

Abstract
In several human pathogens, thyX-encoded flavin-dependent thymidylate synthase (FDTS) catalyzes the last step in the biosynthesis of thymidylate, one of the four DNA nucleotides. ThyX is absent in humans, rendering FDTS an attractive antibiotic target; however, the lack of mechanistic understanding prohibits mechanism-based drug design. Here we report trapping and characterization of two consecutive intermediates, which together with previous crystal structures indicate that the enzyme’s reduced flavin relays a methylene from the folate carrier to the nucleotide acceptor. Furthermore, these results corroborate an unprecedented activation of the nucleotide that involves no covalent modification but only electrostatic polarization by the enzyme’s active site. These findings indicate a mechanism that is very different from thymidylate biosynthesis in humans, underscoring the promise of FDTS as an antibiotic target.
Enzymes involved in DNA biosynthesis are primary targets of chemotherapeutic and antibiotic agents. One such enzyme is thymidylate synthase, or TSase (EC 2.1.1.45). Encoded by the thyA gene (TYMS in humans), TSase produces thymidylate (dTMP), a precursor of one of the DNA bases, thymine. TSase catalyzes the reductive methylation of the nucleotide dUMP with the folate derivative CH2H4fol (Fig. 1). The reaction also produces dihydrofolate (H2fol), which is restored to H4fol for reuse in catalysis by dihydrofolate reductase (DHFR). TSase is successfully targeted by drugs such as 5-fluorouracil and raltitrexed, and DHFR is the target of methotrexate and trimethoprim.
Fig. 1.

Reactions catalyzed by TSase, DHFR, and FDTS. Highlighted are the reducing hydrogen in the TSase reaction (red), methylene (magenta), and nucleotide under study (blue). R = 2'-deoxyribose-5'-phosphate; R' = (p-aminobenzoyl)glutamate.
In several human pathogens, however, thymidylate formation is catalyzed by thyX-encoded flavin-dependent thymidylate synthase, or FDTS (EC 2.1.1.148), which carries out the functions of both TSase and DHFR (Fig. 1) (1, 2). Many pathogens depend solely on thyX for thymine, including all Rickettsia (causing typhus, spotted fever, and other diseases). Pathogens containing both thyX and thyA, such as M. tuberculosis, can synthesize thymidylate through either pathway, and often develop multi-drug resistance. As multi- and extreme-drug-resistance in these pathogens become more common, the addition of an FDTS inhibitor to the cocktail could prove essential for treatment. Further information regarding the prevalence of thyX gene in human pathogens, and its potential as a drug target, is provided in the Supplementary Materials (SM). Hitherto no drugs are known to selectively inhibit FDTS, and the mechanistic intricacies necessary for the rational design of mechanism-based inhibitors are yet to be resolved. FDTS is genetically and structurally dissimilar not only from the canonical TSase and DHFR but from other flavoenzymes (3, 4). The current report provides a much-needed roadmap to better understand FDTS catalysis and to rational design of inhibitory drugs.
To delineate the mechanism of FDTS-catalysis, we report here on the trapped reaction intermediates, i.e., intermediate species that have been chemically modified by a reaction quencher (here, acid or base). Characterization of these trapped compounds revealed a unique nucleotide methylation path. Our previous report on rapid-quenching experiments with T. maritima FDTS (TmFDTS) revealed a significant accumulation of an acid-modified intermediate, identified as 5-hydroxymethyl-dUMP (5). In the current study we employed a basic quencher to stop enzymatic turnover and trap different derivatives of the intermediates. Indeed, with radiolabeled nucleotide, [2-14C]-dUMP, we observed a new radioactive species, distinct from the acid-modified 5-hydroxymethyl-dUMP. The base-modified species was also observed with methylene-labeled folate, [11-14C]-CH2H4fol, indicating that the nucleotide-intermediate acquired the methylene before being trapped by the base (Fig. S1).
The top panel in Figure 2 shows the accumulation and decay of the base-trapped intermediate (blue) with [2-14C]-dUMP as a substrate. Intriguingly, upon base addition, dTMP product (black) is formed even though the flavin is in a reduced state prior to the quencher’s addition (green stopped-flow trace). In the figure's bottom panel, comparison of the time course of the acid- and base-modified intermediate derivatives suggests that at least two different reaction intermediates (I1 and I2) are trapped in acid as 5-hydroxymethyl-dUMP. In base, I1 is trapped in a different chemical form than in acid, and I2 is converted to dTMP (see further discussion of this phenomenon in the SM).
Fig. 2.
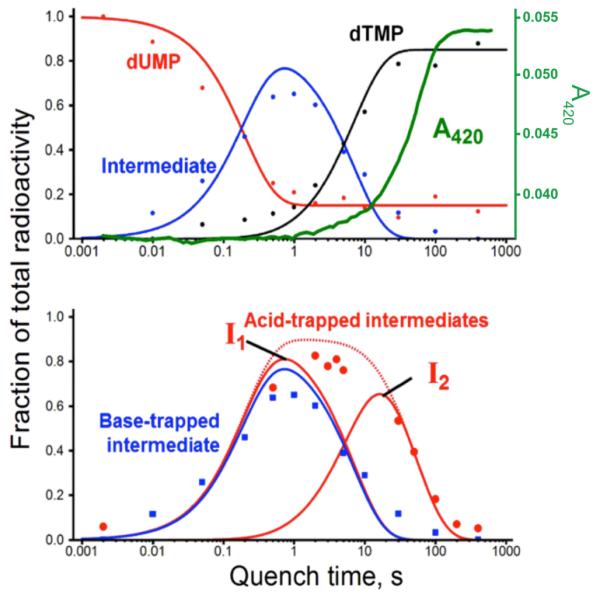
Oxidative half-reaction kinetics of FDTS. (Top panel) Base-quenched data (dots) globally fitted to a single-intermediate model (lines), overlaid with stopped-flow trace at 420 nm (flavin absorbance in green) (6). Each time point was obtained from a radiogram like that shown in Fig. S1A, with dUMP in red, dTMP in black, and intermediate in blue. (Bottom panel) Base-trapped intermediate kinetics (blue) overlaid with acid-trapped intermediate data (red), globally fitted to a two-intermediate model (red curves). The combined total of the two intermediates is shown as a dashed red curve.
What is the base-modified intermediate?
The mass of the base-trapped intermediate was determined to be [M-H]− 705.1212 Da (Fig. S2A). MS/MS of the compound displayed a loss of 320 Da, corresponding to CH2-dUMP (Fig. S2B), which accords with the 14C found in this intermediate when using 14C labeled dUMP or 14CH2H4fol. Hence the [M-H]- 385 ion, corresponding to the mass difference between the trapped species and CH2-dUMP moiety (Fig. S2B), must have belonged to an adduct covalently linked to the trapped nucleotide.
All FDTS mechanisms proposed heretofore (6-9) (e.g., Fig. S3) postulate that the methylene is transferred directly from CH2H4fol to the nucleotide, as in the TSase reaction (10), and the absorbance spectrum of the trapped intermediate (Fig. S4) accords with folate absorbance. Consequently, the most logical candidate for the adduct was the pterin moiety of CH2H4fol. However, when using CH2H4fol radiolabeled at pterin or benzoyl moieties, we found no radioactivity in the base-modified intermediate, ruling out folate as a component of the base-trapped intermediate. Furthermore, the MS/MS ion of 385 Da in the based-trapped intermediate did not match any buffer or protein constituents.
The only remaining part of the quenched FDTS reaction that could have provided the mysterious adduct was an FAD derivative. To investigate, we turned to FDTS reconstituted with isotopically-labeled FAD (11). FDTS reconstituted with FAD labeled at the adenine moiety yielded no labeled intermediate, but the reaction of [7a,8a-3H]-FAD-FDTS produced a tritiated trapped intermediate (Fig. S5B), indicating that the labeled dimethylbenzene portion of isoalloxazine (Fig. S5A) was a part of the trapped species. Additionally, when we used FDTS containing FAD its 13C and 15N at dioxopyrimidine (ring “C” in Fig. S5), i.e., 6-Da heavier than unlabeled FAD, the mass of the isolated trapped intermediate was heavier by only 2 Da than that produced by the unlabeled enzyme. This finding suggests that only two labeled atoms of the dioxopyrimidine ring of the flavin are retained in the base-modified intermediate.
Several NMR experiments were performed to determine the structure of the based-trapped intermediate (Fig. 3, Figs. S6-S7, and Table S2). Critically, the methylene (U7) originating in the CH2H4fol had bonded to C5 of the dUMP moiety and coupled to FC5a and FC4a of the degraded flavin in the NMR HMBC spectrum (Fig. 3). These results and others presented in the SM show that CH2H4fol-derived methylene bridges dUMP and degraded flavin, connecting C5 of dUMP and N5 of the decomposed isoalloxazine moiety. Other structural features of the base-trapped intermediate derivative are described in the SM.
Fig. 3.

Structure of the base-modified FDTS reaction intermediate. (Top panel) Structural components originating in dUMP (red) and CH2H4fol (black) are referred to as “U subunit” and the structural parts derived from FAD (blue) as “F subunit.” Atomic numbering in F subunit follows convention adapted from the original FAD (Fig. S5A). (Bottom panel) Overlay of 1H/13C HMQC (black; red marks folded cross peaks) and HMBC (green) spectra. The cross peaks are labeled with the first letter representing the subunit (U or F). The cross peaks of the HMBC spectrum circled in red unambiguously establish that the CH2H4fol-drived methylene (U7) bridges dUMP and flavin derivative.
The presence of a covalent methylene bridge between dUMP and flavin indicates a novel mechanism dramatically different from earlier mechanisms (6, 7, 9) (Fig. S3). This mechanism, given in Figs. 4 and S8, postulates that N5 of the reduced flavin accepts the activated methylene from the CH2H4fol Schiff base (step 1), then passes it to C5 of the enzyme-polarized dUMP (steps 2 and 3). This mechanism accords well with crystal structures of FDTS complexes with FAD, dUMP, and folates (PDB IDs: 4GT9, 4GTA, 4GTB), in which the isoalloxazine moiety of FAD is sandwiched between the folate ring and uracil (8) (Fig. 4). Thus, while protein-bound flavins typically move electrons from one side of the flavin ring to the other, the FAD of FDTS moves a methylene.
Fig. 4.

Proposed chemical mechanism for FDTS. (Left panel) Methylene (CH2, magenta) is transferred from the N5 of H4fol (green) via N5 of FAD cofactor (gold) to C5 of dUMP substrate (cyan). Oxygens are in red, nitrogens in blue, and the hydride transferred in step 4 in brown. The proposed steps of flavin oxidative half-reaction are numbered. I1 (boxed) is the reaction intermediate that in base is converted to the compound presented in Fig. 3, and in acid undergoes water addition (Fig. S10 and Supplementary Discussion). I2 (boxed) refers to the intermediate that in base forms dTMP, and in acid reacts with water (Fig. S10 and Supplementary Discussion). The “420 nm absorbance” vs. “colorless” labels of flavins relate the 420 nm time-trace (green line in Fig. 3) to the mechanistic model. A rigorous electron-pushing description of this mechanism is presented in Fig. S8. R = 2′-deoxyribose-5′-phosphate, R′ = (p-aminobenzoyl)glutamate, and R"′ = adenosine-5′-pyrophosphate–ribityl. (Right panel) Active site of FDTS (PDB ID 4GTA (8)) in complex with FAD, dUMP, and folinic acid, a stable analogue of the activated CH2H4fol. For clarity some the carbonyl oxygen of folinic acid are not shown, and the structure is inversed to match the orientation of ligands in the left panel. The formyl carbon (magenta) on folinic acid represents the methylene to be transferred. Black arrows mark the methylene transfer trajectory from donor (N5 of folate), to proposed relay atom (N5 of flavin), and final acceptor (C5 of dUMP).
The proposed intermediates I1 and I2 are modified in base to the compound identified in Fig. 3 above and product, respectively. However, both are converted to 5-hydroxymethyl-dUMP in acid. The mechanisms of their degradation are discussed in the SM (pages S4-S5) and presented in Fig. S10.
Why wasn't such a mechanism considered when the structure was solved in 2012
(8)? The reason lies in reports of activity of FDTS reconstituted with 5-deaza-5-carba-FAD (henceforth “5-deaza-FAD”) (7, 9). In 5-deaza-FAD, N5 of the flavin has been replaced with a carbon, which would prohibit several chemical conversions proposed in Fig 4. The identification of the base-modified intermediate prompted us to revisit this finding. We synthesized 5-deaza-FAD and incorporated it into apo-FDTS. In doing so, we found that the procedure used in the past for removing FAD from holo-TmFDTS (salting out) (4, 9) leaves behind variable amounts of FAD, producing the same level of (residual) activity in the salted-out 'apoenzyme' as in 5-deaza-FAD-reconstituted FDTS. By denaturing the apoenzyme with 8 M urea after salting out, completely removing FAD, then refolding and reconstituting with either FAD or 5-deaza-FAD, we found no activity for the apoenzyme, fully recovered activity for FAD-reconstituted enzyme, and no activity for the enzyme reconstituted with 5-deaza-FAD (Fig. S9). This indicates that 5-deaza-FAD-FDTS is actually not active at all (<10−6 at the S/N limit, see SM), removing the conceptual barrier for the mechanism proposed in Fig. 4.
Flavin’s N5-mediated methylene transfer might be rare, but it has been proposed for TrmFO tRNA methyltransferase (12). In TrmFO, however, the uracil is activated via Michael addition of active-site cysteine to C6 of the pyrimidine ring, and a second cysteine activates and delivers the methylene. In FDTSs, no such enzymatic nucleophile is available (7), and the cause of the uracil's initial activation has been a matter of uncertainty. One hypothesis posits that N5 hydride of reduced FAD nucleophilically activates dUMP (7) (Fig. S3A), while another suggests that the enzyme polarizes the pyrimidine ring of dUMP for an attack on the CH2H4fol methylene (6, 9) without a Michael nucleophile (Fig. S3B). The latter chemistry is unprecedented in nucleotide methylation, and the need for pyrimidine activation via Michael addition to its C6 has been emphasized in enzymatic and non-enzymatic systems (6). The only supporting pieces of evidence so far were the absence of deuterated trapped intermediates (6) and UV-vis features consistent with those of reduced flavin (Fig. 2, green trace, and refs (5) and (9)), but these observations can be rationalized by more conventional mechanisms (6). Such nucleotide activation was thus met with healthy skepticism.
The current findings provide the strongest supporting evidence so far for pyrimidine activation via polarization in the reduced enzyme’s active center. Since the methylene-carrier role for flavin’s N5 proposed here (Fig. 4) requires FAD to be reduced during carbon transfer to dUMP, flavin oxidation during substrate activation (Fig. S3A) can be definitively ruled out. The mechanism proposed in Fig. 4 also agrees with the observation (7) that when the TmFDTS reaction is conducted in D2O at near-physiological temperatures, a deuterium is incorporated at C7 of the dTMP product while some deuterium is found at C6 of the product at low temperature (Fig. S8).
In summary, the data presented above dictate a thymidylate biosynthesis mechanism (Fig. 4) in thyX-dependent human pathogens that is fundamentally different from that found in humans, lending hope for development of mechanism-based FDTS inhibitors as a new class of nontoxic antibiotics (see SM).
Supplementary Material
Acknowledgements
We thank Dr. Daniel Roston for assistance with kinetic data fitting, and Mohammad S. Hossain and Prof. Frank W. Foss Jr. for assistance with preparation of 5-deaza-riboflavin. This work was funded by NIH RO1 GM110775 to AK, and fellowship from the Iowa Center of Biocatalysis and Bioprocessing (NIH T32 GM008365) to TVM and KK.
Footnotes
Supplementary Materials
Materials and Methods
Figures S1-S10
Tables S1-S2
Supplementary Discussion
References and Notes:
- 1.Myllykallio H, et al. An alternative flavin-dependent mechanism for thymidylate synthesis. Science. 2002;297:105–107. doi: 10.1126/science.1072113. [DOI] [PubMed] [Google Scholar]
- 2.Leduc D, et al. Two distinct pathways for thymidylate (dTMP) synthesis in (hyper)thermophilic Bacteria and Archaea. Biochem. Soc. Trans. 2004;32:231–235. doi: 10.1042/bst0320231. [DOI] [PubMed] [Google Scholar]
- 3.Lesley SA, et al. Structural genomics of the Thermotoga maritima proteome implemented in a high-throughput structure determination pipeline. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:11664–11669. doi: 10.1073/pnas.142413399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mathews II, et al. Functional analysis of substrate and cofactor complex structures of a thymidylate-synthase complementing protein. Structure (Camb.) 2003;11:677–690. doi: 10.1016/s0969-2126(03)00097-2. [DOI] [PubMed] [Google Scholar]
- 5.Mishanina TV, et al. Trapping of an intermediate in the reaction catalyzed by flavin-dependent thymidylate synthase (FDTS) J. Am. Chem. Soc. 2012;134:4442–4448. doi: 10.1021/ja2120822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mishanina TV, Corcoran JM, Kohen A. Substrate Activation in Flavin-Dependent Thymidylate Synthase. J. Am. Chem. Soc. 2014;136:10597–10600. doi: 10.1021/ja506108b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koehn EM, et al. An unusual mechanism of thymidylate biosynthesis in organisms containing the thyX gene. Nature. 2009;458:919–924. doi: 10.1038/nature07973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koehn EM, et al. Folate binding site of flavin-dependent thymidylate synthase. Proc. Natl. Acad. Sci. U. S. A. 2012;109:15722–15727. doi: 10.1073/pnas.1206077109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conrad JA, Ortiz-Maldonado M, Hoppe SW, Palfey BA. Detection of Intermediates in the Oxidative Half-Reaction of the FAD-Dependent Thymidylate Synthase from Thermotoga maritima: Carbon Transfer without Covalent Pyrimidine Activation. Biochemistry. 2014;53:5199–5207. doi: 10.1021/bi500648n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carreras CW, Santi DV. The catalytic mechanism and structure of thymidylate synthase. Annu. Rev. Biochem. 1995;64:721–762. doi: 10.1146/annurev.bi.64.070195.003445. [DOI] [PubMed] [Google Scholar]
- 11.Mishanina TV, Kohen A. Synthesis and application of isotopically labeled flavin nucleotides. J. Label. Compd Radiopharm. 2015;58:370–375. doi: 10.1002/jlcr.3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamdane D, Argentini M, Cornu D, Golinelli-Pimpaneau B, Fontecave M. FAD/folate-dependent tRNA methyltransferase: flavin as a new methyl-transfer agent. J. Am. Chem. Soc. 2012;134:19739–19745. doi: 10.1021/ja308145p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.