

Abstract
Freshwater fauna are particularly sensitive to environmental change and disturbance. Management agencies frequently use fish and amphibian biodiversity as indicators of ecosystem health and a way to prioritize and assess management strategies. Traditional aquatic bioassessment that relies on capture of organisms via nets, traps and electrofishing gear typically has low detection probabilities for rare species and can injure individuals of protected species. Our objective was to determine whether environmental DNA (eDNA) sampling and metabarcoding analysis can be used to accurately measure species diversity in aquatic assemblages with differing structures. We manipulated the density and relative abundance of eight fish and one amphibian species in replicated 206‐L mesocosms. Environmental DNA was filtered from water samples, and six mitochondrial gene fragments were Illumina‐sequenced to measure species diversity in each mesocosm. Metabarcoding detected all nine species in all treatment replicates. Additionally, we found a modest, but positive relationship between species abundance and sequencing read abundance. Our results illustrate the potential for eDNA sampling and metabarcoding approaches to improve quantification of aquatic species diversity in natural environments and point the way towards using eDNA metabarcoding as an index of macrofaunal species abundance.
Keywords: community ecology, environmental DNA, mesocosm, metabarcoding, species diversity
Introduction
Freshwater fishes and amphibians are particularly sensitive to environmental change and disturbance (Brander 2007; Dudgeon 2010) and include many taxa that are in global decline (Bruton 1995; Stuart et al. 2004). As a result of such sensitivity, the status of local and regional fish and amphibian biodiversity is a useful indicator of changes in total regional biodiversity and ecosystem health (Sala et al. 2005; Xenopoulos et al. 2005; Abell et al. 2008). Additionally, freshwater biodiversity, including that of fishes and amphibians, has direct and indirect value to environmental management agencies and the general public (Giller et al. 2004; Sala et al. 2005). Management agencies frequently use assessments of aquatic biodiversity to prioritize management actions and measure the effectiveness of management efforts (Bailey et al. 2004; Hubert & Quist 2010). Traditionally, aquatic bioassessment has relied on capture or observations of organisms via nets, traps or electrofishing gear (Murphy & Willis 1996; Bonar et al. 2009). However, due to inefficiencies of underwater sampling and the mobility of organisms, detection probabilities for rare species in aquatic environments are frequently low (Bayley & Peterson 2001; Mackenzie & Royle 2005). This limitation can lead to incorrect conclusions that rare species are absent when they are actually present (Gu & Swihart 2004). Because rare species can contribute substantially to overall community richness (Williams 1964; Cao et al. 2001), the potential for underestimating true species richness is often high, unless sampling effort is more extensive than is typically feasible (McDonald 2004; MacKenzie et al. 2005). Novel sampling methods that increase ‘detection per unit effort’, particularly for rare species, have the potential to improve biodiversity estimates by decreasing systematic errors in inference about species richness resulting from low‐detection probabilities for rare species during capture‐based sampling.
Noninvasive genetic sampling is any method of collecting genetic material from biota with minimal disturbance of the actual organism (Beja‐Pereira et al. 2009). Environmental DNA (eDNA) sampling for macroorganisms is a noninvasive genetic method, via the collection of water samples, that takes advantage of shed cellular material suspended in aquatic environments to detect the presence of organisms including rare taxa (Ficetola et al. 2008; Jerde et al. 2011; Takahara et al. 2013). Correspondingly, metabarcoding focuses on the analysis of taxon richness using homologous genes from environmental samples (Taberlet et al. 2012; Thomsen et al. 2012a). Synonyms include metagenetics, ecometagenetics, metasytematics, ecogenomics and environmental barcoding (Taberlet et al. 2012). Metabarcoding approaches are commonly used in the assessment of microorganism species richness but are just emerging as tools for the bioassessment of aquatic macrofauna species richness.
Because eDNA persists in water for days to weeks after organisms are removed from controlled experimental systems, it contains a catalog of species present in the recent past (Dejean et al. 2011; Thomsen et al. 2012b; Barnes et al. 2014). Previous research has illustrated that qPCR and metabarcoding approaches can successfully detect amphibian, fish, bird, insect, crustacean and mammal taxa in freshwater (Thomsen et al. 2012b). Furthermore, eDNA‐metabarcoding approaches have been used successfully to detect eDNA from several species of marine fishes in natural seawater (Thomsen et al. 2012a) and multiple families and genera of marine fishes in a 4.5‐million litre aquarium (Kelly et al. 2014). However, species‐level tests of the eDNA‐metabarcoding approach against comprehensively known communities have not been reported, nor have previous studies been experimentally replicated to test the reproducibility of species detection.
To test the precision in measuring species diversity from eDNA samples with metabarcoding, we conducted a replicated mesocosm experiment designed to determine whether this approach can accurately quantify aquatic biodiversity in known assemblages with differing assemblage structures. Specifically, three research questions were investigated: (i) Can species richness be accurately measured using eDNA metabarcoding in species assemblages with differing species densities and relative abundances? (ii) Is ultrasequencing read abundance positively related to species abundance? (iii) Which species abundance estimator better predicts read abundance, the number of individuals or the species biomass?
Methods
Experimental conditions
The experiment was conducted in twelve, 340‐L, 1‐m diameter circular polyethylene tanks located inside an isolated climate‐controlled solarium at the University of Notre Dame (Notre Dame, IN, USA). Prior to the experiment, the solarium floor and walls, experimental tanks and all equipments were decontaminated via a 10‐min exposure to 10% bleach solution (Prince & Andrus 1992). A mean air temperature of 20.4 ± 0.8 °C and natural summer (August) photoperiod were maintained in the solarium for the duration of the experiment. Experimental tanks were filled with 206 L of groundwater and vigorously aerated for the duration of the experiment. Each tank was covered with 6 μm thick, clear, polyvinyl sheeting to prevent evaporation and contain splashes. Water quality was monitored daily for nitrate concentration, nitrite concentration, ammonia concentration, dissolved oxygen, pH and temperature. All water quality measurements were found to be satisfactory for organisms during the duration of the experiment.
Experimental design and sampling
The experimental design consisted of four experimental treatments of assemblage structure, with three replicates per treatment: (i) high total density and even relative abundance, (ii) low total density and even relative abundance, (iii) high total density and skewed relative abundance and (iv) low total density and skewed relative abundance. This crossed experimental treatment design enabled evaluation of the effects of both density (high vs. low) and relative abundance (even vs. skewed) on species detection. Nine species of aquatic macrofauna were included in each of the experimental treatments: fathead minnow (Pimephales promelas), bluegill (Lepomis macrochirus), common carp (Cyprinus carpio), white sucker (Catostomus commersonii), central stoneroller (Campostoma anomalum), eastern mosquitofish (Gambusia holbrooki), creek chub (Semotilus atromaculatus), blackstripe topminnow (Fundulus notatus) and American bullfrog tadpole (Rana catesbeiana). High‐density treatments contained 90 total individuals per tank. Low‐density treatments contained 36 total individuals per tank. Treatments with even relative abundances contained 10 (high density) or 4 (low density) individuals of each species. The relative abundances of species for treatments with skewed relative abundances (Table 1) were chosen based on a log‐normal distribution that conformed to observed natural occurring fish assemblages (Magurran & Henderson 2003). Individual fish or tadpoles from each mesocosm were weighed (nearest 0.1 g) at the conclusion of each experimental trial (Table 1).
Table 1.
Biomass (g) and number (in parentheses) of each of the nine study species in the experimental mesocosms
| Species | High density, even abundance | Low density, even abundance | High density, skewed abundance | Low density, skewed abundance | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tank 1 | Tank 2 | Tank 3 | Tank 1 | Tank 2 | Tank 3 | Tank 1 | Tank 2 | Tank 3 | Tank 1 | Tank 2 | Tank 3 | |
| Campostoma anomalum | 29 (10) | 36.5 (10) | 32.5 (10) | 19.7 (4) | 17.3 (4) | 15.6 (4) | 69.1 (18) | 72.3 (18) | 15.3 (4) | 21.1 (5) | 19.9 (5) | 7.3 (2) |
| Catostomus commersonii | 22.2 (10) | 17.4 (10) | 20.9 (10) | 8.0 (4) | 7.1 (4) | 10.2 (4) | 7 (4) | 9.0 (4) | 7.3 (4) | 3.1 (2) | 4.4 (2) | 3.5 (2) |
| Cyprinus carpio | 74.8 (10) | 88.4 (10) | 139.1 (10) | 16.3 (4) | 19.0 (4) | 77.2 (4) | 26.4 (5) | 26.6 (5) | 44.2 (5) | 4.4 (2) | 36.2 (2) | 18 (2) |
| Fundulus notatus | 12.6 (10) | 10.8 (10) | 14.0 (10) | 3.7 (4) | 5.0 (4) | 5.4 (4) | 13.1 (7) | 11.1 (7) | 5.5 (4) | 4.2 (3) | 6.7 (3) | 3.8 (2) |
| Gambusia holbrooki | 0.7 (10) | 0.3 (10) | 2.0 (10) | 0.8 (4) | 0.4 (4) | 0.8 (4) | 0.5 (4) | 0.4 (4) | 1.0 (7) | 0.1 (2) | 0.2 (2) | 0.6 (3) |
| Lepomis macrochirus | 10.8 (10) | 12.9 (10) | 16.0 (10) | 4.0 (4) | 4.7 (4) | 3.7 (4) | 5.5 (4) | 6.3 (4) | 19.9 (18) | 1.8 (2) | 2.6 (2) | 6.4 (5) |
| Pimephales promelas | 6.7 (10) | 14.0 (10) | 18.4 (10) | 3.4 (4) | 4.5 (4) | 6.5 (4) | 67.1 (46) | 73.7 (46) | 78.6 (46) | 28.5 (18) | 25.4 (18) | 29.9 (18) |
| Rana catesbeiana | 44.5 (10) | 47.7 (10) | 53.3 (10) | 17.2 (4) | 18.1 (4) | 24.8 (4) | 17.4 (4) | 21.7 (4) | 14.5 (4) | 8.8 (2) | 8.1 (2) | 8.4 (2) |
| Semotilus atromaculatus | 13.4 (10) | 39.6 (10) | 11.1 (10) | 2.7 (4) | 13.1 (4) | 4.9 (4) | 13.6 (4) | 15.9 (4) | 6.6 (4) | 2.9 (2) | 4.8 (2) | 3.4 (2) |
Study species were obtained from commercial sources. Fathead minnow, bluegill and bullfrog tadpoles were procured from Jones Fish Lake Management (Cincinnati, OH, USA). White sucker, central stoneroller, blackstripe topminnow and creek chub were procured from Jonah's Aquarium (Delaware, OH, USA). Eastern mosquitofish were procured from Carolina Biological Supply Company (Burlington, NC, USA). Common carp were procured from Kloubec Koi Farm (Amana, IA, USA).
Prior to each experiment, each species was maintained in a room disconnected from the solarium, in single‐species tanks, for a minimum of 18 days. During that time, all study organisms were fed daily to satiation with frozen brine shrimp (Artemia spp.; San Francisco Bay Brand Inc., Newark, CA, USA) and dried phytoplankton flake food (Brine Shrimp Direct, Ogden, UT, USA). These two diet items were chosen to avoid introducing foreign fish or amphibian DNA into the quarantine tanks and mesocosms. During the quarantine period, each single‐species tank was tested daily for the presence of bluegill (Takahara et al. 2013) and common carp (Turner et al. 2014) eDNA using published qPCR assays to monitor the degradation of eDNA from other species with which any one species may previously have co‐existed. We used bluegill and common carp as indicators of these sources of contamination and assume that the eDNA of other species degraded at rates similar to those of bluegill and common carp. This testing was performed to ensure that the single‐species tanks were indeed monocultures and not contaminated with eDNA from other species and, therefore, could be used to generate controlled mesocosm assemblages. Prior to moving the study organisms to their experimental tanks, a 250‐mL water sample was collected in a Nalgene bottle (cleaned with 10% bleach and then autoclaved prior to use) from each of the single‐species and experimental tanks. These initial water samples were qPCR tested to ensure lack of bluegill and common carp contamination of the experimental and single‐species tanks.
Experimental treatments were randomly assigned to the experimental array of 10 tanks arranged in three rows (3, 4 and 3 tanks per row) with 1.3 m separation between each tank. Due to limited abundances of study organisms and space, only two experimental replicates of the four experimental treatments could be run concurrently. Therefore, the first experimental trial (Trial #1) consisted of two full sets of replicates (eight experimental mesocosms) and two negative control tanks. The second experimental trial (Trial #2) consisted of one set of replicates (four experimental mesocosms) and six negative control tanks. Negative control tanks contained no macrofauna and were maintained in the solarium for the duration of the experiment. For the duration of the experiment, dead study organisms were immediately removed from the experimental tanks and replaced with healthy individuals of the same species from the single‐species tanks to maintain treatment densities. All mesocosm tanks received a daily feeding of brine shrimp and phytoplankton flakes for the duration of each experimental trial. In total, metabarcoding data were collected from 12 experimental mesocosms (three sets of replicates). Five days after transferring the study organisms to the experimental tanks, a single 250‐mL water sample was collected from each tank, including negative control tanks. The 250‐mL water samples collected from the negative control tanks were tested for evidence of cross‐contamination using the common carp and bluegill qPCR assays. Metabarcoding data were collected from the 250‐mL water samples collected from the 12 experimental mesocosms.
Sample processing and extraction
All water samples were vacuum‐filtered, within 1 h of collection, using bleach‐decontaminated 300‐mL filter funnels onto 1.2‐μm pore size Isopore™ polycarbonate membrane filters (EMD Millipore Corporation, Billerica, MA, USA). Filters containing sample retentate were placed in 2‐mL microcentrifuge tubes containing 700 μL of CTAB buffer (Coyne et al. 2005) and stored at −20 °C until further processing. DNA was extracted from mesocosm and negative control tank samples using the same methodology. Before beginning any molecular techniques, counters were wiped with 10% bleach and pipettes were wiped with DNA AWAY; filter tips were used for all pipetting to further reduce contamination risks. DNA extractions followed a CTAB protocol where chloroform dissolves filters and DNA is precipitated in isopropanol and salt (Renshaw et al. 2015).
PCR preparation
As the ongoing focus of our metabarcoding research is broadly based on fishes and amphibians, the design of metabarcoding primer sets was conducted with this long‐term goal in mind. Additionally, assay design and validation occurred prior to the selection of the nine species that are the focus for the current mesocosm experiment and, as such, varied in potential mismatches across taxon (Table S1, Supporting information). In total, six primer sets targeting mitochondrial DNA (mtDNA) were utilized for the metabarcoding assays in the current study: two primer sets (L14735/H15149c and L2513/H2714) were previously described in the literature (Burgener & Hübner 1998; Kitano et al. 2007), while four primer sets were designed de novo and are described here for the first time. Design of the four de novo primer sets targeted conserved regions as determined by two methods. For Ve16S, sequences for the Vertebrata 16S were batch downloaded (8207 sequences) from GenBank and potential primer locations were identified by ecoPrimers (Riaz et al. 2011). The default settings in ecoPrimers were used with the exceptions that one mismatch was allowed for primer matching, 90% of the input sequences had to match proposed primer pairs, and the primer size was set at 21 bp in length. For the other three de novo primer pairs, sequences were downloaded from ogre (Jameson et al. 2003) for the Actinopterygii 12S (Ac12S; 403 sequences), Actinopterygii 16S (Ac16S; 403 sequences), and Amphibia 12S (Am12S; 82 sequences), aligned (clustalw Multiple alignment) and viewed in bioedit (Hall 1999), and conserved priming sites identified by manual visual search. Sequences for all six primer sets are given in Table 2.
Table 2.
Primer sets used for PCR amplification of environmental DNA
| Name | Target gene | Forward primer | Reverse primer | Amplicon length (bp) | Annealing temperatures (°C) AT1, AT2, AT3 | Source |
|---|---|---|---|---|---|---|
| L14912/H15149c | Cyt B | AAAAACCACCGTTGTTATTCAACTA | GCCCCTCAGAATGATATTTGTCCTCA | 413 | 60°, 58°, 55° | Burgener & Hübner (1998) |
| Ac12s | 12s | ACTGGGATTAGATACCCCACTATG | GAGAGTGACGGGCGGTGT | 385 | 63°, 60°, 58° | Current study |
| Am12s | 12s | AGCCACCGCGGTTATACG | CAAGTCCTTTGGGTTTTAAGC | 241 | 65°, 62°, 60° | Current study |
| Ac16s | 16s | CCTTTTGCATCATGATTTAGC | CAGGTGGCTGCTTTTAGGC | 330 | 63°, 60°, 58° | Current study |
| Ve16s | 16s | CGAGAAGACCCTATGGAGCTTA | AATCGTTGAACAAACGAACC | 310 | 65°, 62°, 60° | Current study |
| L2513/H2714 | 16s | GCCTGTTTACCAAAAACATCAC | CTCCATAGGGTCTTCTCGTCTT | 202 | 60°, 58°, 55° | Kitano et al. (2007) |
A 50‐μL PCR volume was used for all six primer sets, with a single reaction per sample per primer set (Table 2). To reduce cross‐contamination between samples, eight‐tube strip tubes with individually attached lids were used instead of 96‐well plates. We used the following recipe: 2.5 μL sterile water, 10 μL 5× high‐fidelity buffer (Bio‐Rad, Hercules, CA, USA), 4 μL 10 mm dNTPs, 1.5 μL 50 mm MgCl2 (Bio‐Rad), 10 μL 10 μ m forward primer, 10 μL 10 μ m reverse primer, 2 μL 2 U/μL iProof Taq (Bio‐Rad) and 10 μL DNA. All PCRs were performed using a Mastercycler Pro S thermocycler (Eppendorf AG, Hamburg, Germany). Annealing temperatures (AT) for all primers are provided in Table 2. A ‘step‐down’ cycling protocol was incorporated to allow for potential mismatches across a range of taxa. Cycling conditions for all primers were (i) 98 °C for 2 min; (ii) 98 °C for 10 s; (iii) AT1 for 20 s; (iv) 72 °C for 30 s; (v) repeat steps 2–4 an additional nine times; (vi) 98 °C for 10 s; (vii) AT2 for 20 s; (viii) 72 °C for 30 s; (ix) repeat steps 6–8 an additional nine times; (x) 98 °C for 10 s; (xi) AT3 for 20 s; (xii) 72 °C for 30 s; (xiii) repeat steps 10–12 an additional 29 times; (xiv) 72 °C for 10 min; (xv) hold at 4 °C.
PCR products were run through a 2% agarose gel, stained with ethidium bromide and visualized on a UV light platform. For several primer sets, we observed slight ‘smearing’ around the expected amplicon. Therefore, to reduce sequencing of PCR artefacts, gel bands were manually cut out with single‐use razor blades, cleaned with the QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany) and eluted from spin columns with 50 μL of Buffer EB. The DNA concentration of the elution was quantified with the Qubit dsDNA BR Assay (Life Technologies, Grand Island, NY, USA).
Illumina library preparation and MiSeq sequencing
Single‐indexed Illumina libraries were prepared with the TruSeq Nano DNA LT Sample Prep Kit (Illumina, San Diego, CA, USA) per the manufacturer's directions. The suggested 200 ng input of DNA was approximated by an input of PCR‐amplified DNA from each marker at the following amounts: Am12s at 31 ng, Ac12s at 37 ng, Ac16s at 34 ng, Ve16s at 34 ng, L2513/H2714 at 31 ng and L14735/H15149c at 37 ng, with the DNA from all six markers combined per sample to be individually indexed as one library and the total volume brought to 50 μL with the addition of resuspension buffer. A different amount of each amplicon was included in each library to account for anticipated PCR bias favouring shorter amplicons during the subsequent PCR enrichment step. Twelve individual libraries, one from each experimental treatment (4) and replicate (3), were pooled and submitted to the Notre Dame Genomics and Bioinformatics Core Facility (GBCF) for paired‐end sequencing on one MiSeq flowcell using a v3 600 cycle kit. Prior to sequencing, the GBCF checked library quality following MiSeq recommendations.
Bioinformatic analysis
Demultiplexing of pooled samples was performed automatically by the illumina miseq reporter 2.5 software based on the 12 unique indices inserted during library preparation. Only reads with ≤1 nucleotide (nt) mismatch from an expected index were retained. Using a custom Unix script, forward and reverse reads were then demultiplexed further based on the forward primer sequences inserted during PCR. Only read pairs without mismatches from an expected forward primer and with the primer occupying the beginning of the read (5′–3′ orientation) were retained. Using clc genomic workbench v7.0.3 (CLC Bio, Aarhus, Denmark) for all subsequent steps, primers were trimmed from all reads. Overlapping paired‐end reads were merged with a minimum score of 10, a mismatch cost of 2 and gap cost of 3. Merged reads were trimmed to a minimum length of 50 nt with a 0.011 quality limit (Phred score of 20). Additionally, trimming functioned to remove any remaining TruSeq adapters. Finally, reads were assigned to mesocosm species by mapping against a single custom reference list containing one reference sequence for each marker for each species. To create the list, a single individual from each of the nine species was euthanized and DNA extracted. All six target amplicons were PCR amplified, Sanger sequenced and submitted to GenBank: Cam. anomalum (KM273807, KM282399, KM282460, KM434929, KM435001, KM523267), Cat. commersonii (KM273808, KM282400, KM282461, KM434930, KM435002, KM523268), Cy. carpio (KM273814, KM282406, KM282467, KM434936, KM435008, KM523272), F. notatus (KM273826, KM282416, KM282478, KM434950, KM435019, KM523285), G. holbrooki (KM273827, KM282417, KM282479, KM434951, KM435020, KM523286), L. macrochirus (KM273836, KM282426, KM282486, KM434959, KM435028, KM523292), P. promelas (KM273855, KM282445, KM282503, KM434978, KM435047, KM523310), R. catesbeiana (KM282504, KM434979, KM435048, KM523312), and S. atromaculatus (KM282512, KM434987, KM435055, KM523318). The only exceptions were both 12S amplicons from the bullfrog tadpole, in which case we relied on GenBank Accession NC_022696, and both 12S amplicons from the creek chub, in which case we relied on GenBank Accession AF_023199y. A read was considered mapped if it met the following criteria: (i) cost of 2, 3, 3 for mismatch, insertion and deletion, respectively, (ii) 99% similarity across 100% of the read length and (iii) mapped to only one species. The number of reads for each primer set was recorded separately for each experimental mesocosm for subsequent statistical analysis.
Statistical analysis
Neither the species specific nor the pooled read abundance data from the 12 mesocosms (9 species × 12 mesocosms = 108 data points) were normally distributed. We were unable to sufficiently transform the data to normality using either a log or square‐root transformation. Therefore, we used robust multiple‐modal (MM)‐estimation in the form of iteratively reweighted least squares regression to identify data outliers and fit linear models to the data. MM‐estimation is a robust statistical estimation technique that affords greater statistical power than classic statistical approaches when applied to non‐normal and heteroscedastic data (Yohai 1987; Erceg‐Hurn et al. 2013). Iteratively reweighted least squares regression identifies outliers in an iterative process that assigns greater weight to central observations (data closely fitting the model at each iteration), while farther observations are weighted less and observations with weights of zero eliminated as outliers. The reweighting process uses a bisquare redescending score function to retain the maximum fraction of outliers that the sample can contain without corrupting the estimate (Yohai 1987). Iteratively reweighted least squares regressions were completed using the lmrob function within the r‐package robustbase. All statistical analyses were conducted in r 2.15.2 (R Core Team 2012). We evaluated the effect of taxa mortality that occurred during the experiment on the biomass vs. read abundance relationship by comparing the results of the regressions when calculated with biomass of standing stock and when calculated with cumulative biomass (mass standing stock and mass of mortalities).
Results
Evaluation of contamination
No bluegill or common carp eDNA was detected during the final testing from the quarantine tanks prior to each experiment. Additionally, no eDNA was amplified from the initial water samples collected from the empty mesocosm tanks (prior to populating with species). Quantitative PCR testing of the eight negative control tanks found only trace quantities of bluegill mtDNA in one tank (1 copy/mL of tank water) and common carp mtDNA in two tanks (1 and 2 copies/mL of tank water) from the second experimental trial. The low quantity and frequency of negative control tank DNA from these two species suggests that cross‐contamination among mesocosms was negligible. It is unlikely that such low amounts of contamination could sufficiently bias the Illumina sequencing data to produce the relationships between biomass and read abundance we observed.
Potential bias in the results due to mismatches across a range of taxon within primer sets was not likely as the per cent identity across the nine species in the current study [as evaluated within primer set (Table S1, Supporting information)], varied by <6% for four of the six metabarcoding primer sets (Ac12S, Ac16S, L2513/H2714 and Ve16S). Additionally, mismatch potential and the coefficient of determination (r 2) between read and biomass numbers did not show a consistent trend. For example, the primer set with the largest mismatch potential, L14735/H15149c, had the highest r 2 between the read and biomass numbers, while the primer set with the smallest mismatch potential, Ac12S, had the second highest r 2 between read and biomass numbers.
Species detection
All fish and amphibian species were detected in all treatments and all mesocosms. Our approach accurately measured the species richness of each species assemblage irrespective of differences in the relative abundances and densities of the constituent species.
The accuracy of species richness measurement varied with primer selection. The number of species detected per mesocosm differed among the six primer sets both within and among mesocosms. In our experimental trials, we detected the full species richness (nine species) in all 12 mesocosms using either the Ac16S or the L2513/H2714 primer sets. The other four primer sets measured at least six of the nine species in each of the 12 mesocosms. Eastern mosquitofish was the most commonly undetected species in the 12 mesocosms, failing to be detected in one mesocosm by the L14912/H15149c primer set, in two mesocosms by the Ac12S primer set and in nine mesocosms by the Ve16S primer set (Table S2, Supporting information). Bullfrog tadpole was not detected in two of the 12 mesocosms by the L14912/H15149c primer set and in three mesocosms by the Am12S primer set (Table S2, Supporting information). Blackstripe topminnow was not detected in three of the 12 mesocosms by the L14912/H15149c primer set (Table S2, Supporting information) and white sucker was not detected in one mesocosm by the L14912/H15149c primer set (Table S2, Supporting information). Excluding the Ac16S and L2513/H2714 primer sets, a minimum of three of the remaining four primer sets was needed to detect all species in all mesocosms.
Raw read counts from the single MiSeq run were normally distributed across libraries (Shapiro–Wilk test = 0.934, P = 0.424). Read abundances for each of the six primer sets ranged from 0 to 92 678 per species per mesocosm (Tables S3–S8, Supporting information). In the skewed abundance mesocosms, the highest abundance species (fathead minnow) in each mesocosm had the greatest number of reads for only one (L2513/H2714) of the six primer sets (Tables S3–S8, Supporting information). A wide range in the number of reads for each species was observed for each of the six primer sets within the even abundance mesocosms (Tables S3–S8, Supporting information).
When analysing the pooled data, we detected significant relationships between (i) species biomass and read abundance for all six primer sets (Fig. 1) and (ii) the number of individuals and read abundance for five of the six primer sets (Fig. 2). Comparison of the r 2 between the two data sets indicated that species biomass (Fig. 1) had more explanatory power than species abundance (Fig. 2) for predicting read abundance for five of the six primer sets. We found the biomass vs. reads regressions to be robust to mortality that occurred during the experiment (Table S9, Supporting information). Both cumulative biomass vs. read abundance (Fig. S1, Supporting information) and standing stock biomass vs. read abundance (Fig. 1) illustrated significant positive relationships with a similar r 2. Among the six primer sets, the number of species with significant relationships ranged from five to eight of the species (Figs 3 and S2–S6, Supporting information). The species that did not show significant linear relationships between species biomass and read abundance varied among the six primer sets with no consistent pattern.
Figure 1.
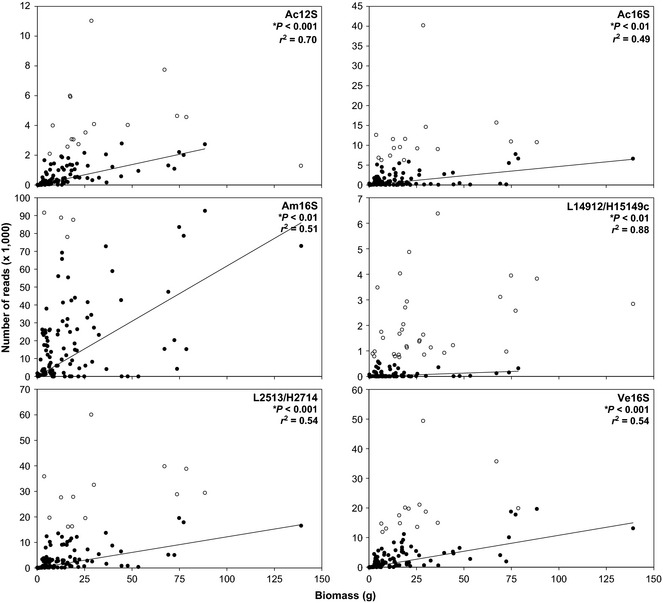
Iteratively reweighted least square regressions of standing stock biomass of all species combined (g) and read abundance of all species combined (number of mapped reads) for each of the six primer sets. Iteratively reweighted least square regression analysis results in fitting the linear model to reweighted data (closed points) exclusive of outliers (open points). Data pooled from all mesocosms (n = 108).
Figure 2.
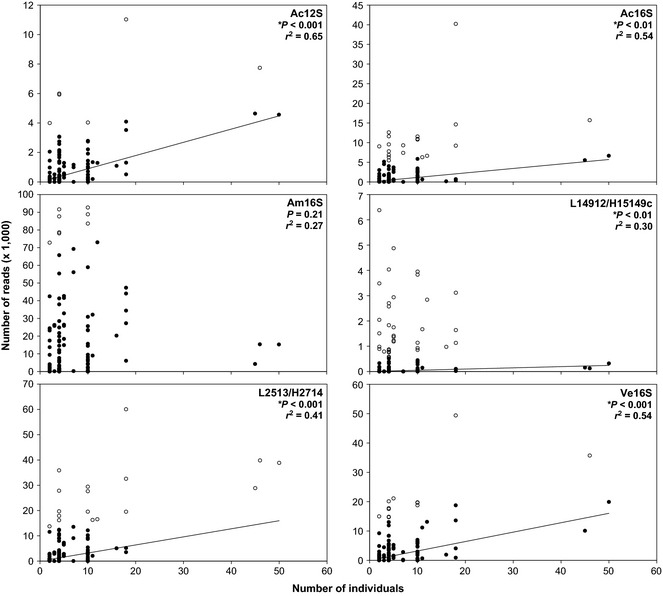
Iteratively reweighted least square regressions of abundance of all species combined (number of individuals) and read abundance of all species combined (number of mapped reads) for each of the six primer sets. Iteratively reweighted least square regression analysis results in fitting the linear model to reweighted data (closed points) exclusive of outliers (open points). Data pooled from all mesocosms (n = 108).
Figure 3.
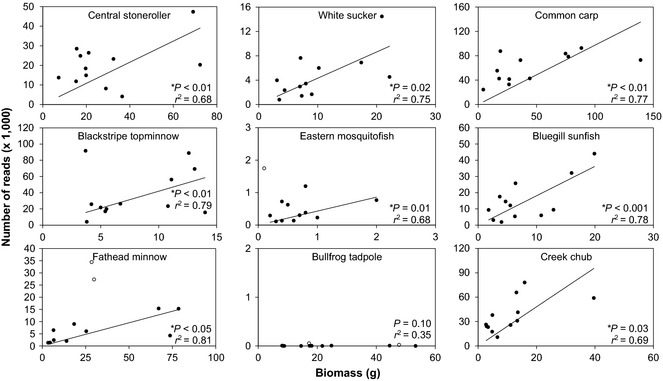
Iteratively reweighted least square regressions of standing stock biomass (g) and read abundance (number of mapped reads) for each species for the Am12s primer (see Figs S1–S5, Supporting information for additional primers]. Iteratively reweighted least square regression analysis results in fitting the linear model to reweighted data (closed points) exclusive of outliers (open points). Data pooled by species from each of the independent mesocosms (n = 12).
Discussion
Our overall objective was to determine the efficacy of using a metabarcoding approach for eDNA samples to measure fish and amphibian species diversity in replicated mesocosms with known and differing assemblage structures. This mesocosm experiment allowed us to evaluate (i) the ability of eDNA metabarcoding to detect the array of vertebrate species placed into the mesocosms, (ii) the effects of skewed species relative abundances and densities on species detection, and (iii) the effects of species abundance and biomass on the number of sequenced reads attributable to each species.
Our results illustrate that the eDNA‐metabarcoding approach employed in this study is capable of (i) detecting all nine vertebrates species placed into the mesocosm, and (ii) accurately measuring species richness of high‐ and low‐density species assemblages with even and skewed relative species abundances. Previous research has suggested that limitations in sequencing depth, differential DNA shedding rates, and preferential PCR amplification have the potential to directionally bias estimates of species richness from eDNA samples (Adams et al. 2013; Deagle et al. 2013; Kelly et al. 2014). Our results suggest that in low to moderately diverse macrofauna species assemblages under mesocosm conditions, rarer species in communities with skewed species abundances can still be detected when analysed via a combined suite of markers. Moreover, our results are consistent with previous findings that indicate primer selection can influence species detection results. Preferential PCR amplification of bony fishes was suggested as a reason for the failed detection of cartilaginous fishes and sea turtles in a previous mesocosm experiment that utilized an eDNA‐metabarcoding approach (Kelly et al. 2014). Similarly, barcode sequence cluster recovery from diverse arthropod samples has been found to be much lower for individual primer sets than when using a combined suite of 11 primer sets (Gibson et al. 2014). In our study, we found that only two of the six primer sets individually detected all of the species present in all of the mesocosms. The L14912/H15149c primer set most commonly failed to detect multiple species in each of the mesocosms. The frequent failure of the L14912/H15149c primer set to detect species may in part be a result of lower total read abundance of the primer set relative to the five other primer sets (Tables S3–S8, Supporting information).
Although we did not have a large gradient in number of individuals of each species, our results for some primers are consistent with other research suggesting lower detectability of rarer species with metabarcoding approaches (Adams et al. 2013; Deagle et al. 2013; Kelly et al. 2014). For some primer sets, a greater percentage of species was detected in the treatments with even relative abundances than in the treatments with skewed abundances. Within the skewed relative abundance treatments, we failed to detect lower relative abundance species (eastern mosquitofish, white sucker and bullfrog tadpole) more frequently than moderate abundance species (blackstripe topminnow). However, all primer sets detected the high‐abundance species in all mesocosms. These results may suggest the potential for high‐abundance species to ‘mask’ detection of low‐abundance species under some conditions with some primer sets.
Such ‘species masking’ could potentially result from multiple interacting mechanisms including: (i) PCR bias towards templates that are more abundant in DNA extracts, (ii) sequencing bias towards amplicons that are more abundant in PCR product, or (iii) lower collection probability for templates that are less abundant in environmental water samples. Previous research on microbial communities has illustrated a similar pattern, where rare species are missed due to limitations in sequencing depth (Adams et al. 2013). Contrary to the effects of relative abundance on species detection, relative biomass of species does not appear to strongly influence the detection of species. Species that were not detected were not always those with the lowest biomass in the mesocosms. In some mesocosms, the species with the 3rd to 6th greatest rank biomasses were the only ones not detected. However, these moderate biomass species were also present in low relative abundances. This result, in addition to research illustrating differences in eDNA production rates between juvenile and adult fish (Maruyama et al. 2014; Klymus et al. 2015), suggests a potential interaction between relative abundance (i.e. number of individuals) and relative biomass that affects eDNA production rates and, therefore, detectability of species when using metabarcoding approaches.
In addition to successfully measuring the species richness of our mesocosms, we observed a positive relationship between species abundance and sequence read abundance. This supports previous laboratory (Porazinska et al. 2010; Thomsen et al. 2012b) and field studies (Takahara et al. 2012; Pilliod et al. 2013; Jane et al. 2015) that denote a positive relationship between species abundance and eDNA concentration. However, the relationship between species abundance and read abundance was modest (r 2 range, 0.30–0.65). The high variability within the relationship could result from differences among species in eDNA production or from a disconnect between read abundance and eDNA concentration that results from error introduced during PCR, PCR cleanup, library preparation and library normalization for sequencing (Amend et al. 2010; Porazinska et al. 2010; Murray et al. 2011). For our study system, species biomass was a better predictor of read abundance than the number of individuals. However, our results are inconsistent with previous research that found a positive linear relationship between rank biomass and rank read abundance using a metabarcoding approach (Kelly et al. 2014). In our experimental mesocosms, the species with the lowest biomass in the mesocosm was frequently, but not always, the species with the lowest number of reads. Similarly, the species with the greatest biomass in the mesocosm was frequently, but not always, the species with the greatest number of reads. The rank read abundance of species with intermediate biomasses in the mesocosms frequently did not correspond to rank biomass. Therefore, our results suggest a more complex relationship. A number of interacting factors may be responsible for the deviation in the relationship between linear rank species biomass and rank read abundance. It is not well understood how species traits such as body size, activity level, metabolism and susceptibility to stress may affect eDNA production. We also assumed that eDNA of all species was homogeneously distributed within each mesocosm tank. We believe this assumption was reasonable because water was well circulated through aeration from bottom to top within each mesocosm for the duration of the experimental trials. However, homogeneous distributions may be less likely in natural ecosystems (Turner et al. 2015).
Positive detection of the entire species assemblage in each of the mesocosms advances practical bioassessment by illustrating the potential for estimation of species richness via eDNA‐metabarcoding approaches. Our successful measurement of the complete species assemblages in all of the mesocosms by only two of the six primer sets suggests that it may be necessary to utilize multiple primer sets when attempting to estimate the entire species richness of natural communities. However, our results also suggest that it may be possible to utilize a relatively small suite of primer sets to successful quantify species richness in diverse communities.
The experimental design and negative controls employed in our experiment enabled us to detect significant positive relationships between species biomass and read abundance. However, the field of eDNA‐metabarcoding research is rapidly developing and recently published literature has indicated the importance of including additional contamination controls in metabarcoding studies (Bohmann et al. 2014; Rees et al. 2014; Thomsen & Willerslev 2015). Among published eDNA studies, considerable variation is apparent in the types and abundance of samples used to control spurious effects (Rees et al. 2014). Due to the inherently sporadic occurrence of DNA contamination (Champlot et al. 2010; Erlwein et al. 2011; Tuke et al. 2011), experimental designs should include multiple negative controls throughout the entire experimental process. Moreover, recent literature suggests that it may be important to sequence negative controls even when there is no PCR product visible in gel electrophoresis (Nguyen et al. 2015; Schnell et al. 2015). Some laboraotory‐derived contamination is nearly inevitable with high‐throughput sequencing technologies, making authentication of taxa detections essential (Thomsen & Willerslev 2015). Both Nguyen et al. (2015) and Schnell et al. (2015) recently illustrated that Illumina sequencing of eDNA‐derived metabarcoding PCR amplicons detects target species contamination in negative controls despite no visible PCR product in gel electrophoresis. Our experimental design included negative control tanks during the experimental trials; however, we did not use our metabarcoding primers to amplify all the negative controls because our qPCR testing provided evidence that contamination was uncommon in the mesocosm experiment and, therefore, an unlikely source of bias in the species biomass and read abundance relationships.
Our analysis of the relationship between species abundance and read abundance illustrates the potential and limitations of estimation of relative species abundance from metabarcoding sequence data. The positive relationship between species biomass and read abundance for the pooled mesocosm data indicates that it may be possible to estimate the relative biomass of species within a controlled species assemblage. However, the relatively weak relationship with high levels of unexplained variation in these small controlled mesocosms suggests that, at best, it will be challenging, and in practice may not be possible, to derive statistically significant relationships between biomass and read abundance in more complex natural ecosystems. Thus, our mesocosm‐level results illustrate the potential for eDNA sampling and metabarcoding approaches in biological community assessment surveys that seek to determine the identities of the organisms present in the community. However, estimation of relative abundance of species from read abundance may not be practical with current approaches.
Traditional capture‐based aquatic bioassessment approaches employed by management agencies for use in prioritizing management actions and evaluating ecological impacts are labour intensive and inherently limited in the effectiveness with which they detect rare species. The results of our experiment indicate that eDNA‐metabarcoding approaches may improve the effectiveness and efficiency of aquatic bioassessment by increasing detection, per unit effort, of rare and low‐abundance species. Therefore, our study represents a potential advancement for ecological assessment and natural resource management by illustrating how eDNA can be used to measure species richness in macrofaunal assemblages.
Our findings also emphasize the need for additional research on the effects of species characteristics and life history traits that may affect eDNA production rates to better understand the relationship between species abundance/biomass and read abundance. Future mesocosm experiments that include additional phylogenetically similar and more diverse species would provide a means to test the ability of metabarcoding approaches to quantify species richness and abundance in more complex ecosystems. A key next step will then be to apply these new approaches to measuring species richness in natural field communities. Regardless, our study illustrates the potential of eDNA sampling to significantly advance bioassessment of natural ecosystems and fundamental ecological research.
This research was part of a larger project aimed at using environmental DNA and metabarcoding approaches to monitor and conserve threatened and endangered freshwater fishes and amphibians. All authors assisted with experimental design. N.T.E. conducted the experiment, analysed the data and wrote the manuscript. B.P.O. performed the bioinformatic analysis with Y.L. and M.E.P. M.A.R. designed the primer sets with C.R.T. and B.P.O. and performed the molecular techniques. C.R.T., C.L.J., A.R.M., M.E.P, G.A.L. and D.M.L. provided guidance during the study and assisted with manuscript writing.
Data accessibility
Detailed descriptions of the number of sequence reads for each of the six primer sets are available as Tables S3–S8 (Supporting information). Iteratively, reweighted least square regressions of biomass and sequence read abundance for Ac12s (Fig. S2, Supporting information), Ac16s (Fig. S3, Supporting information), L14912/H15149c (Fig. S4, Supporting information), L2513/H2714 (Fig. S5, Supporting information) and Ve16s (Fig. S6, Supporting information) primer sets are available as Supporting information. All Illumina MiSeq raw sequence data are available on Dryad doi:10.5061/dryad.r6m04.
Supporting information
Fig. S1 Iteratively reweighted least square regressions of cumulative biomass of all species combined (g) and read abundance of all species combined (number of mapped reads) for each of the six primer sets.
Fig. S2 Iteratively reweighted least square regressions of standing stock biomass (g) and read abundance (number of mapped reads) for each species for the Ac12s primer.
Fig. S3 Iteratively reweighted least square regressions of standing stock biomass (g) and read abundance (number of mapped reads) for each species for the Ac16s primer.
Fig. S4 Iteratively reweighted least square regressions of standing stock biomass (g) and read abundance (number of mapped reads) for each species for the L14912/H15149c primer.
Fig. S5 Iteratively reweighted least square regressions of standing stock biomass (g) and read abundance (number of mapped reads) for each species for the L2513/H2714 primer.
Fig. S6 Iteratively reweighted least square regressions of standing stock biomass (g) and read abundance (number of mapped reads) for each species for the Ve16s primer.
Table S1 Potential mismatches between the six metabarcoding primer pairs and the nine species in the current study.
Table S2 Number of species detected (species with at least one mapped read) in each mesocosm using each of the six genetic primers at a mapping specificity of 99% match to reference sequence at 100% of sequence length.
Table S3 Number of sequence reads from the Ac12s primer that mapped to each study species in each of the experimental treatments and tanks.
Table S4 Number of sequence reads from the Ac16s primer that mapped to each study species in each of the experimental treatments and tanks.
Table S5 Number of sequence reads from the Am12s primer that mapped to each study species in each of the experimental treatments and tanks.
Table S6 Number of sequence reads from the L14912/H15149c primer that mapped to each study species in each of the experimental treatments and tanks.
Table S7 Number of sequence reads from the L2513/H2714 primer that mapped to each study species in each of the experimental treatments and tanks.
Table S8 Number of sequence reads from the Ve16s primer that mapped to each study species in each of the experimental treatments and tanks.
Table S9 Total mortality (number of individuals) of each species over the duration of the 5‐day mesocosm experiement.
Acknowledgements
Funding provided by United States Department of Defense Strategic Environmental Research and Development Program Grant W912HQ‐12‐C‐0073 (RC‐2240). We thank Mary Hupka, Karen Uy, Crysta Gantz and Michael Brueseke for their assistance with maintaining fish populations and eDNA sample processing. We also thank Melissa Stephens and the GBCF for consultation and sequencing. This research was completed in accordance with University of Notre Dame Institutional Animal Care and Use Protocol #16‐015. This is a publication of the University of Notre Dame Environmental Change Initiative.
References
- Abell R, Thieme ML, Revenga C et al (2008) Freshwater ecoregions of the world: a new map of biogeographic units for freshwater biodiversity conservation. BioScience, 58, 403–414. [Google Scholar]
- Adams R, Amend A, Taylor J, Bruns T (2013) A unique signal distorts the perception of species richness and composition in high‐throughput sequencing surveys of microbial communities: a case study of fungi in indoor dust. Microbial Ecology, 66, 735–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amend AS, Seifert KA, Bruns TD (2010) Quantifying microbial communities with 454 pyrosequencing: does read abundance count? Molecular Ecology, 19, 5555–5565. [DOI] [PubMed] [Google Scholar]
- Bailey RC, Norris RH, Reynoldson TB (2004) Bioassessment of Freshwater Ecosystems. Kluwer Academic Publisher, New York, New York. [Google Scholar]
- Barnes MA, Turner CR, Jerde CL et al (2014) Environmental conditions influence eDNA persistence in aquatic systems. Environmental Science & Technology, 48, 1819–1827. [DOI] [PubMed] [Google Scholar]
- Bayley PB, Peterson JT (2001) An approach to estimate probability of presence and richness of fish species. Transactions of the American Fisheries Society, 130, 620–633. [Google Scholar]
- Beja‐Pereira A, Oliveira R, Alves PC, Schwartz MK, Luikart G (2009) Advancing ecological understandings through technological transformations in noninvasive genetics. Molecular Ecology Resources, 9, 1279–1301. [DOI] [PubMed] [Google Scholar]
- Bohmann K, Evans A, Gilbert MTP et al (2014) Environmental DNA for wildlife biology and biodiversity monitoring. Trends in Ecology & Evolution, 29, 358–367. [DOI] [PubMed] [Google Scholar]
- Bonar SA, Hubert WA, Willis DW. (eds) (2009) Standard Methods for Sampling North American Freshwater Fishes. American Fisheries Society, Bethesda, Maryland. [Google Scholar]
- Brander KM (2007) Global fish production and climate change. Proceedings of the National Academy of Sciences of the USA, 104, 19709–19714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruton MN (1995) Have fishes had their chips—the dilemma of threatened fishes. Environmental Biology of Fishes, 43, 1–27. [Google Scholar]
- Burgener M, Hübner P (1998) Mitochondrial DNA enrichment for species identification and evolutionary analysis. Zeitschrift für Lebensmitteluntersuchung und ‐Forschung A, 207, 261–263. [Google Scholar]
- Cao Y, Larsen DP, Thorne RS‐J (2001) Rare species in multivariate analysis for bioassessment: some considerations. Journal of the North American Benthological Society, 20, 144–153. [Google Scholar]
- Champlot S, Berthelot C, Pruvost M et al (2010) An efficient multistrategy DNA decontamination procedure of PCR reagents for hypersensitive PCR applications. PLoS ONE, 5, e13042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne KJ, Handy SM, Demir E et al (2005) Improved quantitative real‐time PCR assays for enumeration of harmful algal species in field samples using an exogenous DNA reference standard. Limnology and Oceanography: Methods, 3, 381–391. [Google Scholar]
- Deagle BE, Thomas AC, Shaffer AK, Trites AW, Jarman SN (2013) Quantifying sequence proportions in a DNA‐based diet study using Ion Torrent amplicon sequencing: which counts count? Molecular Ecology Resources, 13, 620–633. [DOI] [PubMed] [Google Scholar]
- Dejean T, Valentini A, Duparc A et al (2011) Persistence of environmental DNA in freshwater ecosystems. PLoS ONE, 6, e23398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudgeon D (2010) Prospects for sustaining freshwater biodiversity in the 21st century: linking ecosystem structure and function. Current Opinion in Environmental Sustainability, 2, 422–430. [Google Scholar]
- Erceg‐Hurn DM, Wilcox RR, Keselman HJ (2013) Robust statistical estimation In: The Oxford Handbook of Quantitative Methods: Volume 2 Statistical Analysis (ed. Little TD.), pp. 388–406. Oxford University Press, New York, New York. [Google Scholar]
- Erlwein O, Robinson MJ, Dustan S et al (2011) DNA extraction columns contaminated with murine sequences. PLoS ONE, 6, e23484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficetola GF, Miaud C, Pompanon F, Taberlet P (2008) Species detection using environmental DNA from water samples. Biology Letters, 4, 423–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson J, Shokralla S, Porter TM et al (2014) Simultaneous assessment of the macrobiome and microbiome in a bulk sample of tropical arthropods through DNA metasystematics. Proceedings of the National Academy of Sciences of the USA, 111, 8007–8012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giller PS, Hillebrand H, Berninger UG et al (2004) Biodiversity effects on ecosystem functioning: emerging issues and their experimental test in aquatic environments. Oikos, 104, 423–436. [Google Scholar]
- Gu W, Swihart RK (2004) Absent or undetected? Effects of non‐detection of species occurrence on wildlife–habitat models. Biological Conservation, 116, 195–203. [Google Scholar]
- Hall TA (1999) BioEdit: a user‐friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series, 41, 95–98. [Google Scholar]
- Hubert WA, Quist MC. (eds) (2010) Inland Fisheries Management in North America, 3rd edn American Fisheries Society, Bethesda, Maryland. [Google Scholar]
- Jameson D, Gibson AP, Hudelot C, Higgs PG (2003) OGRe: a relational database for comparative analysis of mitochondrial genomes. Nucleic Acids Research, 31, 202–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jane SF, Wilcox TM, McKelvey KS et al (2015) Distance, flow, and PCR inhibition: eDNA dynamics in two headwater streams. Molecular Ecology Resources, 15, 216–227. [DOI] [PubMed] [Google Scholar]
- Jerde CL, Mahon AR, Chadderton WL, Lodge DM (2011) “Sight‐unseen” detection of rare aquatic species using environmental DNA. Conservation Letters, 4, 150–157. [Google Scholar]
- Kelly RP, Port JA, Yamahara KM, Crowder LB (2014) Using environmental DNA to census marine fishes in a large mesocosm. PLoS ONE, 9, e86175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitano T, Umetsu K, Tian W, Osawa M (2007) Two universal primer sets for species identification among vertebrates. International Journal of Legal Medicine, 121, 423–427. [DOI] [PubMed] [Google Scholar]
- Klymus KE, Richter CA, Chapman DC, Paukert C (2015) Quantification of eDNA shedding rates from invasive bighead carp Hypophthalmichthys nobilis and silver carp Hypophthalmichthys molitrix . Biological Conservation, 183, 77–84. [Google Scholar]
- Mackenzie DI, Royle JA (2005) Designing occupancy studies: general advice and allocating survey effort. Journal of Applied Ecology, 42, 1105–1114. [Google Scholar]
- MacKenzie DI, Nichols JD, Sutton N, Kawanishi K, Bailey LL (2005) Improving inferences in population studies of rare species that are detected imperfectly. Ecology, 86, 1101–1113. [Google Scholar]
- Magurran AE, Henderson PA (2003) Explaining the excess of rare species in natural species abundance distributions. Nature, 422, 714–716. [DOI] [PubMed] [Google Scholar]
- Maruyama A, Nakamura K, Yamanaka H, Kondoh M, Minamoto T (2014) The release rate of environmental DNA from juvenile and adult fish. PLoS ONE, 9, e114639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald LL (2004) Sampling rare populations In: Sampling Rare or Elusive Species (ed. Thompson WL.), pp. 11–42. Island Press, Washington, District of Columbia. [Google Scholar]
- Murphy BR, Willis DW. (eds) (1996) Fisheries Techniques, 2nd edn American Fisheries Society, Bethesda, Maryland. [Google Scholar]
- Murray DC, Bunce M, Cannell BL et al (2011) DNA‐based faecal dietary analysis: a comparison of qPCR and high throughput sequencing approaches. PLoS ONE, 6, e25776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen NH, Smith D, Peay K, Kennedy P (2015) Parsing ecological signal from noise in next generation amplicon sequencing. New Phytologist, 205, 1389–1393. [DOI] [PubMed] [Google Scholar]
- Pilliod DS, Goldberg CS, Arkle RS, Waits LP (2013) Estimating occupancy and abundance of stream amphibians using environmental DNA from filtered water samples. Canadian Journal of Fisheries and Aquatic Sciences, 70, 1123–1130. [Google Scholar]
- Porazinska DL, Sung WAY, Giblin‐Davis RM, Thomas WK (2010) Reproducibility of read numbers in high‐throughput sequencing analysis of nematode community composition and structure. Molecular Ecology Resources, 10, 666–676. [DOI] [PubMed] [Google Scholar]
- Prince AM, Andrus L (1992) PCR: how to kill unwanted DNA. BioTechniques, 12, 358–360. [PubMed] [Google Scholar]
- R Core Team (2012) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: ISBN 3‐900051‐07‐0. Available at: http://www.R-project.org/. [Google Scholar]
- Rees HC, Maddison BC, Middleditch DJ et al (2014) Review: The detection of aquatic animal species using environmental DNA—a review of eDNA as a survey tool in ecology. Journal of Applied Ecology, 51, 1450–1459. [Google Scholar]
- Renshaw MA, Olds BP, Jerde CL, McVeigh MM, Lodge DM (2015) The room temperature preservation of filtered environmental DNA samples and assimilation into a phenol–chloroform–isoamyl alcohol DNA extraction. Molecular Ecology Resources, 15, 168–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riaz T, Shehzad W, Viari A, Pompanon F, Taberlet P, Coissac E (2011) ecoPrimers: inference of new DNA barcode markers from whole genome sequence analysis. Nucleic Acids Research, 39, e145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala OE, Vuuren VD, Pereira HM et al (2005) Biodiversity across scenarios In: Ecosystems and Human Well Being: Scenarios, Volume 2, Millennium Ecosystem Assessment (eds Carpenter SR, Pingali PL, Bennett EM, Zurek MB.), pp. 375–410. Island Press, Washington, District of Columbia. [Google Scholar]
- Schnell IB, Bohmann K, Gilbert MT (2015) Tag jumps illuminated—reducing sequence‐to‐sample misidentifications in metabarcoding studies. Molecular Ecology Resources, doi: 10.1111/1755‐0998.12402. [DOI] [PubMed] [Google Scholar]
- Stuart SN, Chanson JS, Cox NA et al (2004) Status and trends of amphibian declines and extinctions worldwide. Science, 306, 1783–1786. [DOI] [PubMed] [Google Scholar]
- Taberlet P, Coissac E, Hajibabaei M, Rieseberg LH (2012) Environmental DNA. Molecular Ecology, 21, 1789–1793. [DOI] [PubMed] [Google Scholar]
- Takahara T, Minamoto T, Yamanaka H, Doi H, Kawabata Z (2012) Estimation of fish biomass using environmental DNA. PLoS ONE, 7, e35868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahara T, Minamoto T, Doi H (2013) Using environmental DNA to estimate the distribution of an invasive fish species in ponds. PLoS ONE, 8, e56584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen PF, Willerslev E (2015) Environmental DNA—an emerging tool in conservation for monitoring past and present biodiversity. Biological Conservation, 183, 4–18. [Google Scholar]
- Thomsen PF, Kielgast J, Iversen LL et al (2012a) Detection of a diverse marine fish fauna using environmental DNA from seawater samples. PLoS ONE, 7, e41732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen PF, Kielgast JOS, Iversen LL et al (2012b) Monitoring endangered freshwater biodiversity using environmental DNA. Molecular Ecology, 21, 2565–2573. [DOI] [PubMed] [Google Scholar]
- Tuke PW, Tettmar KI, Tamuri A, Stoye JP, Tedder RS (2011) PCR master mixes harbour murine DNA sequences. Caveat Emptor!. PLoS ONE, 6, e19953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner CR, Barnes MA, Xu CCY et al (2014) Particle size distribution and optimal capture of aqueous macrobial eDNA. Methods in Ecology and Evolution, 5, 676–684. [Google Scholar]
- Turner CR, Uy KL, Everhart RC (2015) Fish environmental DNA is more concentrated in aquatic sediments than surface water. Biological Conservation, 183, 93–102. [Google Scholar]
- Williams CB (1964) Patterns in the Balance of Nature and Related Problems in Quantitative Ecology. Academic Press, London. [Google Scholar]
- Xenopoulos MA, Lodge DM, Alcamo J et al (2005) Scenarios of freshwater fish extinctions from climate change and water withdrawal. Global Change Biology, 11, 1557–1564. [Google Scholar]
- Yohai VJ (1987) High breakdown‐point and high efficiency robust estimates for regression. Annals of Statistics, 15, 642–656. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Iteratively reweighted least square regressions of cumulative biomass of all species combined (g) and read abundance of all species combined (number of mapped reads) for each of the six primer sets.
Fig. S2 Iteratively reweighted least square regressions of standing stock biomass (g) and read abundance (number of mapped reads) for each species for the Ac12s primer.
Fig. S3 Iteratively reweighted least square regressions of standing stock biomass (g) and read abundance (number of mapped reads) for each species for the Ac16s primer.
Fig. S4 Iteratively reweighted least square regressions of standing stock biomass (g) and read abundance (number of mapped reads) for each species for the L14912/H15149c primer.
Fig. S5 Iteratively reweighted least square regressions of standing stock biomass (g) and read abundance (number of mapped reads) for each species for the L2513/H2714 primer.
Fig. S6 Iteratively reweighted least square regressions of standing stock biomass (g) and read abundance (number of mapped reads) for each species for the Ve16s primer.
Table S1 Potential mismatches between the six metabarcoding primer pairs and the nine species in the current study.
Table S2 Number of species detected (species with at least one mapped read) in each mesocosm using each of the six genetic primers at a mapping specificity of 99% match to reference sequence at 100% of sequence length.
Table S3 Number of sequence reads from the Ac12s primer that mapped to each study species in each of the experimental treatments and tanks.
Table S4 Number of sequence reads from the Ac16s primer that mapped to each study species in each of the experimental treatments and tanks.
Table S5 Number of sequence reads from the Am12s primer that mapped to each study species in each of the experimental treatments and tanks.
Table S6 Number of sequence reads from the L14912/H15149c primer that mapped to each study species in each of the experimental treatments and tanks.
Table S7 Number of sequence reads from the L2513/H2714 primer that mapped to each study species in each of the experimental treatments and tanks.
Table S8 Number of sequence reads from the Ve16s primer that mapped to each study species in each of the experimental treatments and tanks.
Table S9 Total mortality (number of individuals) of each species over the duration of the 5‐day mesocosm experiement.
Data Availability Statement
Detailed descriptions of the number of sequence reads for each of the six primer sets are available as Tables S3–S8 (Supporting information). Iteratively, reweighted least square regressions of biomass and sequence read abundance for Ac12s (Fig. S2, Supporting information), Ac16s (Fig. S3, Supporting information), L14912/H15149c (Fig. S4, Supporting information), L2513/H2714 (Fig. S5, Supporting information) and Ve16s (Fig. S6, Supporting information) primer sets are available as Supporting information. All Illumina MiSeq raw sequence data are available on Dryad doi:10.5061/dryad.r6m04.