

Abstract
Theory predicts that speciation‐with‐gene‐flow is more likely when the consequences of selection for population divergence transitions from mainly direct effects of selection acting on individual genes to a collective property of all selected genes in the genome. Thus, understanding the direct impacts of ecologically based selection, as well as the indirect effects due to correlations among loci, is critical to understanding speciation. Here, we measure the genome‐wide impacts of host‐associated selection between hawthorn and apple host races of Rhagoletis pomonella (Diptera: Tephritidae), a model for contemporary speciation‐with‐gene‐flow. Allele frequency shifts of 32 455 SNPs induced in a selection experiment based on host phenology were genome wide and highly concordant with genetic divergence between co‐occurring apple and hawthorn flies in nature. This striking genome‐wide similarity between experimental and natural populations of R. pomonella underscores the importance of ecological selection at early stages of divergence and calls for further integration of studies of eco‐evolutionary dynamics and genome divergence.
Keywords: Adaptation, experimental genomics, genomics of speciation, Rhagoletis pomonella, speciation‐with‐gene‐flow
Introduction
Over 150 years ago, Darwin (1859) hypothesised that divergent natural selection adapting organisms to novel environments is largely responsible for creating new species. Moreover, theory predicts that ecological divergence and speciation in the presence of gene flow are more likely when reproductive isolation caused by divergent selection to novel habitats transitions from an attribute of individual genes to a collective property of the genome (Barton 1983; Bierne et al. 2011; Feder et al. 2012, 2014; Flaxman et al. 2013, 2014; Tittes & Kane 2014). If true, then a pronounced footprint of selection is predicted between the genomes of speciating organisms, especially those diverging with gene flow. However, demographic history, the contingent nature of mutation, recombination and stochastic processes may play prominent roles in affecting genomic differentiation, obscuring the footprint of selection (Noor & Feder 2006).
Quantifying the impact of selection genome wide is important because, as populations diverge, the effects that individual genes have on reproductive isolation (RI) can become coupled, strengthening barriers to gene flow and promoting speciation (Barton & de Cara 2009). Thus, a critical condition for speciation‐with‐gene‐flow may be the evolution of sufficient adaptive divergence to enable genome‐wide coupling of individual gene effects, which transitions RI from a feature of individual genes to a collective property of the genome (Barton 1983; Feder et al. 2012; Flaxman et al. 2014). If predicated solely on new mutations, this transition could take a long time, during which populations could go extinct or conditions could change without speciation. Thus, a prediction for systems with the potential for speciation‐with‐gene‐flow is that they exhibit large stores of standing variation (Barrett & Schluter 2008) and consequently, show extensive, genome‐wide responses to selection when challenged by divergent ecology. Moreover, discerning the genome‐wide impacts of ecological selection is also relevant to developing a greater understanding of the response (or lack thereof) of populations to rapid environmental change (Noor & Feder 2006; Barrett & Schluter 2008), which is increasingly human mediated.
Genome scans in a range of organisms have revealed extensive genetic differentiation between populations at early stages of ecological divergence (e.g., Lawniczak et al. 2010; Jones et al. 2012; Gagnaire et al. 2013). However, genome scans only provide indirect evidence for selection (Noor & Feder 2006; Noor & Bennett 2009). Verifying selection requires integrating genome scans with experiments on key phenotypes under conditions known to differentially affect populations (Feder et al. 2012). The expectation is that experimental responses should predict the direction and magnitude of divergence observed in nature if selection plays a prominent role in affecting allele frequencies directly through differential survivorship and indirectly through linkage.
Here, we test the degree to which divergent ecological selection affects genomic differentiation between hawthorn and apple‐infesting host races of the fly Rhagoletis pomonella, a model for speciation‐with‐gene‐flow driven by divergent ecology (Bush 1966; Coyne & Orr 2004). Rhagoletis pomonella is a member of a sibling species complex containing numerous geographically overlapping taxa proposed to have radiated in sympatry by adapting to many new host plants from several different plant families (Bush 1966; Berlocher 2000). Rhagoletis flies infest the fruits of their host plants, where host fruits are typically available for a discrete window of time over the growing season and each fly species completes one generation per year. Adult flies meet exclusively on or near the host fruits to mate (Feder et al. 1994), females oviposit into the host fruit, larvae consume the fruit, then burrow into the soil to pupate, and enter a pupal diapause that lasts until the following year (Bush 1966). Thus, phenological matching of fly to host plant fruiting is critical to fly fitness (Feder et al. 1993; Dambroski & Feder 2007).
The most recent example of a host shift driving speciation is the shift of R. pomonella from its native host hawthorn to introduced, domesticated apple, which occurred in the mid‐1800's in the eastern United States (Bush 1966). Genetic and field studies have shown that apple and hawthorn flies represent partially reproductively isolated host races, the hypothesised initial stage of speciation with gene flow. Mark‐recapture studies have demonstrated a gross migration rate of ~ 4% per generation between the races (Feder et al. 1994). Thus, gene flow has been continuous between the fly races since their origin and geographic isolation was not a factor contributing to the build‐up of divergence (Feder et al. 2013). Finally, the bases for divergent ecological selection are known (Feder et al. 1993; Dambroski & Feder 2007), providing the crucial natural history information needed to conduct selection experiments. One key trait that differs between the races is the timing of diapause termination, which varies between the races to match the 3–4 week earlier fruiting time of apple than hawthorn trees (Fig. 1). Rhagoletis emerge from their fruits as late instar larvae and overwinter in the soil in a facultative pupal diapause. The earlier fruiting time of apples therefore results in apple flies having to withstand warmer temperatures for longer periods prior to winter. As a result, natural selection favours increased diapause intensity, or greater recalcitrance to cues that trigger premature diapause termination in apple flies (Tauber et al. 1986; Dambroski & Feder 2007).
Figure 1.
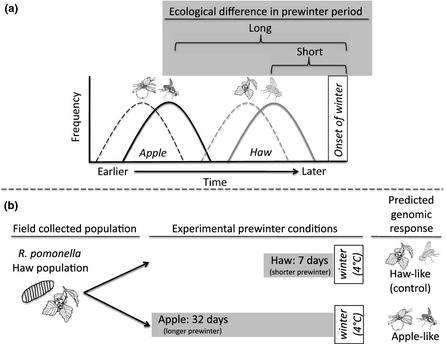
Relationship of diapause life history difference between the R. pomonella host races and divergent ecological rearing conditions imposed in the selection experiment. (a) Fruit on apple trees ripens 3–4 weeks earlier than hawthorn fruit (dashed lines). Apple flies eclose earlier as adults (solid lines) and are exposed to warmer temperatures as pupae in the soil for a longer period of time before winter than hawthorn flies (shaded brackets); (b) In the selection experiment, hawthorn flies were exposed to a short (7‐day) vs. long (32‐day) pre‐winter period to emulate the time difference experienced by hawthorn vs. apple‐fly pupae in nature.
To test the degree to which divergent ecological selection affects genomic differentiation, we performed a ‘selection experiment’ (Fig. 1), exposing flies from the ancestral hawthorn race to environmental conditions experienced by the recently derived apple race. We then determine the extent to which the genome‐wide response of hawthorn flies within a single generation of rearing under apple fly conditions predicts the pattern of divergence between the sympatric host races in nature. We stress that we are quantifying the total genome‐wide impact of selection, which involves both direct effects, where natural selection favours the causal variants underlying selected traits, and indirect effects, where additional loci respond because they are correlated due to linkage disequilibrium with these causal variants (Nielsen 2005; Barrett & Schluter 2008). Thus, the ‘total’ impact of divergent selection (i.e. direct + indirect effects) that we quantify here can involve changes at many loci (Gompert et al. 2014; Soria‐Carrasco et al. 2014).
We exposed ancestral hawthorn fly pupae to warm temperatures for a short 7‐day (‘hawthorn‐like’ control) vs. long 32‐day (‘apple‐like’ experimental) period prior to winter (Fig. 1; see Methods). Allele frequency shifts in the long treatment therefore reflect the within generation genomic response in surviving hawthorn flies to rearing under apple environmental conditions. We then assessed the degree to which the genomic response was concordant to genetic differences in nature by comparing allele frequency shifts in the within‐generation selection experiment to observed differences between the sympatric host races in nature. Previous studies have suggested that host‐related divergence may be widespread in Rhagoletis (Feder et al. 1997; Michel et al. 2010), but were based on only 33 microsatellite and six allozyme markers. Here, we report the first truly genomic dissection of this model system to determine the full extent and magnitude of the genetic footprint of ecological selection.
Materials and methods
Rhagoletis pomonella: field collections and data sets
All flies analysed in the current study were collected as eggs or early instar larvae infesting apple (Malus domestica) or downy hawthorn (Crataegus mollis) fruit from a sympatric site at Grant, MI, USA (43°21′00.17″ N, 85°53′21.98″ W) where the two host trees and flies co‐occur. We generated genotype‐by‐sequencing (GBS) data from three different samples of flies from Grant, MI: (1) mapping families – five single‐pair test crosses made using flies reared to adulthood from larval infested apple fruit to construct a recombination linkage map; (2) selection experiment – adult hawthorn flies equally split by sex that survived the short 7‐day (n = 54 genotyped) and long 32‐day (n = 47) pre‐winter period, both followed by a 30‐week overwinter treatment (Fig. 1); and (3) sympatric natural comparison – adult apple flies equally split by sex that emerged from a 7‐day pre‐winter period followed by a 30‐week overwinter treatment (n = 48), which were compared to the hawthorn flies in the 7‐day pre‐winter treatment above (Fig. 1).
Selection experiment and sympatric comparison
The rationale for the selection experiment was to expose the ancestral R. pomonella population infesting hawthorn (Crataegus mollis) to the ecologically divergent pre‐winter conditions experienced in the earlier fruiting apple (Malus domestica) to test for a genome‐wide response to selection favouring increased diapause intensity (Fig. 1), a critical step in adaptation to the novel apple host plant. To perform the selection experiment, we reared large numbers of outbred flies sampled directly from nature after they emerged as larvae from field‐collected infested fruit and formed puparium under controlled conditions in the laboratory for 7 days (n = 935) or 32 days (n = 981) under a 15/9 h light/dark cycle in a 26 °C constant room temperature (Fig. 1b). We used only flies that pupated in a 3 day window after an initial period of 10 days following field collection to standardise abiotic, non‐host‐related rearing conditions for fly larvae prior to pupariation. The random sampling of flies from a large collection of infested fruit in nature also ensured that many genetically diverse flies were sampled (Rhagoletis can mate 50+ times in their life), resulting in each individual in the selection experiment essentially representing an independent genetic replicate. Following the pre‐winter period, all pupae were then chilled at 4 °C for 30 weeks in a refrigerator to simulate winter, after which time they were placed in a 21 °C incubator with a 14/10 h light/dark cycle. Newly eclosing adults were collected on a daily basis and stored at −80 °C until they were genotyped.
To perform the sympatric comparison, we collected flies from infested apple fruit at Grant, MI site where they co‐occurred with hawthorn flies. Apple‐infesting flies were reared under the same 7‐day pre‐winter conditions as the 7‐day hawthorn fly sample. We have found that the 7‐day treatment imposes minimal selection pressure on R. pomonella flies (Feder et al. 1997; Michel et al. 2010).
Genomic sequence data, assembly and variant SNP calling
For each of the three experimental groups of flies, DNA was isolated and purified from head tissue using Gentra Puregene extraction kits (Qiagen N.V., Venlo, Netherlands). Reduced complexity libraries were then generated for each individual using a restriction enzyme fragment procedure (Parchman et al. 2012). We labelled individual restriction fragments from each fly with a 7 to 10 base pair identification sequence or barcode. DNA sequencing of reduced complexity libraries was performed at the National Center for Genome Research (Santa Fe, NM, USA) using a combination of the Illumina GAII and HiSeq platforms generating 636 million 100 to 124‐bp reads (333 million reads for the sympatric population and selection samples; 303 million for mapping families). We used SeqMan NGen 4.0.0 (DNASTAR) to perform a de novo assembly for a subset (3%) of the sequences using parameters values following Gompert et al. (2012), including a match size of 21, a gap penalty of 40, a mismatch penalty of 15, a match score of 10, a minimum match percentage of 90%, and repeat handling. Roughly 67% (or 13.3 million) sequences in this subset were assembled into 346 000 contigs (mean number of sequences per contig = 56; contig N50 = 109 bp). The expected contig length was 84–109 bp (110–124 bp reads with 8–10 bp barcode and 6 bp associated with the restriction site). After removing low‐quality contigs, we generated a partial ‘pseudo‐reference’ genome from the reads that remained. Contigs were pruned based on their length and content; contigs that were made up of less than 7 reads and contigs that were too far deviated from the expected contig length (less than 78 bp or greater than 113 bp) were also removed. The final reference thus contained 306 000 contigs. We then aligned all sequenced reads to the reference genome using BWA Version 0.6.1‐r104 with the default parameters for the program (Li & Durbin 2009). We discarded all reads from the alignments that did not uniquely map, were not complement reads, or did not align to the beginning of the contig because library preparation should not generate these phenomenon (Parchman et al. 2012). A total of 270 million of the 333 million (81.2%) reads of the population data set and 271 million of the 303 million (89.5%) reads of the mapping family sequences were aligned to the reference genome.
We used custom Perl and Python scripts in concert with GATK version 2.5‐2 (McKenna et al. 2010; DePristo et al. 2011) to locate single‐nucleotide polymorphisms (SNPs) in contigs and to estimate genotype probabilities for each individual at each SNP location. (all custom Perl and Python scripts are available from the authors by request.) We used the default prior in GATK for calling variants and ignored insertion deletion variation. To further prune potential sequence errors from the final data set, we: (1) removed SNPs in which apparent heterozygotes did not follow the expectation of equal allele counts; specifically, we applied a binomial test and discarded all loci where the null hypothesis of P = 0.5, where P is the allele frequency of the more common allele, was rejected with α = 0.05 (Parchman et al. 2012); (2) removed all SNPs that were not in Hardy–Weinberg equilibrium (P ≤ 0.05); (3) removed SNPs that were not bi‐allelic; (4) removed SNPs that had less than 400 reads in the sample populations; and (5) removed SNPs with rare allele frequencies less than 0.05 (minor allele frequency, MAF, < 0.05). We identified 32 455 variable sites with average coverage of 5X and a median coverage of 6X per individual per SNP using these stringent criteria that overlapped between our sympatric comparison and selection experiment. We note that our criteria for editing reads in the study eliminated SNPs present in repetitive sequences and minimised the potential for including reads that possessed missing null alleles.
Tests of significance for allele frequency differences
We used a standard Bayesian model implemented in GATK (McKenna et al. 2010), which incorporates uncertainty due to individual base quality scores and accounts for uneven coverage, to estimate the likelihoods using uniform priors for each of the three possible genotypes for bi‐allelic SNPs. Estimates of SNP allele frequencies (for the alternate allele) for sample populations were calculated from the sequence data as:
where n = the number of individuals, g 1:3 = [0,1,2] (number of alternate allele copies in a homozygote, heterozygote and alternate homozygote, respectively), and p(g j¦D i) were the genotype probabilities for each of the three possible genotypes for the ith individual given the sequence data, D i, as estimated in GATK (Gompert et al. 2012; Parchman et al. 2012). Statistical testing of the allele frequency difference between sample populations was performed through a non‐parametric Monte Carlo approach. For the selection experiment, random samples of n = 54 and n = 47 genotypes for a SNP (the sample sizes in the selection experiment) were drawn with replacement from the pool of 7‐day hawthorn flies. The absolute value of the allele frequency difference between the two random samples was then calculated as above and the process repeated 10 000 to generate a probability distribution to determine if our estimate of the frequency difference was in the upper 95th quantile. For the sympatric comparison, the same procedure was used except n = 54 and n = 48 genotypes for a SNP (the samples sizes in the sympatric comparison) were drawn from the combined pool of 7‐day hawthorn and 7‐day apple flies.
A series of metrics were also calculated for all 32 455 SNPs (Table 1) and for the 2352 mapped markers (Table S1) to summarise various aspects of the allele frequency responses displayed in the selection experiment and genomic divergence between the host races in nature (see Supporting Information for further description).
Table 1.
Genetic response in the selection experiment for all variable SNPs and for subcategories of SNPs displaying significant differences in the selection experiment (sig. sel.), between the host races (sig. races), in both the selection experiment and between the host races (sig. both), and for sets of SNPs in linkage equilibrium (link eq.)
| Locus category | n | |∆ freq| | r | ∆ races | % same |
|---|---|---|---|---|---|
| All SNPs | 32 455 | 0.037 | 0.386 | +0.016 | 63.1 |
| All SNPs sig. sel. | 2245 | 0.104 | 0.691 | +0.045 | 84.1 |
| All SNPs sig. sel. (link eq.) | 162 | 0.093 | 0.712 | +0.049 | 90.5 |
| All SNPs sig. races | 775 | 0.055 | 0.737 | +0.100 | 87.8 |
| All SNPs sig. races (link eq.) | 88 | 0.056 | 0.731 | +0.083 | 84.2 |
| All SNPs sig. both | 154 | 0.116 | 0.961 | +0.138 | 100 |
| All SNPs sig. both (link eq.) | 63 | 0.107 | 0.951 | +0.129 | 100 |
See Table S1 for mapped SNPs. n = number of SNPs per category; ¦∆ freq¦ = mean absolute allele frequency response in selection experiment; r = correlation coefficient between allele frequency response in selection experiment vs. allele frequency difference between host races (P < 10−6 for all r values); ∆ races = mean frequency difference between the host races for alleles increasing in frequency in the selection experiment; % same = percentage of SNPs for which the allele frequency response in the selection experiment changed in the same direction as the difference between the host races.
Building linkage groups
We mapped 2352 of the total 32 455 SNPs to five of the six chromosomes constituting the R. pomonella genome based on five single‐pair test crosses using the program Join Map 4.1 (Kyazma BV, Wageningen, Netherlands) and the major advantage for Dipteran genetics that recombination does not occur in males. Parents for the five test crosses came from apple‐infesting flies collected from the Grant, MI study site and reared to adulthood in the laboratory. Single‐pair crosses were established in small Plexiglas cages supplied with water, a molasses/sugar mixture for food, and store bought red delicious apples for oviposition. Offspring were reared to adulthood using the 7‐day pre‐winter fly husbandry methods described above, with 30 to 42 progeny generated per cross. Parents and offspring were scored for 10 highly variable microsatellites, as per Michel et al. (2010), in addition to SNP markers generated from the Genotyping by Sequencing approach described above. The 10 microsatellites have been previously mapped to chromosomes 1–5 of the R. pomonella genome (Feder et al. 2003; Michel et al. 2010) and provide an anchor for assigning SNPs to linkage groups. Due to the absence of recombination in males, the chromosomal assignment for a variable SNP in male parents could be determined through its non‐random assortment pattern with one of the pairs of microsatellites defining a linkage group. Recombination distance maps among SNPs assigned to the same chromosome were then constructed based on the segregation patterns for variable markers in female parents for each cross using Join Map 4.1. The arrangement of SNPs along each chromosome was inferred with a LOD > 5, theta > 0.4, and the Kosambi mapping function in Join Map. We then used the ‘map integration’ function in Join Map to combine chromosomal maps among crosses. Previous analyses of allozyme, cDNA and microsatellite loci have implied that there is no single linear gene order for each of the five chromosomes consistent with multiple and overlapping chromosome rearrangements distributed across the genome (Feder et al. 2003, 2005; Xie et al. 2008; Michel et al. 2010). Though they segregate in populations, these inversions have a long history dating back to a population of hawthorn flies in the central highlands of Mexico ~ 1.57 MYA (Xie et al. 2008). Thus, recombination distances between SNPs should be viewed in terms of evolutionary map distances of average exchange between markers. The 2352 SNPs were widely distributed across all five chromosomes: ch1 = 271 SNPs, ch2 = 290 SNPs, ch3 = 763 SNPs, ch4 = 441 SNPs and ch5 =587 SNPs (Fig. 2).
Figure 2.
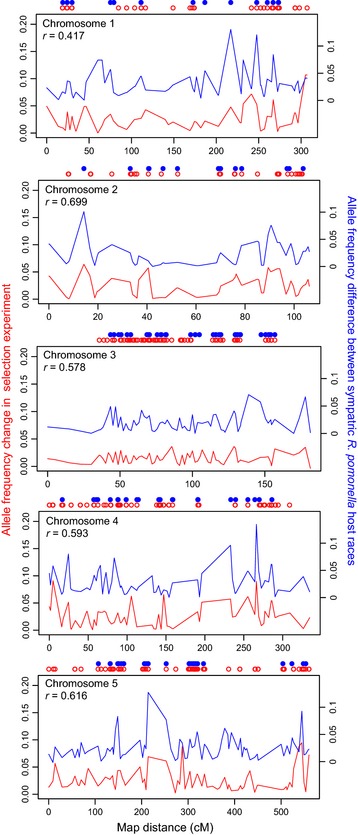
Evidence for genome‐wide effects of selection on divergence between the host races. Genome‐wide sliding window comparison of allele frequency shifts in the selection experiment (red line; left axis) vs. divergence between field‐collected sympatric host races (blue line; right axis) along chromosomes 1–5 (see Methods for details concerning calculation of values). Circles above panels denote SNPs showing statistically significant response in the selection experiment (open red) or difference between the host races (solid blue). Correlation coefficient (r) is reported independently for each chromosome illustrating the association between allele frequency shifts in the selection experiment and allele frequency differences between sympatric host races in nature (all P < 10−4). Note, lines represent sliding window averages taken across the genome in 2 centi‐Morgan intervals around each SNP, not the raw allele frequency differences.
Measuring linkage disequilibrium
To assess the number of potential independent gene regions responding to selection, we estimated Burrow's composite measure of Hardy–Weinberg and linkage disequilibrium (∆) between pairs of variable sites in the 7‐day hawthorn sample (Weir & Cockerham 1978; Weir 1979). Burrow's ∆ does not assume Hardy–Weinberg equilibrium or require phased data, but instead provides a joint metric of intralocus and interlocus disequilibria based solely on genotype frequencies. Thus, ∆ is equivalent to the linkage equilibrium (LD) parameter D under Hardy–Weinberg equilibrium (Weir & Cockerham 1978; Weir 1979). We used a Monte Carlo algorithm to incorporate uncertainty in genotype into our estimates of ∆ (see Nosil et al. 2012). Specifically, we first constructed two‐locus genotypes for each individual fly in the 7‐day hawthorn sampled for each pairwise combination of SNPs tested based on the individual locus likelihoods estimates for the flies. We then calculated ∆ for the sample of genotypes and repeated the process 10 000 times to generate a mean LD estimate. The mean ∆ value was transformed to a standardised correlation coefficient r between SNPs that was tested for significance by a chi‐squared test (Weir 1979). We focused on the 7‐day hawthorn sample to calculate ∆ because this is the sample that determines the independence of the selection response – the LD existing in the ancestral or starting population. We reasoned that determining the number of significantly responding SNPs in LD in the 7‐day hawthorn sample would therefore provide a lower bound estimate of the number of independent loci (or sets of genes) responding to selection in the study, as well as diverging between the host races in nature.
Results
Numbers of SNPs and gene regions responding to selection
In the selection experiment on the ancestral hawthorn race of R. pomonella, overwintering survivorship in the hawthorn‐like 7‐day treatment was 42% and in the apple‐like 32‐day treatment it was 8%. Thus, our within generation selection experiment generated a relative survivorship of ~ 19% (i.e., 8/42) for hawthorn flies raised under the apple‐ vs. hawthorn‐specific pre‐winter conditions. Of the 32 455 SNPs genotyped, a total of 2245 showed significant frequency shifts between the short and long pre‐winter treatments, as determined by Monte Carlo simulations (Table 1). Because of extensive LD in Rhagoletis (Feder et al. 2003), these 2245 SNPs do not provide an estimate of the independent number of gene regions influenced by total selection. Thus, we assessed the number of potential independent gene regions responding to selection using Burrow's composite measure of Hardy–Weinberg and linkage disequilibrium (Δ) between pairs of variable sites in the 7‐day hawthorn sample according to Weir (1979). We determined that the 2245 SNPs exhibiting significant frequency shifts represented 162 different sets whose members were in LD with each other, but in LD with all other SNPs. Because we are quantifying total selection, the pronounced genetic response observed within a single generation in the selection experiment does not require a prohibitive number of selective deaths. In Fig. S1a–c, using a polygenic threshold model of hard selection, we show that it is the statistical detection of the response to selection at a given locus given a finite sample size, rather than the possible number of loci affected by selection, that is the limiting issue in our selection experiment. Thus, after accounting for the table‐wide null expectation of 52 false positives due to type I error, our lower bound estimate finds that a minimum of 110 independent gene regions responded to selection (P < 10−6) (see Supporting Information and Fig. 1a–c for discussion of total number of gene regions under selection).
Genome‐wide response to selection
To determine how physically widespread the response in the selection experiment was across the genome, we constructed a recombination linkage map for Rhagoletis that contained 2352 SNPs generated from five single‐pair test crosses of R. pomonella. A total of 312 (13%) of the 2352 mapped SNPs showed significant frequency shifts in the selection experiment (Table S1). The 312 significant SNPs were dispersed widely across the five major chromosomes of the R. pomonella genome (Fig. 2). Based on our analysis of LD, the 312 significant SNPs could be divided into 125 different independent sets distributed across all five chromosomes (Table S1; P < 10−6 compared to null expectation of 47 SNP sets), which represented 55% of the total 229 independent sets that could be distinguished for all 2352 mapped SNPs. Thus, numerous independent gene regions responded to selection and they were distributed throughout the genome.
The response to selection and host‐related divergence in nature
We next tested whether the response in the selection experiment reflected genomic divergence between hawthorn vs. apple flies in nature. Supporting this hypothesis, we found that the direction and magnitude of allele frequency changes for all 32 455 SNPs in the selection experiment was highly concordant with genetic differences observed between the host races at a sympatric site in Grant, MI site where apple and hawthorn flies co‐occur (r = 0.39, P < 10−6; Table 1, Fig. 2 and Fig. S2). The relationship was more pronounced for SNPs displaying significant differences in the selection experiment (r = 0.69, P < 10−6, n = 2245) and especially so for the 154 loci that were significant in both the selection experiment and between the host races in nature (r = 0.96, P < 10−6, n = 154; Table 1). The results were similar whether mapped markers or all SNPs were considered (Table 1, Table S1). Most strikingly, for all the 154 SNPs showing significant responses and host divergence, the allele that increased in frequency in the hawthorn race after selection due to apple‐like pre‐winter conditions was exactly the same allele in higher frequency in the sympatric apple race in nature (P = (½)154 = 4.4 × 10−47).
Ecological selection and the origin of the apple race of R. pomonella
We then examined the extent to which the single bout of selection on hawthorn flies genetically recreated the derived apple race. We calculated polygenic genotype scores for individual flies across the genome, which we define as the mean proportion of a fly's genome composed of the allele more common in the hawthorn race averaged across the genome (see Supporting Information for more details). For all 32 455 SNPs, the mean SNP frequency for hawthorn flies surviving the apple‐like pre‐winter treatment shifted 39% of the difference between the host races towards apple flies (Fig. 3a). For the 154 SNPs showing significant responses in the selection experiment and host divergence, the shift was 84% (Fig. 3b). These shifts were not simply due to the selection of pure parental ‘apple fly genotypes’ that migrated into the hawthorn population the preceding generation. First, the 8% survivorship of hawthorn flies in the long pre‐winter treatment is twice as high as the migration rate of flies between host plants in the field, as determined from mark and recapture studies (Feder et al. 1994). Second, R. pomonella mate multiply (can be > 50 times over lifetime – Opp et al. 1990), including migrant apple flies (Feder et al. 1994). Thus, pure parental genotypes from alternate hosts will be rare in larvae feeding in apple and hawthorn fruit, which was the generation of flies we used in the selection experiment. This is reflected in the polygenic genotype scores for the control hawthorn population being approximately normal in distribution and not multimodal (solid red line, Fig. 3) as would be expected if there were many pure apple genotypes in the sample.
Figure 3.
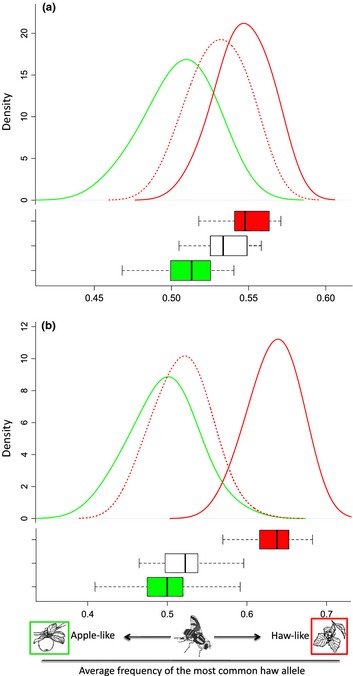
Polygenic response to selection for (a) all 32 455 SNPs and for (b) the 154 SNPs showing significant difference in the selection experiment and in sympatry. Upper panels are the distributions of polygenic genotype scores (i.e. the mean proportion of hawthorn race alleles that an individual's genome possesses) for the ‘selected’ hawthorn host race (dashed red line) compared to the sympatric hawthorn (red) and apple (green) host races. The distribution of the ‘selected’ haw race reflects the degree that lengthening the pre‐winter period genomically shifted the hawthorn towards the apple race. Also shown are box plots illustrating the distribution of genotype scores.
Discussion
Our results demonstrate that: (1) numerous independent gene regions respond to selection within a single generation when challenged by novel environmental conditions, (2) these gene regions are distributed throughout the genome, and (3) the genome‐wide response induced in the selection experiment closely mirrored patterns of divergence in nature. We determined this for a well‐documented model system for rapid divergence‐with‐gene‐flow (< 160 generations) driven by ecological selection (Bush 1966; Feder et al. 1988, 1994) using a comparison of experimental and natural populations. The power of this approach resides not in verifying that a specific locus is under direct selection or its exact genomic location. Rather, the novelty resides in using the large number of SNPs sequenced as replicates to investigate the overall response of loci genome wide to varying ecological conditions. Ongoing gene flow between the apple and hawthorn fly races (4% per generation) will homogenise frequency differences not associated (directly or indirectly) with divergent ecological selection (Slatkin 1985). Moreover, the magnitude of divergence will reflect the combined strength of these direct and indirect effects of selection on SNPs. Thus, the high correspondence between our experimental and natural populations provides dramatic evidence of how extensively diapause life history timing sculpts differentiation across the Rhagoletis genome.
Why is the impact of divergent ecological adaptation so pronounced in Rhagoletis? One contributing factor is the extensive LD in the fly, some of which is due to inversions, requiring additional DNA sequence analysis to resolve (Feder et al. 2003). In the current study, the 32 455 SNPs reduced to 686 independent sets of markers and we detected a minimum number of 110 regions statistically responding to selection above the null expectation, implying that structural features of the genome are limiting recombination and elevating LD. Thus, our findings highlight the complimentary effects that selection and genome structure can have on divergence (Lowry & Willis 2010; Joron et al. 2011). For example, in conceptually similar work in Timema stick‐insects the correspondence between experimental selection and divergence in nature was statistically significant but much weaker than observed in Rhagoletis, likely due to the low LD observed in the stick insect's genome (Gompert et al. 2014; Soria‐Carrasco et al. 2014).
A second factor is the presence of substantial standing genetic variation in R. pomonella, which supports the hypothesis that such stores may define taxa having a greater capacity for speciation with gene flow (Barrett & Schluter 2008). Although host shifts and subsequent host race formation in R. pomonella occurred in sympatry, a portion of the genetic variation involved in race formation has a complex biogeographic history, with periods of allopatry, secondary contact, and gene flow contributing to standing variation subsequently selected upon in sympatric host shifts (Feder et al. 2005; Xie et al. 2008).
Finally, when ecological adaptation involves traits like diapause that can be highly polygenic, selection may more often have genome‐wide consequences. In this regard, regulation of diapause requires a complex coordination of hormonal and molecular cues (Denlinger 2002), natural variation in diapause regulation is often under polygenic control (see Tauber et al. 1986), and microarray studies of R. pomonella have revealed hundreds of loci varying in expression during diapause termination that are potential targets of selection (Ragland et al. 2011). Future work will allow us to determine the identities of the specific loci under selection and quantify the actual strength of divergent selection affecting them in nature.
Despite the extensive divergence between the host races, RI may not yet be a global property of the genome. As this happens, LD is expected to become elevated genome wide, including across chromosomes (Flaxman et al. 2014). Although sets of SNPs within chromosomes showed significant LD within the host races, there was limited LD between SNPs on different chromosomes (see Methods). Another prediction as genomes congeal into distinct species is that populations infesting the same host plant species should genetically cluster with one another across their geographic range of overlap (Powell et al. 2013; Flaxman et al. 2014). However, while allozymes and microsatellites indicate significant local allele frequency differences between apple and hawthorn flies where they co‐occur, the races do not group distinctly from one another across the eastern U.S. (Powell et al. 2013). Indeed, for all 32 455 SNPs, the host races at the Grant, MI site only differed in allele frequencies (maximum difference = 0.258); there was no fixed difference or private allele for any SNP. In comparison, based on 39 microsatellite and allozyme loci, all populations of the sister taxon to R. pomonella that infests flowering dogwood (Cornus florida) cluster distinctly from apple and hawthorn flies (Powell et al. 2013).
In conclusion, divergent ecological selection can have genome‐wide effects even at early stages of speciation. Large stores of standing variation therefore exist in Rhagoletis flies (Michel et al. 2010), which may facilitate the evolution of genome‐wide reproductive isolation and their adaptive radiation with gene flow (Barrett & Schluter 2008). These findings are consistent with recent theory predicting that speciation‐with‐gene‐flow is more likely when the impact of selection transitions from effects on individual genes to a collective property of the genome (Flaxman et al. 2013; Feder et al. 2014) and highlights the important role of standing genetic variation and structural features of the genome in facilitating the rapid response of populations to environmental change.
Authorship
SPE, GJR, GRH, THQP, PN and JLF designed study; JLF performed field collections and selection experiment; SPE generated genomic data; SPE, GJR, LA, SE and JLF analysed data; SPE, GJR, PN and JLF wrote the first draft of the manuscript, and all authors contributed substantially to revisions.
Supporting information
Acknowledgements
This work was supported by the University of Notre Dame's Advanced Diagnostics and Therapeutics Initiative (SPE, JLF) and Environmental Change Initiative (SPE, GJR, JLF), Rice University's Huxley Faculty Fellowship to SPE, NSF grants to SPE, GJR and JLF, and a USDA grant to JLF. PN was supported by the European Research Council (Starter Grant NatHisGen R/129639).
Ecology Letters (2015) 18: 817–825
[The copyright line for this article was changed on 25 June 2015 after original online publication]
References
- Barrett, R.D. & Schluter, D. (2008). Adaptation from standing genetic variation. Trends Ecol. Evol., 23, 38–44. [DOI] [PubMed] [Google Scholar]
- Barton, N.H. (1983). Multilocus clines. Evolution, 37, 454–471. [DOI] [PubMed] [Google Scholar]
- Barton, N.H. & de Cara, M.A.R. (2009). The evolution of strong reproductive isolation. Evolution, 63, 1171–1190. [DOI] [PubMed] [Google Scholar]
- Berlocher, S.H. (2000). Radiation and divergence in the Rhagoletis pomonella species group: inferences from allozymes. Evolution, 54, 543–557. [DOI] [PubMed] [Google Scholar]
- Bierne, N. , Welch, J. , Loire, E. , Bonhomme, F. & David, P. (2011). The coupling hypothesis: why genome scans may fail to map local adaptation genes. Mol. Ecol., 20, 2044–2072. [DOI] [PubMed] [Google Scholar]
- Bush, G.L. (1966). The taxonomy, cytology, and evolution of the genus Rhagoletis in North America (Diptera: Tephritidae). Bull. Mus. Comp. Zool., 134, 431–562. [Google Scholar]
- Coyne, J.A. & Orr, H.A. (2004). Speciation. Sinauer Associates, Sunderland. [Google Scholar]
- Dambroski, H.R. & Feder, J.L. (2007). Host plant and latitude‐related diapause variation in Rhagoletis pomonella: a test for multifaceted life history adaptation on different stages of diapause development. J. Evol. Biol., 20, 2101–2112. [DOI] [PubMed] [Google Scholar]
- Darwin, C. (1859). On the Origin of Species by Means of Natural Selection, or the Preservation of Favoured Races in the Struggle for Life. John Murray, London. [PMC free article] [PubMed] [Google Scholar]
- Denlinger, D.L. (2002). Regulation of diapause. Annu. Rev. Entomol., 47, 93–122. [DOI] [PubMed] [Google Scholar]
- DePristo, M.A , Banks, E. , Poplin, R.E. , Garimella, K. V. , Maguire, J.R. & Hartl, C . et al (2011). A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nat. Genet., 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feder, J.L. , Chilcote, C.A. & Bush, G.L. (1988). Genetic differentiation between sympatric host races of Rhagoletis pomonella . Nature, 336, 61–64. [Google Scholar]
- Feder, J.L. , Hunt, T.A. & Bush, G.L. (1993). The effects of climate, host‐plant phenology and host fidelity on the genetics of apple and hawthorn infesting races of Rhagoletis pomonella . Entomol. Exp. Appl., 69, 117–135. [Google Scholar]
- Feder, J.L. , Opp, S.B. , Wlazlo, B. , Reynolds, K. , Go, W. & Spisak, S. et al (1994). Host fidelity is an effective pre‐mating barrier between sympatric races of the Apple Maggot Fly. Proc. Natl. Acad. Sci. USA, 91, 7990–7994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feder, J.L. , Roethele, J.B. , Wlazlo, B. & Berlocher, S.H. (1997). Selective maintenance of allozyme differences between sympatric host races of the apple maggot fly. Proc. Natl. Acad. Sci. USA, 94, 11417–11421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feder, J.L. , Roethele, J.B. , Filchak, K. , Niedbalski, J. & Romero‐Severson, J. (2003). Evidence for inversion polymorphism related to sympatric host race formation in the apple maggot fly, Rhagoletis pomonella . Genetics, 163, 939–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feder, J.L. , Xie, X. , Rull, J. , Velez, S. , Forbes, A. & Leung, B . et al (2005). Mayr, Dobzhansky, Bush and the complexities of sympatric speciation in Rhagoletis . Proc. Natl. Acad. Sci. USA, 102, 6573–6580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feder, J.L. , Egan, S.P. & Nosil, P. (2012). The genomics of speciation‐with‐gene‐flow. Trends Genet., 28, 342–350. [DOI] [PubMed] [Google Scholar]
- Feder, J.L. , Flaxman, S. , Egan, S.P. , Comeault, A. & Nosil, P. (2013). Geographic mode of speciation and genomic divergence. Annu. Rev. Ecol., Evol. Syst., 44, 73–97. [Google Scholar]
- Feder, J.L. , Nosil, P. , Wacholder, A. , Egan, S.P. , Berlocher, S. & Flaxman, S. (2014). Genome‐wide congealing and rapid transitions across the speci‐ation continuum during speciation with gene flow. J. Hered., 105, 810–820. [DOI] [PubMed] [Google Scholar]
- Flaxman, S.M. , Feder, J.L. & Nosil, P. (2013). Genetic hitchhiking and the dynamic build up of genomic divergence during speciation with gene flow. Evolution, 67, 2577–2591. [DOI] [PubMed] [Google Scholar]
- Flaxman, S.M. , Wacholder, A.C. , Feder, J.L. & Nosil, P. (2014). Theoretical models of the influence of genomic architecture on the dynamics of speciation. Mol. Ecol., 23, 4074–4088. [DOI] [PubMed] [Google Scholar]
- Gagnaire, P.A. , Pavey, S.A. , Normandeau, E. & Bernatchez, L. (2013). The genetic architecture of reproductive isolation during speciation‐with‐gene‐flow in lake whitefish species pairs assessed by rad sequencing. Evolution, 67, 2483–2497. [DOI] [PubMed] [Google Scholar]
- Gompert, Z. , Lucas, L.K. , Nice, C.C. , Fordyce, J.A. , Forister, M.L. & Buerkle, C.A. et al (2012). Genomic regions with a history of divergent selection affect fitness of hybrids between two butterfly species. Evolution, 66, 2167–2181. [DOI] [PubMed] [Google Scholar]
- Gompert, Z. , Comeault, A.A. , Farkas, T.E. , Feder, J.L. , Parchman, T.L. , Buerkle, C.A. et al (2014). Experimental evidence for ecological selection on genome variation in the wild. Ecol. Lett., 17, 369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, F.C. , Grabherr, M.G. , Chan, Y.F. , Russell, P. , Mauceli, E. & Johnson, J. et al (2012). The genomic basis of adaptive evolution in threespine sticklebacks. Nature, 484, 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joron, M. , Frezal, L. , Jones, R.T. , Chamberlain, N.L. , Lee, S.F. & Haag, C.R et al (2011). Chromosomal rearrangements maintain a polymorphic supergene controlling butterfly mimicry. Nature, 477, 203–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawniczak, M.K.N. , Emrich, S. , Holloway, A.K. , Regier, A.P. , Olson, M. & White, B . et al (2010). Widespread divergence between incipient Anopheles gambiae species revealed by whole genome sequences. Science, 330, 512–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. & Durbin, R. (2009). Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry, D.B. & Willis, J.H. (2010). A widespread chromosomal inversion polymorphism contributes to a major life‐history transition, local adaptation, and reproductive isolation. PLoS Biol., 8, e1000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. & Kernytsky, A. et al (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res., 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel, A.P. , Sim, S. , Powell, T. , Nosil, P. & Feder, J.L. (2010). Widespread genomic divergence during sympatric speciation. Proc. Natl. Acad. Sci. USA, 107, 9724–9729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noor, M.A.F. & Bennett, S.M. (2009). Islands of speciation or mirages in the desert? Examining the role of restricted recombination in maintaining species. Heredity, 103, 439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noor, M.A.F. & Feder, J.L. (2006). Genetics of speciation. Nat. Genet., 7, 851–861. [DOI] [PubMed] [Google Scholar]
- Nosil, P. , Gompert, Z. , Farkas, T. , Comeault, A. , Feder, J.L. & Buerkle, C.A. et al (2012). Genomic consequences of multiple speciation processes in a stick insect. Proc. Biol. Sci., 279, 5058–5065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opp, S.B. , Ziegner, J. , Bui, N. & Prokopy, R.J. (1990). Factors influencing estimates of sperm competition in Rhagoletis pomonella (Walsh) (Diptera: Tephritidae). Ann. Entomol. Soc. Am., 83, 521–526. [Google Scholar]
- Parchman, T. , Gompert, Z. , Benkman, C. , Schilkey, F. , Mudge, J. & Buerkle, C.A . (2012). Genome wide association mapping of an adaptive trait in lodgepole pine. Mol. Ecol., 21, 2991–3005. [DOI] [PubMed] [Google Scholar]
- Powell, T.H.Q. , Hood, G.R. , Murphy, M.O. , Heilveil, J.S. , Berlocher, S.H. & Nosil, P. et al (2013). Genetic divergence across the speciation continuum: the transition from host race to species in Rhagoletis . Evolution, 67, 2561–2576. [DOI] [PubMed] [Google Scholar]
- Ragland, G.J. , Egan, S.P. , Feder, J.L. , Berlocher, S.H. & Hahn, D.A. (2011). Developmental trajectories of gene expression reveal candidates for diapause termination: a key life‐ history transition in the apple maggot fly Rhagoletis pomonella . J. Exp. Biol., 214, 3948–3959. [DOI] [PubMed] [Google Scholar]
- Slatkin, M. (1985). Gene flow in natural populations. Annu. Rev. Ecol. Syst., 16, 393–430. [Google Scholar]
- Soria‐Carrasco, V. , Gompert, Z. , Comeault, A.A. , Farkas, T.E. , Parchman, T.L. & Johnson, J.S . et al (2014). Stick insect genomes reveal natural selection's role in parallel speciation. Science, 344, 738–742. [DOI] [PubMed] [Google Scholar]
- Tauber, M.J. , Tauber, C.A. & Masaki, S. (1986). Seasonal Adaptations of Insects. Oxford University Press, New York, N.Y. [Google Scholar]
- Tittes, S. & Kane, N.C. (2014). The genomics of adaptation, divergence and speciation: a congealing theory. Mol. Ecol., 23, 3938–3940. [DOI] [PubMed] [Google Scholar]
- Weir, B.S. (1979) Inferences about linkage disequilibrium. Biometrics, 35, 235–254. [PubMed] [Google Scholar]
- Weir, B.S. & Cockerham, C.C. (1978). Testing hypotheses about linkage disequilibrium with multiple alleles. Genetics, 88, 633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, X. , Michel, A.P. , Schwarz, D. , Rull, J. , Velez, S. & Forbes, A.A. et al (2008). Radiation and divergence in the Rhagoletis pomonella species complex: inferences from DNA sequence data. J. Evol. Biol., 21, 900–913. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials