

Abstract
De novo mutations (DNM) in SYNGAP1, encoding Ras/Rap GTPase‐activating protein SynGAP, have been reported in individuals with nonsyndromic intellectual disability (ID). We identified 10 previously unreported individuals with SYNGAP1 DNM; seven via the Deciphering Developmental Disorders (DDD) Study, one through clinical analysis for copy number variation and the remaining two (monozygotic twins) via a research multi‐gene panel analysis. Seven of the nine heterozygous mutations are likely to result in loss‐of‐function (3 nonsense; 3 frameshift; 1 whole gene deletion). The remaining two mutations, one of which affected the monozygotic twins, were missense variants. Each individual carrying a DNM in SYNGAP1 had moderate‐to‐severe ID and 7/10 had epilepsy; typically myoclonic seizures, absences or drop attacks. 8/10 had hypotonia, 5/10 had significant constipation, 7/10 had wide‐based/unsteady gait, 3/10 had strabismus, and 2/10 had significant hip dysplasia. A proportion of the affected individuals had a similar, myopathic facial appearance, with broad nasal bridge, relatively long nose and full lower lip vermilion. A distinctive behavioral phenotype was also observed with aggressive/challenging behavior and significant sleep problems being common. 7/10 individuals had MR imaging of the brain each of which was reported as normal. The clinical features of the individuals reported here show significant overlap with those associated with 6p21.3 microdeletions, confirming that haploinsufficiency for SYNGAP1 is responsible for both disorders. © 2015 The Authors. American Journal of Medical Genetics Part A Published by Wiley Periodicals, Inc.
Keywords: SYNGAP1, 6p21.3 microdeletion, intellectual disability, epilepsy, syndrome, hypertrichosis, strabismus, hip dysplasia, DDD study, behavioral phenotype
INTRODUCTION
De novo mutations are an important cause of moderate and severe intellectual disability (ID). Heterozygous, de novo loss‐of‐function mutations in SYNGAP1 have been described in 26 individuals to date [Hamdan et al., 2009, 2011a, b; Krepischi et al., 2010; Pinto et al., 2010; Vissers et al., 2010; Zollino et al., 2011; de Ligt et al., 2012; Rauch et al., 2012; Berryer et al., 2013; Carvill et al., 2013; Writzl and Knegt, 2013; Redin et al., 2014]. SYNGAP1 encodes Ras/Rap GTPase‐activating protein SynGAP, which is a major component of the post‐synaptic density that regulates synaptic plasticity and ERK/MAPK signaling probably via N‐methyl‐d‐aspartate (NMDA) receptor activation [Komiyama et al., 2002; Muhia et al., 2010]. SYNGAP1 [603384] has been coded in Online Mendelian Inheritance in Man (OMIM®) as causing mental retardation, autosomal dominant 5 [612621].
In 2009, Hamdan et al. first reported the sequencing of SYNGAP1 in 94 apparently nonsyndromic individuals with intellectual disability; they found de novo mutations in three, thus first‐describing this gene as a cause of nonsyndromic intellectual disability (ID) in humans [Hamdan et al., 2009]. This group subsequently published eight further affected individuals through re‐sequencing predominantly ID cohorts enriched for epilepsy [2011a, b; Berryer et al., 2013]. Carvill et al. performed massively parallel sequencing in 500 individuals with epileptic encephalopathy and identified four patients with de novo SYNGAP1 mutations [Carvill et al., 2013]. Further patients have been described as part of large next generation sequencing studies of individuals with ID [Vissers et al., 2010; de Ligt et al., 2012; Rauch et al., 2012; Redin et al., 2014].
In addition, there have been four individuals with genomic deletions of 6p23.1 involving SYNGAP1, and one with a de novo apparently balanced reciprocal translocation in which one of the breakpoints disrupts SYNGAP1 [Krepischi et al., 2010; Pinto et al., 2010; Klitten et al., 2011; Zollino et al., 2011; Writzl and Knegt, 2013]. Thus the 26 individuals reported to date consist of 21 intragenic mutations, four whole gene deletions, and one translocation. To date, facial images have only been published in six individuals: three in the seminal Hamdan et al. paper, plus three single patients in subsequent papers [Hamdan et al., 2009; Zollino et al., 2011; Rauch et al., 2012; Writzl and Knegt, 2013].
Here, we present molecular and clinical information on 10 previously unreported individuals with de novo mutations in SYNGAP1, most of whom were diagnosed using trio exome sequencing of individuals with undiagnosed developmental disorders. The relatively consistent pattern of clinical features and behavioral anomalies observed in these individuals and in previously reported individuals suggests that there is an emerging SYNGAP1‐associated syndrome.
METHODS
Patient Ascertainment
Seven of the 10 affected individuals were recruited via UK NHS Regional Genetics Services to the Deciphering Developmental Disorders (DDD) study (www.ddduk.org). The eighth individual (7; Table I) was identified as part of routine investigation of ID via a UK NHS paediatric genetics clinic. These eight individuals were seen by the same Paediatric Geneticist (MJP) in addition to the referring Clinical Geneticists. The ninth and tenth individuals are monozygotic twins who were referred for genetic evaluation to the local multi‐disciplinary clinic for children with intellectual disability. See Table I for a summary of the clinical and molecular findings. The Supporting Information online provides additional clinical details.
Table I.
Clinical Features of Ten Previously‐Unreported Patients With SYNGAP1 Haploinsufficiency Reported Herein
| Category/Individual | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| DECIPHER ID | 259041 | 259840 | 258913 | 264135 | 259214 | 259606 | 258536 | 258536 | LEM300469 | LEM300468 |
| Mutation details | ||||||||||
| Genomic coordninates (chr6; hg19) | chr6 g.33406569 CTGTATG>CTG | chr6 g.33400583G>A | chr6 g.33411111C>T | chr6 g.33411093C>T | chr6 g.33400498 AAACGAACGAA> AAACGAA | chr6 g.33411102CT>C | deletion chr6:33201710‐ 33595089 | chr6 g.33411606C>T | chr6 g.33405662T>C | |
| VEP prediction | Transcript; ENST00000418600 frameshift_variant p.LYE517‐519LX | Transcript; ENST00000418600 missense_variant p.R170Q | Transcript; ENST00000418600 stop_gained p.Q928* | Transcript; ENST00000418600 stop_gained p.R922* | Transcript: ENST00000418600 frameshift_variant p.KRTK142‐145KRX | Transcript; ENST00000418600 frameshift_variant p.L925X | multi‐gene deletion; SYNGAP1 plus 18 others | Transcript; ENST00000449372 stop gained p.Q1079* | Transcript; NM_006772.2 missense_variant p.Leu327Pro | |
| Inheritance | de novo | de novo | de novo | de novo | de novo | de novo | de novo | de novo | de novo | de novo |
| Age (years) | 7 | 8 | 7 | 3 | 8 | 12 | 5 | 8 | 14 | 14 |
| Sex | Female | Female | Female | Female | Male | Female | Female | Female | Male | Male |
| Prenatal growth | ||||||||||
| Gestation (weeks) | 40 | 30 | 40 | 41 | 40 | 40 | 41 | 40 | 35 | 35 |
| Birth weight (g) [z score] | 4100 [1.67] | 1360 [0.07] | 3090 [−0.68] | 3600 [0.2] | 3460 [0.21] | 3180 [−0.46] | 3190 [−0.78] | 3650 [0.65] | 2465 [−0.11] | 2460 [−0.12] |
| Head circumference at birth (cm) [z score] | − | − | − | − | − | − | − | − | 32 [−1] | 31.5 [−1.3] |
| NICU admission | No | 4 weeks | No | No | No | No | No | No | − | − |
| Postnatal growth | ||||||||||
| Age when measured (yrs) | 7.1 | 8.0 | 7.1 | 3.1 | 8.1 | 12.1 | 5.1 | 8.1 | 8 y 3 mo | 8 y 3 mo |
| Height (cm) [z score] | 117.5 [−0.82] | 132 [0.85] | 116.4 [−1.03] | 93.5 [−.0.45] | 120 [−1.5] | 131.6 [−2.6] | 103 [−1.4] | 132.5 [0.85] | 119 [−1.8] | 110 [−3.4] |
| Weight (kg) [z score] | 24.8 [0.42] | 37.7 [2.0] | 22.7 [−0.15] | 11.5 [−1.98] | 23.5 [−0.69] | 29.4 [−1.86] | 17.2 [−0.55] | 30.5 [0.89] | 23 [−0.98] | 16 [−4.4] |
| Head circumference (cm) [z score] | 50.7 [−1.6] | 54 [0.81] | 49.5 [−2.6] | 47.4 [−2.5] | 52.2 [−1.1] | 52 [−1.83] | 48.2 [−2.9] | 53 [−0.03] | 52.4 [−0.98] | 52 [−1.2] |
| Facial dysmorphology | Long nose; broad ‐sal bridge; full lower lip | Broad ‐sal bridge; full lower lip | Long nose; broad ‐sal bridge; small ears; full lower lip | Long nose; broad ‐sal bridge; full lower lip | Triangular face; protuberant ears | Long nose; full lower lip | Long nose; broad ‐sal bridge; small ears; full lower lip | Broad ‐sal bridge; full lower lip; ptosis | Deep‐set eyes, high ‐sal bridge; long columella; high‐arched palate | Deep‐set eyes, high ‐sal bridge; long columella; high‐arched palate |
| Neurology, behavior, development | ||||||||||
| Intellectual disability | Moderate | Moderate | Moderate | Moderate | Moderate | Moderate | Moderate | Moderate | Severe | Severe |
| Sat unaided (months) | 20 | 7 | 12 | 24 | 15 | Uncertain | 7 | 12 | 24–36 | 24–36 |
| Walked unaided (months) | 24 | 36 | 60 | NYA | 17 | 24 | 19 | 30 | >60 | >60 |
| Speech | 40–50 single words; occasional two‐word sentences | Several single words; signs | Occasional single word | NYA | ∼200 single words | 20 single words; Points; Signs | Two‐word sentences | Four‐word sentences; echolalia | NYA | NYA |
| Behavior | Aggressive (self & others); routine‐orientated | Aggressive (others); routine‐orientated; obsessions | Aggressive (self & others); routine‐orientated; hand stereotypies | Aggressive (self & others); obsessions (water) | Aggressive (self); obsessions (doors) | Aggressive (others); hand flapping when excited | Aggressive (self & others); obsessions (water) | Autistic spectrum disorder | Routine‐orientated, fasci‐tion with water; laughter outbursts | Routine‐orientated, fasci‐tion with water; laughter outbursts |
| Autism | − | Yes | No | Yes | Yes | Yes | − | No | Yes | Yes |
| Sleep disturbance | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes (not severe) | Yes | Yes |
| Hypotonia | No | Yes | Yes | Yes | Yes | No | Yes | Yes | Yes | Yes |
| Seizures | No | Yes | Yes | Yes | No | Yes | No | Yes | Yes | Yes |
| Seizure age‐of‐onset | − | 6 y | 2 y | 2 y | − | 3 y | − | 5 y | 13 m | 13 m |
| Seizure type | − | Myoclonic; absences | Myoclonic; absences; drop attacks | Absences; drop attacks | − | Head drops & blinking, visually triggered by patterns | − | Absences (possible); drop attacks | Febrile; absences; drop attacks; occasional tonic‐clonic & myoclonic | Febrile; absences; drop attacks; occasional tonic‐clonic & myoclonic |
| Gait | Unsteady gait | Wide‐based gait | Wide‐based gait | − | − | Wide‐based gait | Unsteady gait | − | Ataxic | Ataxic |
| Brain MRI | ND | Normal | Normal | Normal | Normal | Normal | ND | ND | Normal | Normal |
| Skeletal issues | No | Hip dysplasia (unilateral, requiring osteotomy); 5th finger clinodactyly | Pes planus | Pectus excavatum; kyphosis; hip dysplasia (unilateral, requiring open reduction) | Pes planus | No | No | Lordosis | Kyphoscoliosis | Kyphoscoliosis |
| Other issues | Gastro‐esophageal reflux | Constipation; café‐au‐lait patch (arm) | Constipation; unilateral divergent strabismus | Constipation; gastrostomy planned; hirsutism | Café‐au‐lait patch (intercostal) | Constipation; hirsutism | Hirsutism; unilateral divergent strabismus | Constipation; strabismus (repaired) | Progressive lower limb spasticity | Progressive lower limb spasticity; gastrostomy |
Key: NYA, not yet achieved; ND, not done.
Mutation Analysis
For the seven individuals identified via the DDD study, trio‐based exome sequencing was performed on the affected individual and their parents, as previously described [Wright et al., 2014]. Each affected individual has also had a high‐resolution analysis for copy number abnormalities using array‐based comparative genomic hybridization (aCGH). Putative de novo mutations were identified from exome data using DeNovoGear software [Ramu et al., 2013] and were validated using targeted Sanger sequencing.
The eighth individual (7; Table I) was identified as having a ∼0.39Mb deletion of 6p21.32p21.31, via a service ISCA 8 × 60K BlueGnome Array. The ninth and tenth individuals are monozygous twins from Belgium, who were identified through a local multi‐gene panel and were validated using targeted Sanger sequencing.
RESULTS
SYNGAP1 Mutations
The validated de novo mutations are detailed in Table I. There were 10 individuals, but two are monozygotic twins, so we describe eight mutations and one deletion. Three individuals had nonsense mutations; three had frameshift mutations resulting in early stop codons. One individual (2; Table I) had a missense mutation c.509G>A (ENST00000418600); p.Arg170Gln (ENSP00000403636.2). On SIFT analysis; this was labeled “Deleterious” with a score of 0.01 and on PolyPhen analysis “Possibly damaging” with a score of 0.529. This mutated residue lies within the PH domain (Prosite PS50003) of SynGAP. The monozygotic twins (9 and 10; Table I) had a missense mutation c.1081T>C (ENST00000418600); p.Leu327Pro (ENSP00000403636). The SIFT score is 0, “Deleterious”, and the PolyPhen is 0.983, “Probably Damaging”. This mutation lies within the C2 domain, which is required for RapGAP activity. One individual (7; Table I) had a 0.39 Mb genomic deletion, which encompassed the entire SYNGAP1 gene and 18 other genes.
Growth
Birth weight was normal (z score between −2 and 2) for all of the affected individuals. Postnatal growth was normal in five individuals (1, 2, 5, 8 and 9; Table I). Three of 10 had mild microcephaly (z score between −2.5 and −3), with one of these also having weight on a similar centile (4; Table I). Two individuals had significant short stature (Patients 6 and 10; Table I), one of whom was also significantly underweight (Patient 10; Table I).
Development
Global delay in developmental milestones was present in all patients with an unusual temporal sequence seen in some patients. For example, Patient 5 (Table I) did not sit unaided until 15 months, but walked unaided at 17 months. Typically independent walking was achieved in the third year of life with the subsequent gait being wide‐based and unsteady. Language acquisition was highly variable within this group. Expressive language was delayed with most children using a limited vocabulary of single words. None of the affected individuals were toilet‐trained at the time of assessment.
Behavior
Seven of the ten individuals showed general hyperexcitability and aggressive behavior, often directed towards others. A disturbed sleep pattern was reported in all patients, with almost all being treated with, or having had a therapeutic trial of, Melatonin. Anecdotally, families reported a high pain threshold and hyperacusis in some affected individuals.
Neurology
Seven of the 10 individuals had seizures, most commonly complex and generalized, including myoclonic, drop attacks, and absences. Congenital central hypotonia was common. Most of the affected individuals required ankle splints and/or Piedro boots to aid with walking. Seven individuals have had MR imaging of their brains and in each patient this was reported as normal.
Facial Features
The facial appearance of the ten affected individuals is shown in Figure 1. The most common shared facial characteristics are almond‐shaped palpebral fissures, which slant downwards slightly. All but one had a mildly myopathic appearance. An open‐mouthed appearance, with a relatively full lower lip vermilion was common, as was a low‐hanging columella. 7/10 had relatively long noses (sometimes with under‐development of the ala nasi); 6/10 had relatively long ears with protuberant lobes; 4/10 had relatively deep‐set eyes; and one had a degree of ptosis. There was no obvious difference in facial appearance between the deletion and intragenic mutation patients.
Figure 1.
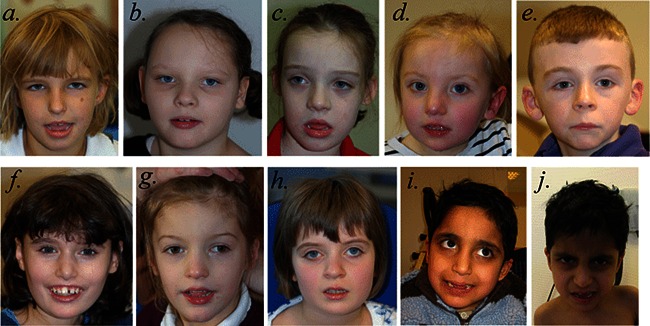
Faces of individuals with SYNGAP1 haploinsufficiency. Facial photographs of Patient 1 at 7 years, 3 months (a); Patient 2 at 8 years, 2 months (b); Patient 3 at 7 years, 9 months (c); Patient 4 at 3 years, 2 months (d); Patient 5 at 8 years, 4 months (e); Patient 6 at 12 years, 10 months (f); Patient 7 at 5 years, 7 months (g); Patient 8 at 8 years, 7 months (h); and Patients 9 and 10 at 8 years, 3 months (i and j). The most common shared facial characteristics are almond‐shaped palpebral fissures, which slant downwards slightly. With the exception of Patient 5 (e), the others have a mildly myopathic appearance, with an open mouth and relatively full lower lip. Patients 1 (a), 3 (c), 4 (d), 6 (f), 7 (g) and 9 (i) and 10 (j) have relatively long noses; Patients 2 (b), 4 (d), 5 (e), 6 (f), 7 (g) and 8 (h) have relatively long ears with protuberant lobes. Patients 1 (a), 6 (f) and 9 (i) and 10 (j) were thought to have relatively deep‐set eyes and Patient 8 (h) has a degree of ptosis. Patient 6 (f) has a missing central incisor due to trauma. We do not believe that Patient 7 (g), the only deletion patient in this series, differs significantly in appearance from the others.
Other Features
Five of the individuals had constipation, requiring medical therapies; three individuals had fine hirsutism, especially noticeable over limbs and spine; two had significant hip dysplasia, requiring surgical management; three had a kyphosis or kyphoscoliosis; and one had a pectus excavatum (Table I).
DISCUSSION
SYNGAP1 was originally reported as causing non‐syndromal intellectual disability [Hamdan et al., 2009]. Supplementary Table I summarizes the available clinical data on the 26 individuals who have been reported to date with presumed causative mutations in SYNGAP1 or deletions or translocations involving this gene [Hamdan et al., 2009, 2011a, b; Krepischi et al., 2010; Pinto et al., 2010; Vissers et al., 2010; Cook, 2011; Klitten et al., 2011; Zollino et al., 2011; Clement et al., 2012; de Ligt et al., 2012; Rauch et al., 2012; Berryer et al., 2013; Carvill et al., 2013; Writzl and Knegt, 2013; Dyment et al., 2014; O'Roak et al., 2014; Redin et al., 2014]. De novo mutations in this gene are undoubtedly a significant cause of intellectual disability, accounting for 0.62% of all the patients in the DDD Study [Wright et al., 2014] and major contributors to other cohorts that have been studied (Supplementary Table II).
The original designation of the phenotype associated with SYNGAP1 haploinsufficiency as non‐syndromal is understandable given the generally normal antenatal growth parameters and the relative normality of post‐natal growth. In addition, all patients have a moderate‐to‐severe intellectual disability with few structural anomalies reported on brain imaging. The genomic pathology is also remarkably consistent with almost all patients having heterozygous, de novo, loss of function mutations. The associated genetic mechanism is very likely to be haploinsufficiency given the similarity of the intragenic mutations with the whole gene deletions.
Although there is wide variability in the type and severity of the clinical features associated with SYNGAP1 haploinsufficiency, some aspects of the phenotype show a level of consistency that suggests SYNGAP1 haploinsufficiency may be associated with a clinically recognizable syndrome. The seizure type and the behavioral phenotype were relatively consistent in our cohort. Myoclonic, absence and drop attack seizures are typical, both in the reported individuals and those presented in this paper. General hyperexcitability, sleep disturbance and aggressive behavior, often directed towards others, are common features in our cohort and are mentioned in some of the previously reported patients. Clearly these distressing behavioral components of the phenotype require further investigation. Facial photographs were not available in most of the previous reports, but in the cohort presented here a subtle but consistent facial appearance is suggested, although further observations will be required to determine if this is in any way discriminative. The pattern of growth may also be helpful in making a clinical diagnosis. Six of 18 reported patients with postnatal head circumferences recorded, and 3/10 of the patients reported here, had measurements of two standard deviations below the mean for their age. A mild postnatal microcephaly is clearly over‐represented in this group.
In our cohort, 8/10 patients had previously been investigated for Angelman syndrome. There are some similarities with this condition, although we believe that they are clinically distinguishable. Nevertheless, we believe that SYNGAP1 should also be added to the expanding list of differential diagnoses for Angelman syndrome or patients presenting with Angelman‐like features.
CONCLUSION
SYNGAP1 has previously been described as presenting in a non‐syndromal manner. Mutations in this gene have been found to be a relatively‐common cause of intellectual disability in large‐scale massively parallel sequencing studies, where subjects are usually recruited because a clinical syndromal diagnosis has not previously been made. It is arguable whether the term non‐specific may be more appropriate to many subjects recruited into such studies, who most likely represent a heterogeneous mix of those genuinely non‐syndromal, but also of some syndromes more subtle in their associations and/or dysmorphology. For SYNGAP1 we consider discriminative features in individuals with moderate‐to‐severe ID to be the characteristic facial features, seizure type and behavioral phenotype (generalized hyper‐excitability, sleep disturbance and a propensity to aggression). It is not yet clear if hypotonia, hip dysplasia, strabismus, wide‐based/unsteady gait, fine hirsutism (limbs and spine), and significant constipation are helpful discriminators. Some patients have microcephaly, but growth parameters are generally within the normal range.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supporting Information Table S1: Summarizes the available clinical data on the 26 individuals who have been previously‐reported with presumed causative mutations in SYNGAP1, deletions or translocations involving this gene.
Supporting Information Table S2: Summarizes the SYNGAP1 pick‐up rate in NGS studies in which SYNGAP1 patients have previously been reported.
Supplementary Materials.
ACKNOWLEDGMENTS
The DDD study presents independent research commissioned by the Health Innovation Challenge Fund [grant number HICF‐1009‐003], a parallel funding partnership between the Wellcome Trust and the Department of Health, and the Wellcome Trust Sanger Institute [grant number WT098051]. The views expressed in this publication are those of the author(s) and not necessarily those of the Wellcome Trust or the Department of Health. The study has UK Research Ethics Committee approval (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12 granted by the Republic of Ireland REC). The research team acknowledges the support of the National Institute for Health Research, through the Comprehensive Clinical Research Network.
MJP would also like to thank Professor Jill Clayton‐Smith for her comments on the dysmorphology of the UK patients, Dr Meena Balasubramanian for reading drafts of this paper, and Professor Jacques Michaud for his help and encouragement in the development of this project. VB, DL and IM would like to thank the Fonds Marguerite‐Marie Delacroix, for the research grant provided to DL to achieve his PhD thesis on epileptic encephalopathies, and the Institut de Recherche Scientifique en Pathologie et Génétique for its financial support.
We would like to thank all the local clinicians and other healthcare professionals involved with these children. Finally, we would of course especially like to thank the families of the children described here, for collaborating with this project and consenting to this publication.
Parker MJ, Fryer AE, Shears DJ, Lachlan KL, McKee SA, Magee AC, Mohammed S, Vasudevan PC, Park S‐M, Benoit V, Lederer D, Maystadt I, study D, FitzPatrick DR. 2015. De novo, heterozygous, loss‐of‐function mutations in SYNGAP1 cause a syndromic form of intellectual disability. Am J Med Genet Part A 167A:2231–2237.
The copyright line for this article was changed on 20 May 2016 after original online publication.
Conflicts of interest: none.
REFERENCES
- Berryer MH, Hamdan FF, Klitten LL, Moller RS, Carmant L, Schwartzentruber J, Patry L, Dobrzeniecka S, Rochefort D, Neugnot‐Cerioli M, Lacaille JC, Niu Z, Eng CM, Yang Y, Palardy S, Belhumeur C, Rouleau GA, Tommerup N, Immken L, Beauchamp MH, Patel GS, Majewski J, Tarnopolsky MA, Scheffzek K, Hjalgrim H, Michaud JL, Di Cristo G. 2013. Mutations in SYNGAP1 cause intellectual disability, autism, and a specific form of epilepsy by inducing haploinsufficiency. Hum Mutat 34:385–394. [DOI] [PubMed] [Google Scholar]
- Carvill GL, Heavin SB, Yendle SC, McMahon JM, O'Roak BJ, Cook J, Khan A, Dorschner MO, Weaver M, Calvert S, Malone S, Wallace G, Stanley T, Bye AM, Bleasel A, Howell KB, Kivity S, Mackay MT, Rodriguez‐Casero V, Webster R, Korczyn A, Afawi Z, Zelnick N, Lerman‐Sagie T, Lev D, Moller RS, Gill D, Andrade DM, Freeman JL, Sadleir LG, Shendure J, Berkovic SF, Scheffer IE, Mefford HC. 2013. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet 45:825–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement JP, Aceti M, Creson TK, Ozkan ED, Shi Y, Reish NJ, Almonte AG, Miller BH, Wiltgen BJ, Miller CA, Xu X, Rumbaugh G. 2012. Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell 151:709–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook EHJ. 2011. De novo autosomal dominant mutation in SYNGAP1. Autism Res 4:155–156. [DOI] [PubMed] [Google Scholar]
- de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, Vulto‐van Silfhout AT, Koolen DA, de Vries P, Gilissen C, del Rosario M, Hoischen A, Scheffer H, de Vries BB, Brunner HG, Veltman JA, Vissers LE. 2012. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 367:1921–1929. [DOI] [PubMed] [Google Scholar]
- Dyment DA, Tetreault M, Beaulieu CL, Hartley T, Ferreira P, Chardon JW, Marcadier J, Sawyer SL, Mosca SJ, Innes AM, Parboosingh JS, Bulman DE, Schwartzentruber J, Majewski J, Tarnopolsky M, Boycott KM. 2014. Whole‐exome sequencing broadens the phenotypic spectrum of rare pediatric epilepsy: A retrospective study. Clin Genet 2014. doi: 10.1111/cge.12464. PMID: 25046240. [DOI] [PubMed] [Google Scholar]
- Hamdan FF, Daoud H, Piton A, Gauthier J, Dobrzeniecka S, Krebs MO, Joober R, Lacaille JC, Nadeau A, Milunsky JM, Wang Z, Carmant L, Mottron L, Beauchamp MH, Rouleau GA, Michaud JL. 2011a. De novo SYNGAP1 mutations in nonsyndromic intellectual disability and autism. Biol Psychiatry 69:898–901. [DOI] [PubMed] [Google Scholar]
- Hamdan FF, Gauthier J, Araki Y, Lin DT, Yoshizawa Y, Higashi K, Park AR, Spiegelman D, Dobrzeniecka S, Piton A, Tomitori H, Daoud H, Massicotte C, Henrion E, Diallo O, Shekarabi M, Marineau C, Shevell M, Maranda B, Mitchell G, Nadeau A, D'Anjou G, Vanasse M, Srour M, Lafreniere RG, Drapeau P, Lacaille JC, Kim E, Lee JR, Igarashi K, Huganir RL, Rouleau GA, Michaud JL. 2011b. Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am J Hum Genet 88:306–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan FF, Gauthier J, Spiegelman D, Noreau A, Yang Y, Pellerin S, Dobrzeniecka S, Cote M, Perreau‐Linck E, Carmant L, D'Anjou G, Fombonne E, Addington AM, Rapoport JL, Delisi LE, Krebs MO, Mouaffak F, Joober R, Mottron L, Drapeau P, Marineau C, Lafreniere RG, Lacaille JC, Rouleau GA, Michaud JL. 2009. Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N Engl J Med 360:599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klitten LL, Moller RS, Nikanorova M, Silahtaroglu A, Hjalgrim H, Tommerup N. 2011. A balanced translocation disrupts SYNGAP1 in a patient with intellectual disability, speech impairment, and epilepsy with myoclonic absences (EMA). Epilepsia 52:e190–e193. [DOI] [PubMed] [Google Scholar]
- Komiyama NH, Watabe AM, Carlisle HJ, Porter K, Charlesworth P, Monti J, Strathdee DJ, O'Carroll CM, Martin SJ, Morris RG, O'Dell TJ, Grant SG. 2002. SynGAP regulates ERK/MAPK signaling, synaptic plasticity, and learning in the complex with postsynaptic density 95 and NMDA receptor. J Neurosci 22:9721–9732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krepischi AC, Rosenberg C, Costa SS, Crolla JA, Huang S, Vianna‐Morgante AM. 2010. A novel de novo microdeletion spanning the SYNGAP1 gene on the short arm of chromosome 6 associated with mental retardation. Am J Med Genet A 152A:2376–2378. [DOI] [PubMed] [Google Scholar]
- Muhia M, Yee BK, Feldon J, Markopoulos F, Knuesel I. 2010. Disruption of hippocampus‐regulated behavioural and cognitive processes by heterozygous constitutive deletion of SynGAP. Eur J Neurosci 31:529–543. [DOI] [PubMed] [Google Scholar]
- O'Roak BJ, Stessman HA, Boyle EA, Witherspoon KT, Martin B, Lee C, Vives L, Baker C, Hiatt JB, Nickerson DA, Bernier R, Shendure J, Eichler EE. 2014. Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nat Commun 5:5595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, Almeida J, Bacchelli E, Bader GD, Bailey AJ, Baird G, Battaglia A, Berney T, Bolshakova N, Bolte S, Bolton PF, Bourgeron T, Brennan S, Brian J, Bryson SE, Carson AR, Casallo G, Casey J, Chung BH, Cochrane L, Corsello C, Crawford EL, Crossett A, Cytrynbaum C, Dawson G, de Jonge M, Delorme R, Drmic I, Duketis E, Duque F, Estes A, Farrar P, Fernandez BA, Folstein SE, Fombonne E, Freitag CM, Gilbert J, Gillberg C, Glessner JT, Goldberg J, Green A, Green J, Guter SJ, Hakonarson H, Heron EA, Hill M, Holt R, Howe JL, Hughes G, Hus V, Igliozzi R, Kim C, Klauck SM, Kolevzon A, Korvatska O, Kustanovich V, Lajonchere CM, Lamb JA, Laskawiec M, Leboyer M, Le Couteur A, Leventhal BL, Lionel AC, Liu XQ, Lord C, Lotspeich L, Lund SC, Maestrini E, Mahoney W, Mantoulan C, Marshall CR, McConachie H, McDougle CJ, McGrath J, McMahon WM, Merikangas A, Migita O, Minshew NJ, Mirza GK, Munson J, Nelson SF, Noakes C, Noor A, Nygren G, Oliveira G, Papanikolaou K, Parr JR, Parrini B, Paton T, Pickles A, Pilorge M, Piven J, Ponting CP, Posey DJ, Poustka A, Poustka F, Prasad A, Ragoussis J, Renshaw K, Rickaby J, Roberts W, Roeder K, Roge B, Rutter ML, Bierut LJ, Rice JP, Salt J, Sansom K, Sato D, Segurado R, Sequeira AF, Senman L, Shah N, Sheffield VC, Soorya L, Sousa I, Stein O, Sykes N, Stoppioni V, Strawbridge C, Tancredi R, Tansey K, Thiruvahindrapduram B, Thompson AP, Thomson S, Tryfon A, Tsiantis J, Van Engeland H, Vincent JB, Volkmar F, Wallace S, Wang K, Wang Z, Wassink TH, Webber C, Weksberg R, Wing K, Wittemeyer K, Wood S, Wu J, Yaspan BL, Zurawiecki D, Zwaigenbaum L, Buxbaum JD, Cantor RM, Cook EH, Coon H, Cuccaro ML, Devlin B, Ennis S, Gallagher L, Geschwind DH, Gill M, Haines JL, Hallmayer J, Miller J, Monaco AP, Nurnberger JIJ, Paterson AD, Pericak‐Vance MA, Schellenberg GD, Szatmari P, Vicente AM, Vieland VJ, Wijsman EM, Scherer SW, Sutcliffe JS, Betancur C. 2010. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466:368–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramu A, Noordam MJ, Schwartz RS, Wuster A, Hurles ME, Cartwright RA, Conrad DF. 2013. DeNovoGear: De novo indel and point mutation discovery and phasing. Nat Methods 10:985–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, Albrecht B, Bartholdi D, Beygo J, Di Donato N, Dufke A, Cremer K, Hempel M, Horn D, Hoyer J, Joset P, Ropke A, Moog U, Riess A, Thiel CT, Tzschach A, Wiesener A, Wohlleber E, Zweier C, Ekici AB, Zink AM, Rump A, Meisinger C, Grallert H, Sticht H, Schenck A, Engels H, Rappold G, Schrock E, Wieacker P, Riess O, Meitinger T, Reis A, Strom TM. 2012. Range of genetic mutations associated with severe non‐syndromic sporadic intellectual disability: An exome sequencing study. Lancet 380:1674–1682. [DOI] [PubMed] [Google Scholar]
- Redin C, Gerard B, Lauer J, Herenger Y, Muller J, Quartier A, Masurel‐Paulet A, Willems M, Lesca G, El‐Chehadeh S, Le Gras S, Vicaire S, Philipps M, Dumas M, Geoffroy V, Feger C, Haumesser N, Alembik Y, Barth M, Bonneau D, Colin E, Dollfus H, Doray B, Delrue MA, Drouin‐Garraud V, Flori E, Fradin M, Francannet C, Goldenberg A, Lumbroso S, Mathieu‐Dramard M, Martin‐Coignard D, Lacombe D, Morin G, Polge A, Sukno S, Thauvin‐Robinet C, Thevenon J, Doco‐Fenzy M, Genevieve D, Sarda P, Edery P, Isidor B, Jost B, Olivier‐Faivre L, Mandel JL, Piton A. 2014. Efficient strategy for the molecular diagnosis of intellectual disability using targeted high‐throughput sequencing. J Med Genet 51:724–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers LE, de Ligt J, Gilissen C, Janssen I, Steehouwer M, de Vries P, van Lier B, Arts P, Wieskamp N, del Rosario M, van Bon BW, Hoischen A, de Vries BB, Brunner HG, Veltman JA. 2010. A de novo paradigm for mental retardation. Nat Genet 42:1109–1112. [DOI] [PubMed] [Google Scholar]
- Wright CF, Fitzgerald TW, Jones WD, Clayton S, McRae JF, van Kogelenberg M, King DA, Ambridge K, Barrett DM, Bayzetinova T, Bevan AP, Bragin E, Chatzimichali EA, Gribble S, Jones P, Krishnappa N, Mason LE, Miller R, Morley KI, Parthiban V, Prigmore E, Rajan D, Sifrim A, Swaminathan GJ, Tivey AR, Middleton A, Parker M, Carter NP, Barrett JC, Hurles ME, FitzPatrick DR, Firth HV. 2014. Genetic diagnosis of developmental disorders in the DDD study: A scalable analysis of genome‐wide research data. Lancet 385:1305–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Writzl K, Knegt AC. 2013. 6p21.3 microdeletion involving the SYNGAP1 gene in a patient with intellectual disability, seizures, and severe speech impairment. Am J Med Genet A 161A:1682–1685. [DOI] [PubMed] [Google Scholar]
- Zollino M, Gurrieri F, Orteschi D, Marangi G, Leuzzi V, Neri G. 2011. Integrated analysis of clinical signs and literature data for the diagnosis and therapy of a previously undescribed 6p21. 3 deletion syndrome. Eur J Hum Genet 19:239–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supporting Information Table S1: Summarizes the available clinical data on the 26 individuals who have been previously‐reported with presumed causative mutations in SYNGAP1, deletions or translocations involving this gene.
Supporting Information Table S2: Summarizes the SYNGAP1 pick‐up rate in NGS studies in which SYNGAP1 patients have previously been reported.
Supplementary Materials.