

Summary
Background
Glucocorticoids (GCs) are first‐line treatment for keloid disease (KD) but are limited by high incidence of resistance, recurrence and undesirable side‐effects. Identifying patient responsiveness early could guide therapy.
Methods
Nineteen patients with KD were recruited at week 0 (before treatment) and received intralesional steroids. At weeks 0, 2 and 4, noninvasive imaging and biopsies were performed. Responsiveness was determined by clinical response and a significant reduction in vascular perfusion following steroid treatment, using full‐field laser perfusion imaging (FLPI). Responsiveness was also evaluated using (i) spectrophotometric intracutaneous analysis to quantify changes in collagen and melanin and (ii) histology to identify changes in epidermal thickness and glycosaminoglycan (GAG) expression. Biopsies were used to quantify changes in glucocorticoid receptor (GR) expression using quantitative reverse transcriptase polymerase chain reaction, immunoblotting and immunohistochemistry.
Results
At week 2, the FLPI was used to separate patients into steroid responsive (n = 12) and nonresponsive groups (n = 7). All patients demonstrated a significant decrease in GAG at week 2 (P < 0·05). At week 4, responsive patients exhibited significant reduction in melanin, GAG, epidermal thickness (all P < 0·05) and a continued reduction in perfusion (P < 0·001) compared with nonresponders. Steroid‐responsive patients had increased GR expression at baseline and showed autoregulation of GR compared with nonresponders, who showed no change in GR transcription or protein.
Conclusions
This is the first demonstration that keloid response to steroids can be measured objectively using noninvasive imaging. FLPI is a potentially reliable tool to stratify KD responsiveness. Altered GR expression may be the mechanism gating therapeutic response.
Short abstract
What's already known about this topic?
Steroids are used as first‐line treatment for keloid disease, but response is variable with apparent steroid responders and nonresponders.
It remains unclear whether steroid responsiveness is due to an intrinsic difference in the mechanism of the glucocorticoid receptor (GR) action or reduced sensitivity to the steroid itself.
What does this study add?
We show for the first time the utility of noninvasive imaging techniques in stratifying steroid responsiveness in patients treated with steroids.
GR expression increased significantly in keloid tissue. Increased GR expression also correlated with an increased response to steroid treatment.
Steroid responders show a significant decrease in GR transcript and protein 2 weeks after steroid treatment (P < 0·05), which was not evident in steroid‐resistant patients.
Keloid disease (KD) is a fibroproliferative disorder of unknown aetiopathogenesis. Lesions arise as a result of skin injury or infection, although it may not be possible to identify the causative event, and lesions typically extend beyond the original wound boundary, commonly recurring after excision.1 These scars have an aesthetic, physical and psychological effect on the patient and adversely impact on patients’ quality of life.2
Management of keloid disease is ill defined and a variety of treatment strategies have been recommended. The international guidelines on management of keloid scarring have recommended the use of corticosteroids as a first‐line therapy.3 However, not all keloid scars respond to steroid treatment and there is a 50% recurrence rate.3 Most treatment algorithms require multiple serial injections of intralesional steroid, which are not only painful but can also cause a number of steroid‐induced side‐effects, including skin atrophy, hypopigmentation, telangiectasia, ulceration and, rarely, Cushing syndrome.4 The partial response to steroid therapy in a significant number of patients and the commonly reported side‐effects indicate that intralesional steroid injections should be used with caution. Thus, an early, objective assessment of steroid responsiveness would be very useful.
Synthetic steroids are commonly used in the management of KD and include dexamethasone, methylprednisolone and triamcinolone (TAC).5, 6 Steroids used to treat KD act by decreasing inflammation, reducing lesional volume and erythema.7 In vitro studies have shown that certain changes are induced in lesions treated with corticosteroids, including reduction in the quantity of extracellular matrix proteins, suppression of the pro‐α1 collagen gene, and reduction in both vascular endothelial growth factor and angiogenesis.7, 8, 9, 10, 11 Changes in the architecture of KD have been documented with an improvement in the organization of collagen bundles as well as a degeneration of the collagen nodules.12
Triamcinolone and other glucocorticoids (GCs) are reliant upon the functioning of the glucocorticoid receptor (GR), a member of the nuclear receptor superfamily.6 GR is expressed in most cell types and is essential for regulating a range of physiological processes, most notably immunity and metabolism.6 To date, little is known about the underlying mechanisms causing steroid resistance in KD, and there are currently no methods to objectively track steroid responses longitudinally.
Materials and methods
Patient recruitment and tissue collection
Ethical approval for this study was obtained from the South Manchester Research Ethics Committee, Manchester, U.K. The study adhered to Declaration of Helsinki principles and written informed consent was obtained from all patients. Twenty patients (Fig. 1) with untreated keloids were recruited from the University Hospital of South Manchester National Health Service Foundation Trust. A full medical history was taken and the scar photographed. Baseline noninvasive imaging of the scar was performed using spectrophotometric intracutaneous analysis (SIAscopy)13, 14, 15 (Astron Clinica, Cambridge, U.K.) and full‐field laser perfusion imaging (FLPI; Moor Instruments, Axminster, U.K.)13, 14, 16, 17, 18 (Fig. 2). Following imaging, a 4‐mm punch biopsy of the keloid scar was taken under local anaesthetic (week 0). Subsequently, participants underwent treatment with an intralesional injection of 10 mg ml−1 TAC, using a fine bore needle, inserted horizontally into the keloid tissue and gradually withdrawn while simultaneously injecting until the scar became uniformly blanched (to normalise for different scar volumes) with a maximum dose of 5 mg at any one site.
Figure 1.

(a) Experimental design. qRT‐PCR, quantitative reverse transcriptase polymerase chain reaction; GR IHC, glucocorticoid receptor immunohistochemistry; GAG, glycosaminoglycan. (b) Patient demographics. M, male; F, female.
Figure 2.
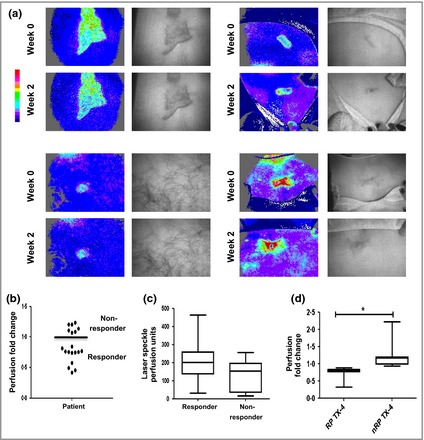
Flux profiling to stratify patient responses to steroid treatment. Keloids from 19 patients were imaged using full‐field laser perfusion imaging. In each case, perfusion of keloid scar was normalized to the surrounding normal tissue. (a) Representative flux profiles are shown. Areas of high flux are red and low flux are blue. (b) Steroid‐dependent change in perfusion at week 2. Those who demonstrated reductions (below the line) in flux were designated responsive patients (RPs) and those who did not (above the line) were designated nonresponsive patients (nRPs). (c) Perfusion at week 0 for RPs (n = 12) and nRPs (n = 7). (d) Perfusion at week 4 for RPs and nRPs. *P < 0·05.
Nineteen patients (one declined further follow‐up) returned for evaluation at two and 4 weeks when scars were assessed using noninvasive imaging and a further 4‐mm punch biopsy was taken from the same scar. Biopsies were bisected and stored in the appropriate medium according to experimental requirements. Samples were stored in compliance with the Human Tissue Act of 2004.
Full‐field laser perfusion imaging
Laser Doppler imaging, such as FLPI, is a validated technique which utilizes a monochromatic laser. Dynamic components of the skin, e.g. red blood cells, cause variations in the laser speckle contrast and a shift in the laser wavelength. Based on these changes, the instrument is able to assess dermal blood flow.19, 20
The FLPI was positioned 40 cm from the scar and used to take 10 separate images over 10 s and the average flux per second was calculated. Perfusion of the scar was normalized to the surrounding uninvolved skin (defined as skin without pathology which was at least 10 cm away from the scar of interest). Fold change was calculated post‐treatment at week 2 compared with the level before treatment. Perfusion was also measured at week 4 to determine if reductions in perfusion continued or returned to baseline levels.
Spectrophotometric intracutaneous analysis
Changes in collagen and melanin were measured using SIAscopy as described previously.13, 14 SIAscopy measures the absorption and reflection of visible and infrared light and can penetrate the skin to a depth of 2 mm. The reflected light is analysed giving quantitative data regarding the concentration of melanin and collagen within the skin.21, 22 The SIAscope was placed within the centre of the scar for 10 s and applied under gentle pressure to avoid blanching of the skin.
Histology
Tissues were fixed in buffered formalin and dehydrated through serial alcohol prior to immersion in xylene and wax embedding. Three sections were used per biopsy. Tissue sections (4‐μm thick) were dewaxed, and then stained with haematoxylin and eosin.23 Images were taken using the Keyence BioImager (Keyence, Itasca, IL, U.S.A.) and randomized. Six representative measurements of the epidermis were taken per section, using Image J (Wayne Rasband, NIH, Bethesda, MD, U.S.A.), and averaged.
Further tissue sections (4‐μm thick) were dewaxed, stained with 0·25% Alcian blue 8GX (diluted in 0·5 mol L−1 HCl; 60 min). Serial images were analysed with Image J, and image intensity/5 μm from the dermal epidermal junction extending 300 μm into the dermis was calculated. Data was plotted using Microsoft Excel (Microsoft Corporation, Redmond, WA, U.S.A.) and GraphPad Prism 4.0 (GraphPad Software, La Jolla, CA, U.S.A.).
Quantitative reverse transcriptase polymerase chain reaction
Biopsies were homogenized using TissueLyser II (Qiagen, Venlo, the Netherlands) in Trizol. RNA extraction, cDNA synthesis and quantitative reverse transcriptase polymerase chain reaction (qRT‐PCR) was carried out as described previously.24, 25 Each sample was analysed for both target (GR) and reference genes [RPL32 (60S ribosomal protein L32) and SDHA (succinate dehydrogenase complex, subunit A)] (Table 1). The raw data for each reaction was analysed by using the comparative cycle threshold value (C T value) for both the reference genes and the candidate target gene and fold change was calculated. For comparison, normal skin samples (n = 5) were obtained from patients without keloids and compared with keloid lesions before treatment.
Table 1.
List of primary antibodies used in this study
| Antibody | Host species | Isotype | Clone | In‐cell Western dilution | IHC‐dilution | Source |
|---|---|---|---|---|---|---|
| Antiglucocorticoid receptor α | Rabbit | IgG | – | – | 1 : 50 | Thermo Scientific |
| Antiglucocorticoid receptor | Mouse | IgG1 | 41 | 1 : 2000 | ‐ | BD Bioscience |
| Anti‐α‐tubulin | Mouse | IgG1 | DMA1 | 1 : 5000 | ‐ | Sigma‐Aldrich |
IHC, immunohistochemistry.
Immunoblotting
Snap‐frozen biopsy sections were homogenized in radio‐immunoprecipitation assay lysis buffer (containing protease‐ and phosphatase‐inhibitor cocktails; Sigma‐Aldrich, St Louis, MO, U.S.A.) and cleared by centrifugation, and protein concentrations were determined by the Bradford protein assay (BioRad, Hemel Hempstead, U.K.).26 Twenty μg of protein was electrophoresed on 4–12% Tris glycine gels (Invitrogen, Carlsbad, CA, U.S.A.) and transferred to 0·5‐μm nitrocellulose membranes. Membranes were blocked (NaCl 0·15 mol L−1, 1% dried milk, 0·1% Tween 20) and incubated in primary antibody overnight (mouse monoclonal GR antibody‐clone 41, diluted 1 : 2000; BD Biosciences, San Jose, CA, U.S.A) (Table 2). Membranes were washed (88 mmol L−1 Tris pH 7·8, 0·25% dried milk, 0·1% Tween 20) and incubated with anti‐mouse horse radish peroxidase (1 : 5000 dilution). Immunoreactive signals were visualized using chemiluminescent substrate (GE Healthcare, Little Chalfont, U.K.) reagent and band density quantified using image J.
Table 2.
List of secondary antibodies used in this study
| Antibody | Host species | Isotype | Active against | Source |
|---|---|---|---|---|
| ECL Mouse, horseradish peroxidase‐linked whole Ab | Sheep | IgG | Mouse | GE Healthcare |
| Vectastain Elite ABC Kit | Goat | IgG | Rabbit | Vector Laboratories |
Immunohistochemistry
Four‐micron‐thick tissue sections were dewaxed, rehydrated and permeabilized with 0·5% Triton X‐100 in Tris buffered saline (TBS) before quenching in 0·1% hydrogen peroxidase for 30 min. Sections were blocked for 1 h in 3% goat serum in TBS and primary GR antibody (Thermo Scientific, Waltham, MA, U.S.A.) (Table 1) applied overnight at 4 °C. Slides were washed with TBS and then incubated with goat anti‐rabbit‐biotin (ABC Elite kit; Vector Laboratories, Peterborough, U.K.) (Table 3) for 30 min and subsequently washed with TBS for 5 min. Avidin‐biotin reagent was applied for 30 min and washed again with TBS for 5 min before Vector SG® chromogen (Vector Laboratories) was applied. Counterstaining of the nucleus was carried out using Nuclear Fast Red (Vector Laboratories). Sections were randomized, blinded and then graded on a five point scale of 0 = no staining to 4 = maximal GR staining.27
Table 3.
List of primers used in this study
| Gene/primer | Gene ID | Sequence 5′ to 3′ | Amplicon size (bp) |
|---|---|---|---|
| GR‐L | nm_001018077.1 | GGGTGGAGATCATATAGACAATCAA | 94 |
| GR‐R | nm_001018077.1 | ACATGCAGGGTAGAGTCATTCTC | 94 |
| RPL32 – L | nm_000994.3 | GAAGTTCCTGGTCCACAACG | 76 |
| RPL32 – R | nm_000994.3 | GAGCGATCTCGGCACAGTA | 76 |
| SDHA – L | nm_004168.2 | AGAAGCCCTTTGAGGAGCA | 88 |
| SDHA – R | nm_004168.2 | CACGGGTCTATATTCCAGAGTGA | 88 |
Statistical analysis
Statistical analysis was carried out using SPSS 16·0 (IBM; Armonk, NY, U.S.A.). Kruskal–Wallis one‐way anova was used to calculate the statistical difference between time points within responders and nonresponders. Mann–Whitney U‐tests were used to calculate differences between responders and nonresponders at specific time points and statistical differences between the keloid lesion, clinically normal skin and skin of patients without keloids for qRT‐PCR. Graphs were plotted using GraphPad 5.0 (GraphPad Software).
Results
Clinical findings
As summarized in Fig. 1a, keloid scars from 20 patients were imaged by noninvasive imaging, punch‐biopsied and injected with intralesional steroid (one patient declined further involvement following the initial biopsy). Fold changes in perfusion pre‐ and poststeroid treatment were calculated at week 2 (Fig. 2a) for the 19 remaining participants (demographics in Fig. 1b). Twelve of the 19 patients showed a decrease in vascular perfusion (Fig. 2b) and they were designated as responsive, with the remaining seven being non‐responsive (Fig. 2b).
Baseline perfusion between both responsive patients (RPs) and nonresponsive patients (nRPs) showed no statistically significant difference; however, a decrease in perfusion in RPs was still evident at week 4 (Fig. 2d; Table 4).
Table 4.
Baseline comparison of responders and nonresponders
| Week 0 [median (IQR)] | |||
|---|---|---|---|
| Responders | Nonresponders | Significance between groupsa | |
| Melanin | 147·7 (98·66–431·4) | 218·0 (35·11–639·6) | P = NS |
| Collagen | 225·6 (160·8–272·7) | 282·5 (196·7–392·3) | P = NS |
| Glycosaminoglycan | 11 130 (10 370–14 490) | 12 000 (11 430–12 450) | P = NS |
| Epidermal thickness | 766·2 (628·2–880·9) | 585·5 (461·5–697·0) | P = NS |
| FLPI | 203·9 (138·1–261·7) | 154·7 (34·97–197·9) | P = NS |
| Glucocorticoid receptor transcription | 0·02668 (0·01651–0·5966) | 0·01425 (0·009958–0·01726) | P = 0·0312 |
| Glucocorticoid receptor immunohistochemistry | 2·5 (1·875–2·714) | 1·278 (1·107–1·792) | P = 0·0176 |
| Glucocorticoid receptor western blot | 0·6420 (0·4324–0·7379) | 0·3419 (0·3301–0·5797) | P = 0·2286 |
IQR, interquartile range; FLPI, full‐field laser perfusion imaging. aMann–Whitney U‐test.
In vivo evaluation of glucocorticoid responsiveness in keloid scars using noninvasive imaging
SIAscopy was used to measure changes in collagen and melanin. Baseline comparisons at week 0 showed no significant difference between RPs and nRPs in either collagen or melanin, implying similar phenotypic characteristics of scars in both groups (Fig. 3b and d).
Figure 3.
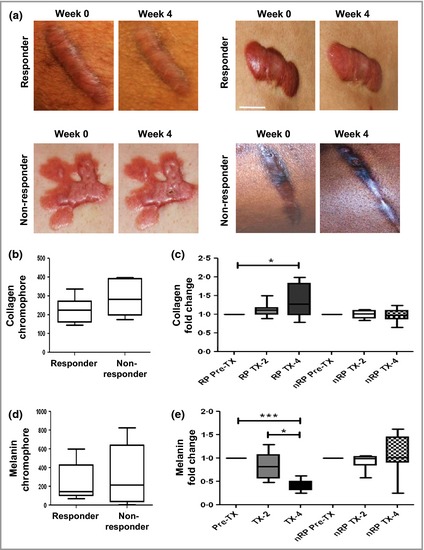
Clinical response to intralesional steroid injections. (a) Representative photographs of keloid scars of responsive patients (RPs) and nonresponsive patients (nRPs) at week 0 and week 4. Scale bar 2 cm. (b–e) Spectrophotometric intracutaneous analysis was used to quantify collagen and melanin chromophores. (b) Collagen chromophore at week 0 for RPs (n = 12) and nRPs (n = 7). (c) Steroid‐dependent change in collagen chromophore at weeks 2 and 4. (d) Melanin chromophore at week 0 for RP (n = 12) and nRP (n = 7). (e) Steroid‐dependent fold change in melanin chromophore at weeks 2 and 4. *P < 0·05.
Responsive patients demonstrated a significant decrease in melanin at weeks 2 and 4 (P < 0·05). In contrast, no significant change was observed in nRPs at either time point (Fig 3e). Compared with nRPs, RPs showed a significant decrease in melanin at week 4 (P < 0·05) (Fig. 3; Tables 5 and 6).
Table 5.
Clinical responsiveness and glucocorticoid receptor expression
| Proteins analysed | Week 2 | Week 4 | ||
|---|---|---|---|---|
| Responders | Nonresponders | Responders | Nonresponders | |
| Melanin | 0·82 (0·57–1·06) P = NS | 0·95 (0·80–2·3) P = NS | 0·43 (0·32–0·50) P = 0·002 | 1·00 (0·37–1·4) P = NS |
| Collagen | 1·12 (1·005–1·17) P = NS | 0·96 (0·88–1·01) P = NS | 1·27 (0·99–1·83) P = 0·019 | 0·87 (0·94–1·04) P = NS |
| Glycosaminoglycan | 0·87 (0·79–0·91) P = 0·013 | 0·83 (0·81–0·96) P = NS | 0·90 (0·871–0·92) P = 0·013 | 0·89 (0·82–0·96) P = 0·026 |
| Epidermal thickness |
0·91 (0·69–1·22) P = NS |
0·91 (0·74–1·31) P = NS | 0·74 (0·57–0·83) P = 0·015 | 1·57 (0·81–1·98) P = NS |
| Glucocorticoid receptor qRT‐PCR | 0·72 (0·48–0·91) P = 0·005 | 0·76 (0·62–1·18) P = NS | 0·85 (0·66–0·90) P = 0·03 | 1·05 (0·89–1·41) P = NS |
| Glucocorticoid receptor protein | 0·59 (0·47–0·67) P = 0·023 | 0·75 (0·73–0·81) P = NS | 0·95 (0·94–1·10) P = NS | 0·60 (0·43–1·68) P = NS |
| Glucocorticoid receptor immunostaining |
0·65 (0·4755–0·895) P = 0·00357 |
1·19804 (1·000·614–1·50521) P = NS | 1·28209 (0·9094–1·8750) P = NS | 0·9482 (0·6438–1·4119) P = NS |
qRT‐PCR, quantitative reverse transcriptase polymerase chain reaction.
Table 6.
Mann–Whitney U‐test comparing responders and nonresponders at weeks 2 and 4 for all parameters
| Proteins analysed | Week 2 responders vs. nonresponders | Week 4 responders vs. nonresponders |
|---|---|---|
| Melanin | P = 0·4943 | P = 0·0140 |
| Collagen | P = 0·1471 | P = 0·0093 |
| Glycosaminoglycan | P = 0·8857 | P = 0·6324 |
| Epidermal thickness | P = 1·000 | P = 0·0381 |
| Glucocorticoid receptor qRT‐PCR | P = 0·3414 | P = 0·0229 |
| Glucocorticoid receptor western blot | P = 0·0571 | P = 0·7000 |
| Glucocorticoid receptor immunostaining | P = 0·0012 | P = 0·3290 |
qRT‐PCR, quantitative reverse transcriptase polymerase chain reaction.
RPs demonstrated an increase in collagen at weeks 2 and 4 (P < 0·05), which was not evident in nRPs (Fig. 3c). A significant increase in collagen was seen in RPs at week 4 compared with nRPs (P < 0·05) (Fig. 3c; Tables 5 and 6).
Histological assessment of glucocorticoid responsiveness in keloid scars
Alcian blue staining was used to quantify glycosaminoglycan (GAG) expression as a measure of histological responsiveness. There was no significant difference between the two groups prior to steroid treatment (Fig 4a). RPs demonstrated a significant decrease in GAG staining at both weeks 2 and 4 (P < 0·05) compared with before treatment. In nRPs, there was a significant reduction only at week 2 (P < 0·05) (Fig. 4b).
Figure 4.

Histological measurement of epidermal thickness and glycosaminoglycan (GAG) content. (a, b) Alcian blue staining was used to quantify GAGs. (a) GAG content at week 0 for responsive patients (RPs) (n = 12) and nonresponsive patients (nRPs) (n = 7). (b) Steroid‐dependent change in GAG content at weeks 2 and 4. (c–e) Sections were stained with haematoxylin and eosin, and epidermal thickness quantified at 10 different positions across each section. (c) Representative images are shown. White line indicates epidermal thickness. (d) Epidermal thickness at week 0 for RPs (n = 12) and nRPs (n = 7). (e) Steroid‐dependent change in epidermal thickness at weeks 2 and 4. *P < 0·05.
Epidermal atrophy is a well‐known side‐effect of GCs.28 Epidermal thickness (ET), was quantified in sections stained with haematoxylin and eosin and showed no significant difference between RPs and nRPs at week 0 (Fig. 4 c,d). In RPs, ET did not change at week 2 but showed a significant decrease by week 4 (P < 0·05); nRPs showed no significant change at either time point (Fig. 4 c,d,e; Tables 5 and 6).
Glucocorticoid receptor expression in responders vs. nonresponders
Immunohistochemistry revealed that GR staining was highest in the epidermal layer, with strong staining in fibroblasts of the papillary dermis layer, and minimal staining in the reticular dermis layer (Fig. 5a). This ratio was consistent across all sections examined. Scoring sections on a four‐point scale suggested that GR expression was significantly higher in RPs at week 0 compared with nRPs (P < 0·05) (Fig. 5b; Tables 5 and 6). RPs demonstrated a significant reduction in GR protein at week 2 (P < 0·05), which returned to baseline by week 4, whereas nRPs did not show any significant difference (Fig. 5c).
Figure 5.
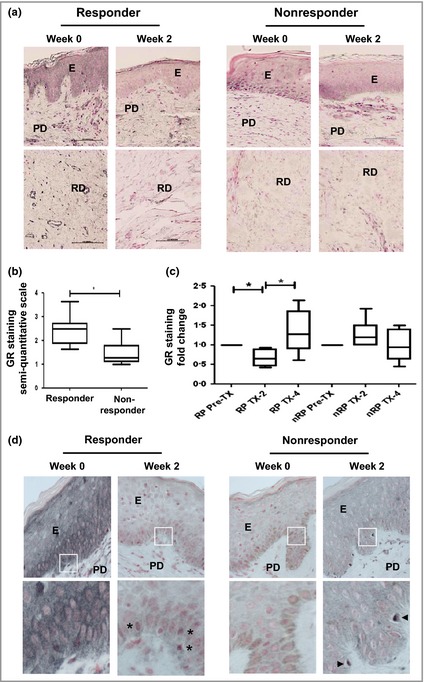
Immunohistological analysis of glucocorticoid receptor (GR) expression and localization. (a–d) Sections were immune‐labelled with a GR‐specific antibody (brown) and the nuclei counterstained (pink). (a) Representative images from both groups are shown. Original magnification ×20; E, epidermis; PD, papillary dermis; RD, reticular dermis. (b) GR expression at week 0 for responsive patients (n = 12) and nonresponsive patients (n = 7). (c) Steroid‐dependent change in GR expression at weeks 2 and 4. (d) Subcellular GR staining in the epidermis. Higher magnification (×40) images are shown below. Asterisks indicate nuclear GR localization. Arrows indicate immune cell infiltration. *P < 0·05.
Closer examination of GR subcellular localization suggested that in RPs at week 0, GR had a heterogeneous subcellular distribution localizing to the nucleus of some cells and the cytoplasm in others. At week 2, GR was identifiable only in cell nuclei, a marker of activation (Fig. 5d, indicated by asterisks). In nRPs, GR showed a comparable heterogeneous distribution, but again there was evidence of immune cell infiltration after steroid treatment (Fig 5d, indicated by arrowheads).
To more readily quantify GR expression, GR transcription was measured by qRT‐PCR. At week 0, consistent with immunohistological analysis, RPs had significantly higher levels of GR transcription than nRPs (Fig 6a). RPs also had a significant decrease in GR transcription at weeks 2 and 4 (P < 0·01 and P < 0·05, respectively). nRPs showed no significant change at any time point (Fig. 6b; Tables 5 and 6). This finding was further supported by immunoblotting (Fig. 6c), which showed increased GR protein in RPs at week 0 (Fig. 6d), and significant downregulation of GR protein in RPs (P < 0·05) but not nRPs at week 2 (Fig. 6e). Interestingly, a significant increase in GR transcription was seen in keloid lesions compared with skin from patients without keloids (P < 0·01), implying that GR expression is upregulated in RPs rather than downregulated in nRPs (Fig. 6f).
Figure 6.

Quantification of glucocorticoid receptor (GR) transcription and protein. (a, b, f) GR transcription was measured by quantitative polymerase chain reaction. (a) GR transcription at week 0 for responsive patients (RPs) (n = 12) and nonresponsive patients (nRPs) (n = 7). (b) Steroid‐dependent change in GR transcription at weeks 2 and 4. (c–d) GR protein expression was measured by immunoblotting using a GR‐specific antibody. (c) A representative immunoblot is shown. Tubulin is shown as a loading control. (d) GR expression at week 0 for RPs (n = 12) and nRPs (n = 7). (e) Steroid‐dependent change in GR expression at weeks 2 and 4. (f) GR transcription in keloid (n = 19) and nonkeloid scars (n = 5). *P < 0·05.
Discussion
This study has used, for the first time, both objective noninvasive and experimental techniques to distinguish between GC‐responsive keloid cases and nonresponders. FLPI was used to measure change in perfusion as an early and sensitive marker of response to steroid treatment that would pre‐date any clinical improvement.13, 29 Clinically, RPs demonstrated a significant decrease in melanin and an increase of collagen, which was not seen in nRPs. This increase in collagen was unexpected, as steroids are known to decrease collagen turnover. It was postulated that the increase in collagen detected by SIAscopy was correlated to epidermal thinning. The SIAscopy is limited by its ability to penetrate to 2 mm into the skin. In RPs, we have demonstrated epidermal thinning and, as such, SIAscopy can penetrate deeper into the papillary dermis and therefore detect a greater volume of collagen. In addition, steroid inhibition of GAG may contribute to the apparent increase in collagen by increasing the density of collagen fibres within the dermis.30, 31
RPs showed a significant decrease in melanin. This is consistent with clinical observations that steroids cause hypopigmentation, particularly in darker‐skinned individuals.12, 32 GC inhibition of melanogenesis has been attributed to a number of cellular mechanisms and the arrest of the melanocyte cell cycle.33 Melanocytes express high levels of GR and when incubated with TAC produce a 30% reduction in cell growth.34
Glucocorticoids mediate their action through the GR, yet a paucity of information exists on GR in both keloid and healthy, normal skin. Gadson et al. demonstrated that receptors derived from keloid tissue do not differ from those derived from healthy tissue in terms of steroid specificity, dissociation constant, total number of binding sites or nuclear binding of GR complexes.34 We have reported that GR expression in RPs before treatment was significantly more than in nRPs and that levels significantly decreased in both mRNA and protein post‐GC treatment. Through a process of autoregulation, GCs downregulate steady‐state expression of GR in both cell lines and tissue.35, 36, 37, 38, 39 HeLa cells treated long term with dexamethasone showed a 50% downregulation in the level of the GR in both protein and mRNA levels.40 This decrease in both mRNA and protein seen previously implies that an essential component in the downregulation of GR protein lies at the level of mRNA.36
We have shown that GR levels in RPs were significantly higher than nRPs at baseline. A decrease in the expression of GR in RPs has been reported in asthma and systemic lupus.41, 42 GR beta (shown to affect steroid resistance) is an alternate isoform, which differs from GR alpha at its carboxy terminal.43 Oakley et al. demonstrated that expression of GR beta in cells inhibits GR‐mediated gene expression and is a dominant negative inhibitor of GR alpha.42 Thus, the reduced expression of GR in nRPs and the lack of GR downregulation following GC treatment may represent the presence of GR beta. The expression of this isoform has been seen in mRNA from cultured keloid fibroblasts.24
Interestingly, GR transcription was significantly higher in KD than in skin from patients without keloids. Supporting our results, Syed et al. showed that cultured keloid fibroblasts expressed higher levels of GR than normal skin fibroblasts. In recent years, several papers have highlighted that keloid disease represents an inflammatory condition associated with increased expression of B cells, T cells and mast cells.44 In addition, expression of transforming growth factors and nuclear factor kappa beta (NF‐κB), known to mediate inflammation, is known to be elevated in keloid fibroblasts compared with normal skin fibroblasts.45, 46 An in vivo study by Schottelius et al. showed an increased expression of GR in inflammatory bowel disease and hypothesized an intrinsic mechanism to counteract elevated levels of NF‐κB by downregulating its expression through interaction of GR with NF‐κB‐p65 and upregulating the expression of NF‐κB inhibitory molecule.24, 47
In clinical practice, keloids are treated by a monthly injection of TAC until resolution of the clinical symptoms of the KD. However, a number of patients show resistance to treatment which is not only frustrating for the clinician but is painful with potential undesirable side‐effects for the patient. Steroids should ideally be employed in patients in whom responsiveness is demonstrated. Nonresponders should be considered for alternative treatments.48, 49 In conclusion, we here demonstrate, for the first time, that keloid responsiveness to GCs can be objectively evaluated and is related to both the initial baseline level of GR and its downregulation. Early identification of nonresponders using noninvasive imaging is a promising tool that may stop the clinician from treating unresponsive cases and can prevent the potentially harmful side‐effects of GC therapy. Alternative and more effective treatment regimens may be offered to keloid cases that are considered to be nonresponders to GC therapy.
Funding sources
The authors would like to acknowledge the Royal College of Surgeons of England and the British Association of Dermatologists for partial funding of the project.
Conflicts of interest
None declared.
References
- 1. Shih B, Garside E, McGrouther DA, Bayat A. Molecular dissection of abnormal wound healing processes resulting in keloid disease. Wound Repair Regen 2010; 18:139–53. [DOI] [PubMed] [Google Scholar]
- 2. Tosa M, Murakami M, Hyakusoku H. Effect of lidocaine tape on pain during intralesional injection of triamcinolone acetonide for the treatment of keloid. J Nippon Med Sch 2009; 76:9–12. [DOI] [PubMed] [Google Scholar]
- 3. Mustoe TA, Cooter RD, Gold MH et al International clinical recommendations on scar management. Plast Reconstr Surg 2002; 110:560–71. [DOI] [PubMed] [Google Scholar]
- 4. Roques C, Téot L. The use of corticosteroids to treat keloids: a review. Int J Low Extrem Wounds 2008; 7:137–45. [DOI] [PubMed] [Google Scholar]
- 5. Niessen FB, Spauwen PH, Schalkwijk J, Kon M. On the nature of hypertrophic scars and keloids: a review. Plast Reconstr Surg 1999; 104:1435–58. [DOI] [PubMed] [Google Scholar]
- 6. McMaster A, Ray DW. Drug insight: selective agonists and antagonists of the glucocorticoid receptor. Nat Clin Pract Endocrinol Metab 2008; 4:91–101. [DOI] [PubMed] [Google Scholar]
- 7. Wu WS, Wang FS, Yang KD et al Dexamethasone induction of keloid regression through effective suppression of VEGF expression and keloid fibroblast proliferation. J Invest Dermatol 2006; 126:1264–71. [DOI] [PubMed] [Google Scholar]
- 8. Kauh YC, Rouda S, Mondragon G et al Major suppression of pro‐alpha1(I) type I collagen gene expression in the dermis after keloid excision and immediate intrawound injection of triamcinolone acetonide. J Am Acad Dermatol 1997; 37:586–9. [DOI] [PubMed] [Google Scholar]
- 9. Chen D, Bao W, Wang Q. [Immunological regulations of dendritic cell in abnormal scarring tissue]. Zhonghua Zheng Xing Wai Ke Za Zhi 2001; 17:282–4 (in Chinese). [PubMed] [Google Scholar]
- 10. Alaish SM, Yager DR, Diegelmann RF, Cohen IK. Hyaluronic acid metabolism in keloid fibroblasts. J Pediatr Surg 1995; 30:949–52. [DOI] [PubMed] [Google Scholar]
- 11. Meyer LJ, Russell SB, Russell JD et al Reduced hyaluronan in keloid tissue and cultured keloid fibroblasts. J Invest Dermatol 2000; 114:953–9. [DOI] [PubMed] [Google Scholar]
- 12. Al‐Attar A, Mess S, Thomassen JM et al Keloid pathogenesis and treatment. Plast Reconstr Surg 2006; 117:286–300. [DOI] [PubMed] [Google Scholar]
- 13. Perry DM, McGrouther DA, Bayat A. Current tools for noninvasive objective assessment of skin scars. Plast Reconstr Surg 2010; 126:912–23. [DOI] [PubMed] [Google Scholar]
- 14. Ud‐Din S, Perry D, Giddings P et al Electrical stimulation increases blood flow and haemoglobin levels in acute cutaneous wounds without affecting wound closure time: evidenced by non‐invasive assessment of temporal biopsy wounds in human volunteers. Exp Dermatol 2012; 21:758–64. [DOI] [PubMed] [Google Scholar]
- 15. Shih B, Sultan MJ, Chaudhry IH et al Identification of biomarkers in sequential biopsies of patients with chronic wounds receiving simultaneous acute wounds: a genetic, histological, and noninvasive imaging study. Wound Repair Regen 2012; 20:757–69. [DOI] [PubMed] [Google Scholar]
- 16. Serov A, Steinacher B, Lasser T. Full‐field laser Doppler perfusion imaging and monitoring with an intelligent CMOS camera. Opt Express 2005; 13:3681–9. [DOI] [PubMed] [Google Scholar]
- 17. Serov A, Lasser T. High‐speed laser Doppler perfusion imaging using an integrating CMOS image sensor. Opt Express 2005; 13:6416–28. [DOI] [PubMed] [Google Scholar]
- 18. Binzoni T, Van De Ville D. Full‐field laser‐Doppler imaging and its physiological significance for tissue blood perfusion. Phys Med Biol 2008; 53:6673. [DOI] [PubMed] [Google Scholar]
- 19. Moor Instruments . Full Field Laser Perfusion Imager User Manual. Millwey, UK: Moor Instruments, 2002. [Google Scholar]
- 20. Leutenegger M, Martin‐Williams E, Harbi P et al Real‐time full field laser Doppler imaging. Biomed Opt Express 2011; 2:1470–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Biocompatibles . Siametrics User Guide. Farnham, Surrey, UK: Biocompatibles, 2001. http://www.biomedicalits.com.au/medx%20documentation/SIAMETRICS-user-guide.pdf (last accessed 5 January 2015). [Google Scholar]
- 22. Govindan K, Smith J, Knowles L et al Assessment of nurse‐led screening of pigmented lesions using SIAscope. J Plast Reconstr Aesthet Surg 2007; 60:639–45. [DOI] [PubMed] [Google Scholar]
- 23. Syed F, Ahmadi E, Iqbal SA et al Fibroblasts from the growing margin of keloid scars produce higher levels of collagen I and III compared with intralesional and extralesional sites: clinical implications for lesional site‐directed therapy. Br J Dermatol 2011; 164:83–96. [DOI] [PubMed] [Google Scholar]
- 24. Syed F, Bayat A. Superior effect of combination vs. single steroid therapy in keloid disease: a comparative in vitro analysis of glucocorticoids. Wound Repair Regen 2013; 21:88–102. [DOI] [PubMed] [Google Scholar]
- 25. Bagabir R, Syed F, Paus R, Bayat A. Long‐term organ culture of keloid disease tissue. Exp Dermatol 2012; 21:376–81. [DOI] [PubMed] [Google Scholar]
- 26. Matthews L, Berry A, Tersigni M et al Thiazolidinediones are partial agonists for the glucocorticoid receptor. Endocrinology 2009; 150:75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Watson RE, Craven NM, Kang S et al A short‐term screening protocol, using fibrillin‐1 as a reporter molecule, for photoaging repair agents. J Invest Dermatol 2001; 116:672–8. [DOI] [PubMed] [Google Scholar]
- 28. El Madani HA, Tancréde‐Bohin E, Bensussan A et al In vivo multiphoton imaging of human skin: assessment of topical corticosteroid‐induced epidermis atrophy and depigmentation. J Biomed Opt 2012; 17:026009. [DOI] [PubMed] [Google Scholar]
- 29. Sommer A, Veraart J, Neumann M, Kessels A. Evaluation of the vasoconstrictive effects of topical steroids by laser‐Doppler‐perfusion‐imaging. Acta Derm Venereol 1998; 78:15–18. [DOI] [PubMed] [Google Scholar]
- 30. Smith TJ. Dexamethasone regulation of glycosaminoglycan synthesis in cultured human skin fibroblasts. Similar effects of glucocorticoid and thyroid hormones. J Clin Invest 1984; 74:2157–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gebhardt C, Averbeck M, Diedenhofen N et al Dermal hyaluronan is rapidly reduced by topical treatment with glucocorticoids. J Invest Dermatol 2010; 130:141–9. [DOI] [PubMed] [Google Scholar]
- 32. Kumar P, Adolph S. Hypopigmentation along subcutaneous veins following intrakeloid triamcinolone injection: a case report and review of literature. Burns 1998; 24:487–8. [DOI] [PubMed] [Google Scholar]
- 33. Slominski A, Tobin DJ, Shibahara S, Wortsman J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol Rev 2004; 84:1155–228. [DOI] [PubMed] [Google Scholar]
- 34. DiSorbo DM, Harris NA, Nathanson L. Effect of triamcinolone acetonide on tyrosinase activity in a human melanoma cell line. Cancer Res 1984; 44:1752–5. [PubMed] [Google Scholar]
- 35. Gadson PF, Russell JD, Russell SB. Glucocorticoid receptors in human fibroblasts derived from normal dermis and keloid tissue. J Biol Chem 1984; 259:11236–41. [PubMed] [Google Scholar]
- 36. Burnstein KL, Bellingham DL, Jewell CM et al Autoregulation of glucocorticoid receptor gene expression. Steroids 1991; 56:52–8. [DOI] [PubMed] [Google Scholar]
- 37. Svec F, Rudis M. Glucocorticoids regulate the glucocorticoid receptor in the AtT‐20 cell. J Biol Chem 1981; 256:5984–7. [PubMed] [Google Scholar]
- 38. Lacroix A, Bonnard GD, Lippman ME. Modulation of glucocorticoid receptors by mitogenic stimuli, glucocorticoids and retinoids in normal human cultured T cells. J Steroid Biochem 1984; 21:73–80. [DOI] [PubMed] [Google Scholar]
- 39. Schlechte JA, Ginsberg BH, Sherman BM. Regulation of the glucocorticoid receptor in human lymphocytes. J Steroid Biochem 1982; 16:69–74. [DOI] [PubMed] [Google Scholar]
- 40. Shimojo M, Hiroi N, Yakushiji F et al Differences in down‐regulation of glucocorticoid receptor mRNA by cortisol, prednisolone and dexamethasone in HeLa cells. Endocr J 1995; 42:629–36. [DOI] [PubMed] [Google Scholar]
- 41. Du J, Li M, Zhang D et al Flow cytometry analysis of glucocorticoid receptor expression and binding in steroid‐sensitive and steroid‐resistant patients with systemic lupus erythematosus. Arthritis Res Ther 2009; 11:R108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cho YJ, Lee KE. Decreased glucocorticoid binding affinity to glucocorticoid receptor is important in the poor response to steroid therapy of older‐aged patients with severe bronchial asthma. Allergy Asthma Proc 2003; 24:353–8. [PubMed] [Google Scholar]
- 43. Oakley RH, Sar M, Cidlowski JA. The human glucocorticoid receptor beta isoform. Expression, biochemical properties, and putative function. J Biol Chem 1996. Apr 19; 271:9550–9. [DOI] [PubMed] [Google Scholar]
- 44. Bagabir R, Byers RJ, Chaudhry IH et al Site‐specific immunophenotyping of keloid disease demonstrates immune upregulation and the presence of lymphoid aggregates. Br J Dermatol 2012; 167:1053–66. [DOI] [PubMed] [Google Scholar]
- 45. Makino S, Mitsutake N, Nakashima M et al DHMEQ, a novel NF‐kappaB inhibitor, suppresses growth and type I collagen accumulation in keloid fibroblasts. J Dermatol Sci 2008; 51:171–80. [DOI] [PubMed] [Google Scholar]
- 46. Messadi DV, Doung HS, Zhang Q et al Activation of NFκB signal pathways in keloid fibroblasts. Arch Dermatol Res 2004; 296:125–33. [DOI] [PubMed] [Google Scholar]
- 47. Schottelius A, Wedel S, Weltrich R et al Higher expression of glucocorticoid receptor in peripheral mononuclear cells in inflammatory bowel disease. Am J Gastroenterol 2000; 95:1994–9. [DOI] [PubMed] [Google Scholar]
- 48. O'Brien L, Pandit A. Silicon gel sheeting for preventing and treating hypertrophic and keloid scars. Cochrane Database Syst Rev 2006; CD003826. [DOI] [PubMed] [Google Scholar]
- 49. Kontochristopoulos G, Stefanaki C, Panagiotopoulos A et al Intralesional 5‐fluorouracil in the treatment of keloids: an open clinical and histopathologic study. J Am Acad Dermatol 2005; 52:474–9. [DOI] [PubMed] [Google Scholar]