improved survival was seen with enzalutamide in patients progressing after chemotherapy and abiraterone |
| Schmid et al. 64 | Docetaxel and abiraterone prior to enzalutamide | Compassionate use programme | 35 | 10 | Consecutive use of enzalutamide and abiraterone after taxane‐based chemotherapy shows a modest clinical activity |
| Thomson et al. 65 | Docetaxel and abiraterone prior to enzalutamide | Expanded access programme | 23 | 39 | Enzalutamide appears to show only modest activity after failure of docetaxel and abiraterone |
| Sandhu et al. 66 | Abiraterone prior to enzalutamide | Retrospective study | 23 | 17 (four out of 23 patients) | Sequential enzalutamide in patients with CRPC post‐abiraterone showed only modest activity |
| Vera‐Badillo et al. 67 | Docetaxel and abiraterone prior to enzalutamide | Retrospective study | 26 | 27 | Modest clinical activity seen with enzalutamide treatment who progressed on docetaxel and abiraterone |
| Cheng et al. 68 | Abiraterone and/or docetaxel prior to enzalutamide | Retrospective study | 310 | 24 (65 out of 274 patients with prior abiraterone and/or docetaxel) | Enzalutamide activity was blunted after abiraterone and after docetaxel; among patients with primary resistance to abiraterone, a subset was sensitive to subsequent enzalutamide |
| Enzalutamide → abiraterone or cabazitaxel | |||||
| Noonan et al. 69 | Enzalutamide prior to abiraterone + prednisone | Retrospective | 30 | 3 | Abiraterone treatment was associated with a modest response rate and brief duration of effect in patients who progressed after enzalutamide |
| Loriot, et al. 70 | Docetaxel and enzalutamide prior to abiraterone + prednisolone | Retrospective | 38 | 8 | Abiraterone plus prednisolone had modest antitumour activities in patients with CRPC pre‐treated with docetaxel and enzalutamide |
| Pezaro et al. 71 | Docetaxel, abiraterone and/or enzalutamide prior to cabazitaxel | Retrospective | 59 | 39 | Significant cabazitaxel activity was reported in patients with CRPC progressing after docetaxel and abiraterone or enzalutamide |
NR, not reported.
Mechanisms of Resistance to Enzalutamide
In general, studies that have investigated the sequencing of hormonal therapies have shown that with any treatment a reduced response is expected with each subsequent line of therapy. Recent research, primarily in patients who progressed on chemotherapy and abiraterone, suggests that one marker for resistance to AR‐targeted therapies is AR‐V7, a constitutively active AR splice variant 72. Based on the currently available data, detection of AR‐V7 in circulating tumour cells from patients with CRPC may be associated with resistance to enzalutamide and abiraterone 72. In a small Phase II prospective study evaluating the effect of enzalutamide in blood and bone marrow, the presence of an AR‐V7 variant was associated with primary resistance to enzalutamide 73. In addition, in vivo studies have shown that cabazitaxel remained highly effective in enzalutamide‐resistant tumours in castrated mice and demonstrated superior antitumour activity compared with docetaxel. These findings suggest that there could be cross‐resistance in the AR pathway between enzalutamide and docetaxel, but not with cabazitaxel, in CRPC 74.
The results of the studies summarised in this section should be interpreted with caution, as they present analyses of small, retrospective case series, typically involving populations of patients previously progressing on more than two previous therapies and in different stages of mCRPC. None of these reports were based on studies specifically designed to assess the efficacy of sequential treatments. Larger, prospective sequencing and combination studies are required to assess the impact of treatments and to define optimal sequencing strategies for patients with mCRPC as the treatment paradigm continues to evolve.
Conclusions
Progress is being made in the treatment of mCRPC with increased understanding of the underlying molecular mechanisms involved in CRPC disease progression. This improved understanding is being applied to rationally designed pharmacological treatment options with novel mechanisms of action, e.g. enzalutamide, for patients with mCRPC. Enzalutamide inhibits three steps in the AR signalling pathway and positive results of Phase III clinical trials validate the AR signalling pathway as a therapeutic target in CRPC 49, 50. Enzalutamide is administered orally, can be taken with or without food, and does not require concomitant administration of steroids. Enzalutamide has not shown signs of liver toxicity and does not require specific monitoring 50.
Prostate cancer is a markedly heterogeneous disease, with potential for multiple mechanisms of resistance to castration. Despite the clinical benefits obtained with currently available drugs for mCRPC, patients will ultimately become resistant and disease progression will eventually occur. Better understanding of drug resistance mechanisms and evaluation of treatment combinations are areas of ongoing research that have, and will continue to, become more important as the treatment options for CRPC evolve and expand.
Funding/Support and Role of the Sponsor
The authors participated in reviewing the paper. Enzalutamide is co‐developed by Medivation, Inc., and Astellas Pharma, Inc. This article was reviewed by each company for medical and scientific accuracy, but the authors had full control of the content and incorporation of comments from the companies was left up to their discretion.
Author Contributions
J.S takes responsibility for the integrity and accuracy of the paper. J.S has read and approved the final version of this manuscript. J.M.F. passed away during development of this manuscript.
Conflicts of Interest
J.S. has received speaker honoraria from Astellas, Sanofi Aventis, Ipsen, and Janssen Oncology.
Abbreviations
- ADT
androgen‐deprivation therapy
- AE
adverse event
- AKT
protein kinase B
- AR
androgen receptor
- (m)CRPC
(metastatic) castration‐resistant prostate cancer
- CXCL8
chemokine ligand 8
- CYP17A1
cytochrome P450 17A1
- DBD
DNA‐binding domain
- DHT
dihydrotestosterone
- EMT
epithelial–mesenchymal transition
- HR
hazard ratio
- LBD
ligand‐binding domain
- NTD
N‐terminal domain
- OS
overall survival
- PI3K
phosphoinositide‐3‐kinase
- PTEN
phosphatase and tensin homolog
- rPFS
radiographic progression‐free survival
Acknowledgements
The authors would like to thank Amineh Zafarani, PhD, at Ogilvy Healthworld and Joy Ramos, PhD, at Complete HealthVizion for assistance with writing and revising the draft manuscript based on detailed discussion and feedback from both authors, and Lauren Smith at Complete HealthVizion for copyediting assistance. Writing and copyediting assistance was funded by Astellas Pharma, Inc. and Medivation, Inc.
Professor John M. Fitzpatrick (1948–2014) contributed to the manuscript before passing away.
References
- 1. Gelmann EP. Molecular biology of the androgen receptor. J Clin Oncol 2002; 20: 3001–15 [DOI] [PubMed] [Google Scholar]
- 2. Hu R, Denmeade SR, Luo J. Molecular processes leading to aberrant androgen receptor signaling and castration resistance in prostate cancer. Expert Rev Endocrinol Metab 2010; 5: 753–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Quigley CA, De Bellis A, Marschke KB, el‐Awady MK, Wilson EM, French FS. Androgen receptor defects: historical, clinical, and molecular perspectives. Endocr Rev 1995; 16: 271–321 [DOI] [PubMed] [Google Scholar]
- 4. Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev 2004; 25: 276–308 [DOI] [PubMed] [Google Scholar]
- 5. Knudsen KE, Kelly WK. Outsmarting androgen receptor: creative approaches for targeting aberrant androgen signaling in advanced prostate cancer. Expert Rev Endocrinol Metab 2011; 6: 483–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Scher HI, Heller G. Clinical states in prostate cancer: toward a dynamic model of disease progression. Urology 2000; 55: 323–7 [DOI] [PubMed] [Google Scholar]
- 7. Horwich A, Hugosson J, de Reijke T, Wiegel T, Fizazi K, Kataja V. Prostate cancer: ESMO Consensus Conference Guidelines 2012. Ann Oncol 2013; 24: 1141–62 [DOI] [PubMed] [Google Scholar]
- 8. Heidenreich A, Bastian PJ, Bellmunt J et al. Guidelines on prostate cancer: European Association of Urology. Available at: http://www.uroweb.org/gls/pdf/09_Prostate_Cancer_LR.pdf. 2013. Accessed December 2014
- 9. Attar RM, Takimoto CH, Gottardis MM. Castration‐resistant prostate cancer: locking up the molecular escape routes. Clin Cancer Res 2009; 15: 3251–5 [DOI] [PubMed] [Google Scholar]
- 10. Ryan CJ, Tindall DJ. Androgen receptor rediscovered: the new biology and targeting the androgen receptor therapeutically. J Clin Oncol 2011; 29: 3651–8 [DOI] [PubMed] [Google Scholar]
- 11. Higano CS, Crawford ED. New and emerging agents for the treatment of castration‐resistant prostate cancer. Urol Oncol 2011; 29: S1–8 [DOI] [PubMed] [Google Scholar]
- 12. Kirby M, Hirst C, Crawford ED. Characterising the castration‐resistant prostate cancer population: a systematic review. Int J Clin Pract 2011; 65: 1180–92 [DOI] [PubMed] [Google Scholar]
- 13. Mostaghel EA, Montgomery B, Nelson PS. Castration‐resistant prostate cancer: targeting androgen metabolic pathways in recurrent disease. Urol Oncol 2009; 27: 251–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen CD, Welsbie DS, Tran C et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med 2004; 10: 33–9 [DOI] [PubMed] [Google Scholar]
- 15. Visakorpi T, Hyytinen E, Koivisto P et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet 1995; 9: 401–6 [DOI] [PubMed] [Google Scholar]
- 16. Watson PA, Chen YF, Balbas MD et al. Constitutively active androgen receptor splice variants expressed in castration‐resistant prostate cancer require full‐length androgen receptor. Proc Natl Acad Sci USA 2010; 107: 16759–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Attard G, Swennenhuis JF, Olmos D et al. Characterization of ERG, AR and PTEN gene status in circulating tumor cells from patients with castration‐resistant prostate cancer. Cancer Res 2009; 69: 2912–8 [DOI] [PubMed] [Google Scholar]
- 18. Koivisto P, Kononen J, Palmberg C et al. Androgen receptor gene amplification: a possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res 1997; 57: 314–9 [PubMed] [Google Scholar]
- 19. Hu R, Dunn TA, Wei S et al. Ligand‐independent androgen receptor variants derived from splicing of cryptic exons signify hormone‐refractory prostate cancer. Cancer Res 2009; 69: 16–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sun S, Sprenger CC, Vessella RL et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest 2010; 120: 2715–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang X, Morrissey C, Sun S et al. Androgen receptor variants occur frequently in castration resistant prostate cancer metastases. PLoS One 2011; 6: e27970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cottard F, Asmane I, Erdmann E, Bergerat JP, Kurtz JE, Céraline J. Constitutively active androgen receptor variants upregulate expression of mesenchymal markers in prostate cancer cells. PLoS One 2013; 8: e63466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Richards J, Lim AC, Hay CW et al. Interactions of abiraterone, eplerenone, and prednisolone with wild‐type and mutant androgen receptor: a rationale for increasing abiraterone exposure or combining with MDV3100. Cancer Res 2012; 72: 2176–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Morgan TM, Koreckij TD, Corey E. Targeted therapy for advanced prostate cancer: inhibition of the PI3K/Akt/mTOR pathway. Curr Cancer Drug Targets 2009; 9: 237–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maxwell PJ, Coulter J, Walker SM et al. Potentiation of inflammatory CXCL8 signalling sustains cell survival in PTEN‐deficient prostate carcinoma. Eur Urol 2013; 64: 177–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. de Bono JS, Oudard S, Ozguroglu M et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration‐resistant prostate cancer progressing after docetaxel treatment: a randomised open‐label trial. Lancet 2010; 376: 1147–54 [DOI] [PubMed] [Google Scholar]
- 27. Petrylak DP, Tangen CM, Hussain MH et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med 2004; 351: 1513–20 [DOI] [PubMed] [Google Scholar]
- 28. Tannock IF, de Wit R, Berry WR et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med 2004; 351: 1502–12 [DOI] [PubMed] [Google Scholar]
- 29. Fitzpatrick JM, de Wit R. Taxane mechanisms of action: potential implications for treatment sequencing in metastatic castration‐resistant prostate cancer. Eur Urol 2014; 65: 1198–204 [DOI] [PubMed] [Google Scholar]
- 30. Darshan MS, Loftus MS, Thadani‐Mulero M et al. Taxane‐induced blockade to nuclear accumulation of the androgen receptor predicts clinical responses in metastatic prostate cancer. Cancer Res 2011; 71: 6019–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Prostate Cancer Trialists’ Collaborative Group . Maximum androgen blockade in advanced prostate cancer: an overview of the randomised trials. Lancet 2000; 355: 1491–8 [PubMed] [Google Scholar]
- 32. Kelly WK, Slovin S, Scher HI. Steroid hormone withdrawal syndromes. Pathophysiology and clinical significance. Urol Clin North Am 1997; 24: 421–31 [DOI] [PubMed] [Google Scholar]
- 33. Tran C, Ouk S, Clegg NJ et al. Development of a second‐generation antiandrogen for treatment of advanced prostate cancer. Science 2009; 324: 787–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Miyamoto H, Yeh S, Wilding G, Chang C. Promotion of agonist activity of antiandrogens by the androgen receptor coactivator, ARA70, in human prostate cancer DU145 cells. Proc Natl Acad Sci USA 1998; 95: 7379–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Small EJ, Halabi S, Dawson NA et al. Antiandrogen withdrawal alone or in combination with ketoconazole in androgen‐independent prostate cancer patients: a phase III trial (CALGB 9583). J Clin Oncol 2004; 22: 1025–33 [DOI] [PubMed] [Google Scholar]
- 36. Schweizer MT, Antonarakis ES. Abiraterone and other novel androgen‐directed strategies for the treatment of prostate cancer: a new era of hormonal therapies is born. Ther Adv Urol 2012; 4: 167–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. de Bono JS, Logothetis CJ, Molina A et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med 2011; 364: 1995–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ryan CJ, Smith MR, de Bono JS et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med 2013; 368: 138–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. XTANDI (enzalutamide) . Summary of Product Characteristics, 2013. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002639/WC500144996.pdf. Accessed April 2015
- 40. XTANDI (enzalutamide) . Prescribing Information, 2014. Available at: https://www.astellas.us/docs/12A005-ENZ-WPI.PDF. Accessed April 2015
- 41. Jung ME, Ouk S, Yoo D et al. Structure‐activity relationship for thiohydantoin androgen receptor antagonists for castration‐resistant prostate cancer (CRPC). J Med Chem 2010; 53: 2779–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen Y, Clegg NJ, Scher HI. Anti‐androgens and androgen‐depleting therapies in prostate cancer: new agents for an established target. Lancet Oncol 2009; 10: 981–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fizazi K, Massard C, Bono P et al. Activity and safety of ODM‐201 in patients with progressive metastatic castration‐resistant prostate cancer (ARADES): an open‐label phase 1 dose‐escalation and randomised phase 2 dose expansion trial. Lancet Oncol 2014; 15: 975–85 [DOI] [PubMed] [Google Scholar]
- 44. Moilanen A, Riikonen R, Oksala R et al. ODM‐201 ‐New Generation Androgen Receptor Inhibitor with Excellent Antiandrogenic and Antitumor Activity in Nonclinical Models of CRPC. Poster P376 presented at the European Cancer Congress 2013
- 45. Clegg NJ, Wongvipat J, Joseph JD et al. ARN‐509: a novel antiandrogen for prostate cancer treatment. Cancer Res 2012; 72: 1494–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rathkopf DE, Morris MJ, Fox JJ et al. Phase I study of ARN‐509, a novel antiandrogen, in the treatment of castration‐resistant prostate cancer. J Clin Oncol 2013; 31: 3525–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Smith MR, Liu G, Shreeve SM et al. A randomized double‐blind, comparative study of ARN‐509 plus androgen deprivation therapy (ADT) versus ADT alone in nonmetastatic castration‐resistant prostate cancer (M0‐CRPC): the SPARTAN trial. J Clin Oncol 2014; 32: abstr TPS5100 [Google Scholar]
- 48. Balbas MD, Evans MJ, Hosfield DJ et al. Overcoming mutation‐based resistance to antiandrogens with rational drug design. Elife 2013; 2: e00499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Beer TM, Armstrong AJ, Rathkopf DE et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med 2014; 371: 424–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Scher HI, Fizazi K, Saad F et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med 2012; 367: 1187–97 [DOI] [PubMed] [Google Scholar]
- 51. Scher HI, Beer TM, Higano CS et al. Antitumour activity of MDV3100 in castration‐resistant prostate cancer: a phase 1‐2 study. Lancet 2010; 375: 1437–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fizazi K, Scher HI, Miller K et al. Effect of enzalutamide on time to first skeletal‐related event, pain, and quality of life in men with castration‐resistant prostate cancer: results from the randomised, phase 3 AFFIRM trial. Lancet Oncol 2014; 15: 1147–56 [DOI] [PubMed] [Google Scholar]
- 53. Sternberg CN, de Bono JS, Chi KN et al. Improved outcomes in elderly patients with metastatic castration‐resistant prostate cancer treated with the androgen receptor inhibitor enzalutamide: results from the phase III AFFIRM trial. Ann Oncol 2014; 25: 429–34 [DOI] [PubMed] [Google Scholar]
- 54. Merseburger AS, Scher HI, Bellmunt J et al. Enzalutamide in European and North American men participating in the AFFIRM trial. BJU Int 2015; 115: 41–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Saad F, de Bono J, Shore N et al. Efficacy outcomes by baseline prostate‐specific antigen quartile in the AFFIRM trial. Eur Urol 2014; 67: 223–30 [DOI] [PubMed] [Google Scholar]
- 56. Badrising S, van der Noort V, van Oort IM et al. Clinical activity and tolerability of enzalutamide (MDV3100) in patients with metastatic, castration‐resistant prostate cancer who progress after docetaxel and abiraterone treatment. Cancer 2014; 120: 968–75 [DOI] [PubMed] [Google Scholar]
- 57. Bournakis E, Gyftaki R, Kafantari E et al. Enzalutamide (ENZA) in Heavily Pretreated Patients with Bone Metastatic Castration Resistant Prostate Cancer (mCRPC) Resistant to Androgen Biosynthesis Inhibitor (ABI) Treatment – the Hellenic Experience of the Name Patient Access Program (NPAP). Poster P413 presented at the European Cancer Congress, 2013
- 58. Scholz MC, Lam RY, Turner JS, Chau KN, Becker LK, Felarca CU. Enzalutamide in men with prostate cancer resistant to docetaxel and abiraterone. J Clin Oncol 2014; 32: abstr 247 [Google Scholar]
- 59. Caffo O, De Giorgi U, Fratino L et al. Clinical outcomes of castration‐resistant prostate cancer treatments administered as third or fourth line following failure of docetaxel and other second‐line treatment: results of an Italian Multicentre Study. Eur Urol 2014; Epub ahead of print, doi: 10.1016/j.eururo.2014.10.014 [DOI] [PubMed] [Google Scholar]
- 60. Schrader AJ, Boegemann M, Ohlmann CH et al. Enzalutamide in castration‐resistant prostate cancer patients progressing after docetaxel and abiraterone. Eur Urol 2014; 65: 30–6 [DOI] [PubMed] [Google Scholar]
- 61. Bianchini D, Lorente D, Rodriguez‐Vida A et al. Antitumour activity of enzalutamide (MDV3100) in patients with metastatic castration‐resistant prostate cancer (CRPC) pre‐treated with docetaxel and abiraterone. Eur J Cancer 2014; 50: 78–84 [DOI] [PubMed] [Google Scholar]
- 62. Thomsen FB, Røder MA, Rathenborg P, Brasso K, Borre M, Iversen P. Enzalutamide treatment in patients with metastatic castration‐resistant prostate cancer progressing after chemotherapy and abiraterone acetate. Scand J Urol 2014; 48: 268–75 [DOI] [PubMed] [Google Scholar]
- 63. Brasso K, Thomsen FB, Schrader AJ et al. Enzalutamide antitumour activity against metastatic castration‐resistant prostate cancer previously treated with docetaxel and abiraterone: a multicentre analysis. Eur Urol 2014; Epub ahead of print, doi: 10.1016/j.eururo.2014.07.028 [DOI] [PubMed] [Google Scholar]
- 64. Schmid SC, Geith A, Boker A et al. Enzalutamide after docetaxel and abiraterone therapy in metastatic castration‐resistant prostate cancer. Adv Ther 2014; 31: 234–41 [DOI] [PubMed] [Google Scholar]
- 65. Thomson D, Charnley N, Parikh O. Enzalutamide after failure of docetaxel and abiraterone in metastatic castrate‐resistant prostate cancer. Eur J Cancer 2014; 50: 1040–1 [DOI] [PubMed] [Google Scholar]
- 66. Sandhu GS, Parikh RA, Appleman LJ, Friedland D. Enzalutamide after abiraterone in patients with metastatic castrate‐resistant prostate cancer (mCRPC). J Clin Oncol 2014; 32: 240 [Google Scholar]
- 67. Vera‐Badillo FE, Leibowitz‐Amit R, Templeton A et al. Clinical activity of enzalutamide against metastatic castration resistant prostate cancer (mCRPC) in patients who have progressed on abiraterone acetate: the Princess Margaret experience. J Clin Oncol 2014; 32: 159 [Google Scholar]
- 68. Cheng HH, Gulati R, Azad A et al. Activity of enzalutamide in men with metastatic castration‐resistant prostate cancer is affected by prior treatment with abiraterone and/or docetaxel. Prostate Cancer Prostatic Dis 2015; 18: 122–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Noonan KL, North S, Bitting RL, Armstrong AJ, Ellard SL, Chi KN. Clinical activity of abiraterone acetate in patients with metastatic castration‐resistant prostate cancer progressing after enzalutamide. Ann Oncol 2013; 24: 1802–7 [DOI] [PubMed] [Google Scholar]
- 70. Loriot Y, Bianchini D, Ileana E et al. Antitumour activity of abiraterone acetate against metastatic castration‐resistant prostate cancer progressing after docetaxel and enzalutamide (MDV3100). Ann Oncol 2013; 24: 1807–12 [DOI] [PubMed] [Google Scholar]
- 71. Pezaro CJ, Omlin AG, Altavilla A et al. Activity of cabazitaxel in castration‐resistant prostate cancer progressing after docetaxel and next‐generation endocrine agents. Eur Urol 2014; 66: 459–65 [DOI] [PubMed] [Google Scholar]
- 72. Antonarakis ES, Lu C, Wang H et al. AR‐V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med 2014; 371: 1028–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Efstathiou E, Titus M, Wen S et al. Molecular characterization of enzalutamide‐treated bone metastatic castration‐resistant prostate cancer. Eur Urol 2015; 67: 53–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. van Soest RJ, de Morrée ES, Kweldam CF et al. Targeting the androgen receptor confers in vivo cross‐resistance between enzalutamide and docetaxel, but not cabazitaxel, in castration‐resistant prostate cancer. Eur Urol 2015; 67: 981–5 [DOI] [PubMed] [Google Scholar]