

Summary
CTEN/TNS4 is a member of the Tensin gene family. It localizes to focal adhesions and induces cell motility. The mechanisms regulating Cten expression are unclear although we have shown regulation by Kras in the colon and pancreas. In normal mammary cell lines, it is reportedly upregulated by epidermal growth factor receptor (EGFR) and STAT3 signalling and upregulation is accompanied by downregulation of Tensin 3 (Tensin switch). In this study, we investigated the roles of EGFR and STAT3 signalling in the regulation of Cten in colorectal cancer (CRC). In addition, we investigated calpain – a regulator of focal adhesion‐associated proteins whose relevance to Cten has not been investigated. CRC cell lines were stimulated with epidermal growth factor (EGF). This resulted in an increase in Cten and Tensin 3 protein. Kras was knocked down and this resulted in downregulation of Cten and Tensin 3. We next investigated the role of STAT3 signalling. Activation and knockdown of STAT3 resulted in downregulation and upregulation, respectively, of Cten. Inhibition of calpain resulted in upregulation of both Cten and Tensin 3. As the regulators of Cten also seemed to regulate Tensin 3, we tested the interaction between Cten and Tensin 3. Cten was forcibly expressed or knocked down resulting, respectively, in upregulation and downregulation of Tensin 3. We conclude that in CRC, Cten is upregulated by EGFR and Kras but downregulated by STAT3. We show that calpain may be a negative regulator of Cten and that a Tensin switch does not occur and, if anything, Cten stabilizes Tensin 3.
Keywords: calpain, Cten, EGF, Kras, STAT3, Tensin 3
Cten is a member of the Tensin protein family which comprises tensins 1‐4. These play pivotal roles in key cellular processes including adhesion, migration, proliferation, differentiation and apoptosis. Tensins 1‐3 are highly homologous and contain an N‐terminal actin‐binding domain (ABD) which maintains contact to the cytoskeleton. At the C‐terminus, they have a phosphotyrosine‐binding (PTB) domain (enabling binding to β‐integrin tails) and a Src homology 2 (SH2) domain (enabling participation in signalling activity). However, Cten (also called Tensin 4) is a more recently evolved member which lacks the ABD (and therefore cannot bind to the actin cytoskeleton) yet still retains the integrin binding and signalling domains (Lo & Lo 2002). In contrast to the other tensins, Cten expression is normally restricted mainly to the prostate and placenta although dysregulation occurs in a number of different types of cancer (Sasaki et al. 2003; Albasri et al. 2011b; Chen et al. 2013). In most cancers, Cten acts as an oncogene and its expression is upregulated although in prostate cancer it appears to act as a tumour suppressor (Lo & Lo 2002; Albasri et al. 2011b; Al‐Ghamdi et al. 2013). In CRC, Cten expression is associated with advanced Dukes’ stage, poor prognosis and distant metastasis (Albasri et al. 2011a).
Cten appears to confer properties of ‘stemness’ and enhanced motility to tumour cells when it is upregulated, and it is emerging as a potentially important player in the development of cancer (Liao et al. 2009). The mechanisms regulating Cten are however poorly described, and as protein‐stabilizing mutations or amplification of CTEN in cancers has not been reported, it is fair to assume that changes in Cten dosage occur as a consequence of changes in upstream signalling pathways. In both normal and tumour breast cells, it has been shown that Cten is positively regulated by EGFR and STAT3 signalling pathways respectively (Katz et al. 2007; Barbieri et al. 2010). A ‘Tensin switch’ has also been described in which EGF‐induced Cten displaces Tensin 3 from integrin binding at focal adhesion regions thereby promoting cell migration (Katz et al. 2007). We have previously shown that Cten is regulated by Kras in both colorectal and pancreatic cancers (Al‐Ghamdi et al. 2011). Other studies have suggested that Cten may be under the regulation of a number of other growth factors and cytokines such as platelet‐derived growth factor (PDGF), fibroblast growth factor (FGF2), nerve growth factor (NGF), insulin‐like growth factor (IGF‐1), transforming growth factor β (TGF‐β), interleukin‐6 (IL‐6) and IL‐13 (Hung et al. 2013).
The regulation of Cten is poorly understood although published data suggest that signalling through the EGFR/Kras pathway may be a common mechanism of Cten upregulation. However, a recent study has shown that Cten is regulated by EGF in the CRC cell line SW480 (Hung et al. 2013). As this cell line harbours an activating KRAS mutation, the induction of Cten by EGF is unlikely to be through the canonical EGFR/Kras pathway and suggests that Kras‐independent mechanisms may exist. This observation together with the fact that Cten regulation is poorly understood in the colon led us to further investigate mechanisms of Cten regulation. We have investigated the EGFR/Kras pathway and the STAT3 signalling pathway. In addition, we have investigated the role of calpain (which regulates the turnover of a number of focal adhesion proteins) in regulating Cten and whether a Tensin switch occurs in CRC (Selliah et al. 1996; Franco et al. 2004; Cortesio et al. 2011).
Experimental procedures
Cell culture, transfection and treatments
CRC cell lines HCT116, RKO, SW480, SW620, C32 and DLD1 were a gift from Professor Ian Tomlinson. Cell lines were validated by mutation profiling as previously described (Seth et al. 2009). Cells were kept at 37°C in an atmosphere containing 5% CO2. The cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, Thermo Fisher Scientific, Massachusetts, United States) supplemented with 10% foetal bovine serum (FBS) and 1% penicillin/streptomycin.
Cells were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. For overexpression of Cten, 5 μg of a CMV promoter‐driven expression construct containing green fluorescent protein (GFP)‐tagged Cten (GFP‐Cten) together with 10 μl Lipofectamine was added to cells at 60–70% confluency. The empty vector (without Cten) was used as the negative control. For gene knockdown, cells were transfected with small interfering RNA (siRNA) targeted to STAT3 (UGGCCCAAUGGAAUCAGCUACAGCA), Kras (CAGGGUGUUGAUGAUGCCUUCUAUA), Cten (AAGAGUAACUGUACCACGAGACCCG) or, as a negative control, luciferase (CAGUGUAGUAGUCGUUUC). For transfection, cells were grown to 40–50% confluency and siRNA duplexes were added at a final concentration of 100 nM together in 5–10 μl Lipofectamine. Transfection reagents were replaced after 6 h and cells harvested 24 and 48 h post‐transfection for overexpression and gene knockdown respectively.
For EGF‐stimulation experiments, cells were starved for 24 h in serum‐free DMEM (supplemented with penicillin/streptomycin) prior to stimulation. At 60–70% confluency, cells were treated with 0–80 ng/ml recombinant EGF (Invitrogen) also under serum‐free conditions and cells harvested after a 24 h incubation period.
Cells were stimulated with IL‐6 (ImmunoTools) at a concentration of 20 ng/ml and harvested after a 24‐h incubation period.
For calpain‐inhibition studies, to assess the bioactivity of calpeptin, cells were treated, in serum‐free media, with 100 μM calpeptin (Calbiochem, Darmstadt, Germany) or DMSO in triplicate and incubated for 30 min at 37°C. Cells were harvested by trypsinization and resuspended in 500 μl serum‐free media and the cell suspension divided into 2 and treated with either 0.08 nM CMAC, t‐BOC‐L‐leucyl‐l‐methionine amide (Calbiochem) or DMSO. Fluorescence was measured at 37°C using the FLUOstar Omega plate reader.
Western blotting
Whole cell extracts were prepared using RIPA lysis buffer (Thermo Scientific) supplemented with phosphatase and protease inhibitor (Thermo Scientific, Thermo Fisher Scientific, Massachusetts, United States) and quantified using the Pierce BCA Protein Assay Kit (Thermo Scientific). A total of 50 μg of protein was run on a NuPAGE 4–12% Bis‐Tris gel (Invitrogen) using the NuPAGE Western blot system (Invitrogen) and transferred to a PVDF membrane using the Trans‐Blot Semi‐Dry Transfer Cell (Bio‐Rad, California, United States). Membranes were probed overnight at 4°C with the following antibodies: Cten 1:10,000 (Sigma), β‐actin 1:50,000 (Sigma, Sigma‐Aldrich, Missouri, United States), Tensin 3 1:1,000 (Sigma), STAT3 1:100 (Abcam, Cambridge, UK) and Kras 1:250 (Abcam) in 5% milk/TPBS. Membranes were incubated with the appropriate horseradish peroxidase‐conjugated anti‐mouse or anti‐rabbit secondary antibody (Sigma) for 1 h at room temperature and subsequent to this incubated with ECL Prime detection solution (Amersham, GE Healthcare, Buckinghamshire, UK) to allow visualization. Densitometric analysis of the bands was carried out using ImageJ software. Pixel counts for each protein of interest were normalized to β‐actin (data included in the supporting information).
Quantitative RT‐PCR
RNA extraction was performed using the Mammalian Total RNA Miniprep kit (Sigma) according to the manufacturer's protocol and quantified using the NanoDrop system. A total of 1 μg of RNA was reverse‐transcribed using 0.5 μg random hexamers (Thermo Scientific), 0.5 mM dNTPs (Promega, Wisconsin, United States) and 200 U M‐MLV reverse transcriptase (Promega) using the GeneAmp PCR System 9700 (Applied Biosystems, Thermo Fisher Scientific, Massachusetts, United States). Gene expression changes were determined by SYBR Green qPCR using the AB7500 Fast system (Applied Biosystems). Primers sequences used were as follows: Tensin 3 (F) GTTGAAAGGGTGCTCGAATGA, Tensin 3 (R) GAACTTTCTGCTATTTCCTCCAATG (Cao et al. 2012), HPRT (F) AAATTCTTTGCTGACCTGCT, HPRT (R) TCCCCTGTTGACTGGTCATT, Cten (F) GCGTCTGCTCAGAATCGAC and Cten (R) GATGAGGAAGTGTCGGATGAG. The run cycle comprised a hold stage at 95°C for 2 min, cycling stage 40X (95°C for 3 s, annealing temperature for 30 s) and a melt curve stage 95°C for 15 s, 50°C for 1 min, 95°C for 15 s, 95°C for 15 s and 50°C for 15 s. Reactions were determined as having similar efficiencies, and the ΔΔCt method was used for gene quantification.
Transwell migration assay
A total of 600 μl of DMEM (20% FBS) was added to the outer wells of the Transwell plate (8 μm pore size) (Corning, New York, United States) and 100,000 cells in DMEM (10% FBS) added in triplicate to the Transwell insert. The plate was incubated at 37°C for 48 h after which cells migrated through to the bottom of the outside well were manually counted.
Statistical analysis
Results were tested for a normal distribution, and the unpaired t‐test or analysis of variance (anova) statistical tests were applied using GraphPad Prism (version 6).
Results
EGF stimulation upregulates Cten and Tensin 3 protein
It has been shown that in untransformed breast cells, Cten may be under the regulation of the EGFR signalling pathway and it may be accompanied by a Tensin switch (Katz et al. 2007). We investigated whether this was also the case in CRC. A total of 4 cell lines with differing Kras mutation status and differing levels of Cten expression were stimulated with EGF. Two cell lines – C32 (Kras WT) and SW480 (Kras mutant) – have moderate levels of endogenous Cten expression, and these both showed a dose‐dependent increase in Cten levels following EGF stimulation (Figure 1a). RKO (Braf mutant) did not express any endogenous Cten, and stimulation with EGF did not induce expression. The cell line SW620 (Kras mutant) showed high endogenous Cten expression and responded with only a slight increase in Cten expression following stimulation with EGF at a low concentration. Stimulation at high concentrations resulted in a reduction in expression. It had been assumed that in Kras mutant cell lines, there would be a lack of effect (and indeed, these were initially included as negative controls); however, as seen in SW480, this was found not to be the case (Figure 1a). To investigate whether this effect was occurring at the transcriptional level, Cten mRNA was quantified using quantitative real‐time PCR (qRT‐PCR) in the cell lines where there was a change of protein. This showed that Cten message expression remained consistent upon stimulation across all EGF concentrations used (Figure 1b) irrespective of Cten protein levels.
Figure 1.
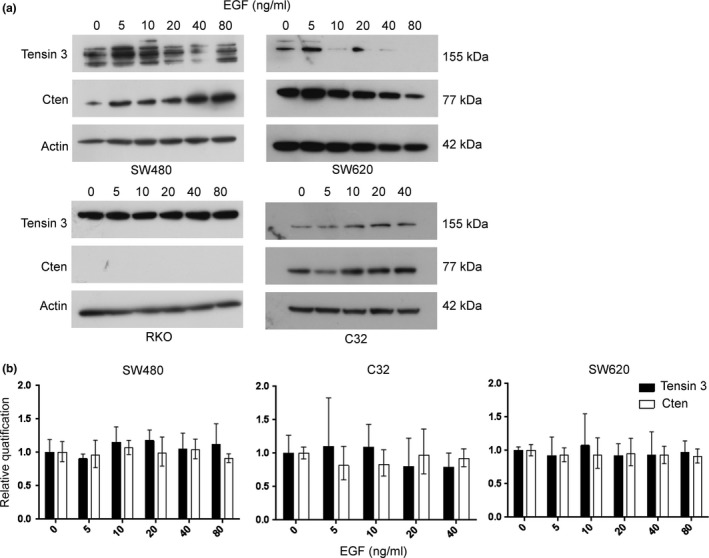
Cten and Tensin 3 are regulated by EGF at the protein level. (a) Stimulation with EGF (0–80 ng/ml) was performed in SW480, SW620, RKO and C32 cell lines and Cten and Tensin 3 protein expression determined by Western blot. Cten expression increased in C32 and SW480 in response to EGF, but a Tensin switch was shown not to occur. Results are representative of at least 2 experimental replicates. (b) Tensin 3 and Cten expression levels were determined by qRT‐PCR in C32, SW480 and SW620 and shown to remain consistent following stimulation with EGF (0–80 ng/ml) (all P > 0.05 n = 3).
Our data suggest that when Cten is not expressed, stimulation with EGF cannot induce expression. When the Cten levels are low, EGF stimulation can cause further upregulation through both Kras‐dependent and Kras‐independent means and probably through post‐transcriptional stabilization of Cten protein. To investigate whether a Tensin switch mechanism operates in CRC, Tensin 3 expression was also investigated. In response to stimulation with EGF, Tensin 3 expression remained consistent at the mRNA level, but protein expression was variable and in most cases reflected the level of Cten protein (Figure 1). Thus, any changes seen in Tensin 3 were probably due to post‐transcriptional regulation, but importantly, we did not observe a Tensin switch following EGF treatment of CRC cell lines.
Stimulation with EGF Increases Cell Migration in Colorectal Cancer
To demonstrate that the upregulation of Cten by EGF was functionally relevant, a Transwell migration assay was performed following stimulation with EGF together with knockdown of Cten (Figure 2). Here, stimulation of C32 cells with 20 ng/ml EGF increased cell migration, and this was abolished on knockdown of Cten. This concludes that the upregulation of Cten by EGF is functionally relevant.
Figure 2.
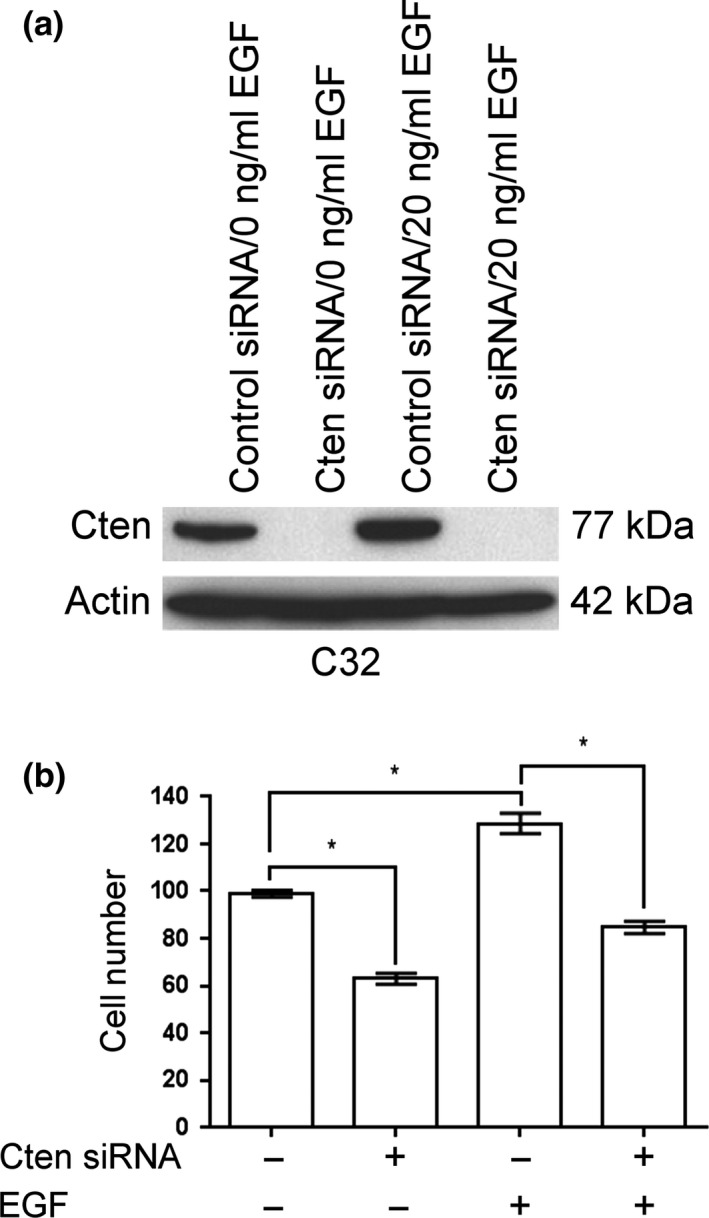
Stimulation with EGF increases cell migration through Cten. (a) Stimulation with EGF caused an upregulation of Cten in the C32 cell line. Cten was successfully knocked down using siRNA duplexes. (b) The Transwell migration assay showed that knockdown of Cten was associated with a decrease in cell migration (P = 0.0002). Stimulation with EGF led to increased cell migration (P = 0.0028) which was abolished on knockdown of Cten (P = 0.0009) (* = P < 0.05).
Kras Modulates Cten and Tensin 3 Protein and mRNA
Previously, we have shown that in pancreatic cancer and CRC cell lines, Cten is to some degree under the regulation of Kras. To further investigate the relationship between Cten and Tensin 3, we knocked down Kras in the Kras mutant cell lines SW620 and DLD1 (Al‐Ghamdi et al. 2011). As expected, knockdown of Kras resulted in a reduction in Cten protein level in both cell lines (Figure 3a). This was accompanied by a decrease in Tensin 3 expression thereby confirming that a Tensin switch does not occur in the colon. Unexpectedly, however, the levels of Cten mRNA were reduced when Kras was knocked down. This was further complicated by an increase in mRNA levels for Tensin 3 even though protein levels had decreased (Figure 3b). We have previously demonstrated the functional relevance of the Kras‐Cten signalling axis (Al‐Ghamdi et al. 2011).
Figure 3.
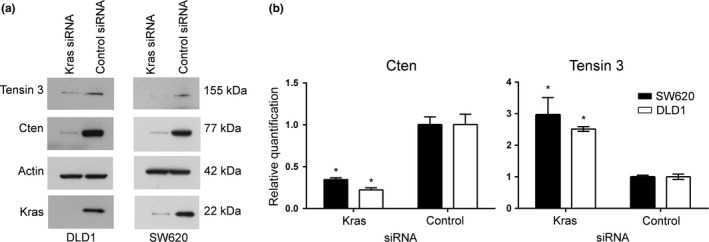
A Tensin switch occurs at the mRNA level but not at the protein level on knockdown of Kras. (a) Western blot analysis showed a decrease in both Cten and Tensin 3 protein expression following knockdown of Kras. Results are representative of at least 2 experimental replicates. (b) Following knockdown of Kras, Cten expression was reduced in SW620 (P = 0.0003) and DLD1 (P = 0.0004) as determined by qRT‐PCR. Simultaneously, an increase in Tensin 3 was observed (SW620, P = 0.0033; DLD1, P < 0.0001, n = 3) (* = P < 0.05).
Cten and Tensin 3 are negatively regulated by calpain
Cten is among the proteins that make up focal adhesion complexes. Calpain has been described to regulate focal adhesion turnover through focal adhesion protein degradation (Franco et al. 2004; Chan et al. 2010; Cortesio et al. 2011; Storr et al. 2011). We therefore sought to investigate whether calpain regulated Cten expression. SW620 cells were treated with calpeptin, a calpain inhibitor. Results showed that Cten expression increased when the cells were treated with calpeptin compared to the DMSO carrier‐treated controls. These data suggest that calpain could negatively regulate Cten (Figure 4). Consistent with our earlier data regarding the absence of a Tensin switch in CRC, we found that calpeptin treatment also led to an increase in Tensin 3.
Figure 4.
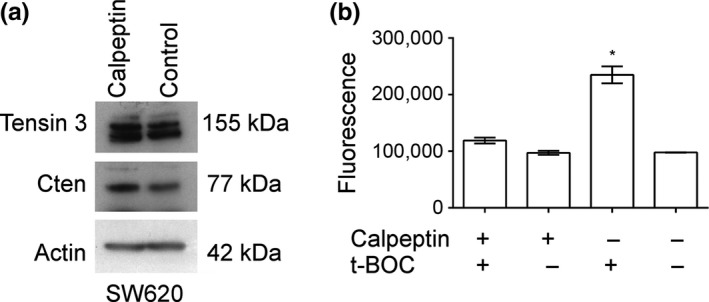
Calpeptin treatment positively regulated Cten and Tensin 3 expression. (a) The inhibition of calpain was associated with an increase in both Cten and Tensin 3 protein expression. Western blot is representative of 2 replicates. (b) calpain was successfully inhibited by calpeptin as shown by a reduction in fluorescence when used in conjunction with the calpain substrate CMAC, t‐BOC‐L‐leucyl‐l‐methionine amide (t‐BOC) (P = 0.0002, n = 3) (* = P < 0.05).
Cten may directly regulate Tensin 3
As the changes in levels of Tensin 3 expression appeared to mirror the changes in Cten expression in our experiments, we investigated the possibility that there may be a direct interaction between the proteins. Cten was forcibly expressed in the cell line HCT116 which does not express Cten. The levels of Tensin 3 protein increased on overexpression of Cten (Figure 5). We performed the reciprocal experiment of knockdown of Cten in SW620, and this resulted in a decrease in Tensin 3. Together, the data suggest that Cten positively regulates Tensin 3.
Figure 5.
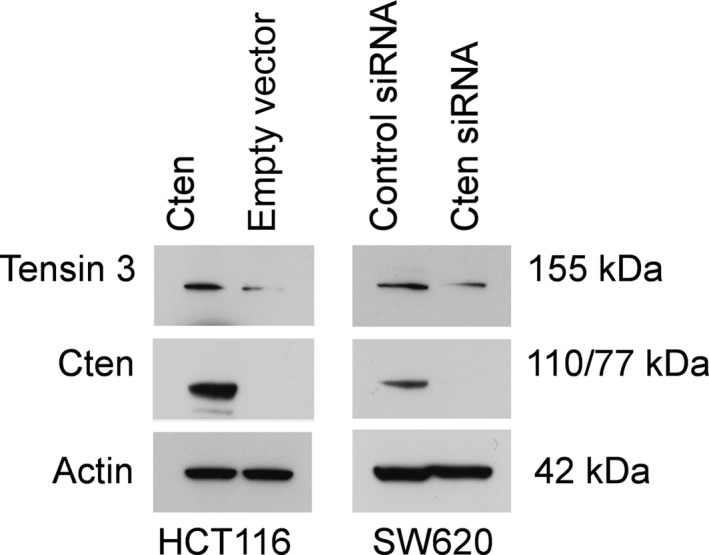
Cten regulates Tensin 3. Overexpression of Cten in HCT116 increased Tensin 3 protein expression as determined by Western blot. The reciprocal of this was true on siRNA‐mediated knockdown of Cten in SW620 (* = P < 0.05).
STAT3 negatively regulates Cten
STAT3 has been shown to positively regulate Cten in mouse tumour‐derived breast cells, but this has yet to be investigated in other tissues. To investigate the role of STAT3 signalling in the regulation of Cten in CRC, we initially knocked down STAT3 in DLD1 and SW620. This resulted in an increase in the levels of Cten expression (Figure 6a). To validate these data, we also stimulated cells with interleukin‐6 (IL‐6). This is a cytokine that is known to induce STAT3 expression and activation (Bromberg 2001). SW620 cells were stimulated with IL‐6, which led to an upregulation of STAT3 and an associated downregulation of Cten (Figure 6b). In contrast to that previously shown in breast tumour cells, we can conclude, based on these results, that STAT3 negatively regulates Cten.
Figure 6.
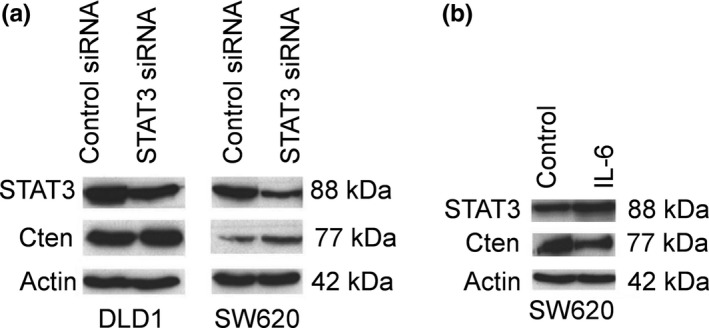
STAT3 negatively regulates Cten. (a) Knockdown of STAT3 in SW620 and DLD1 cell lines increased Cten protein expression. (b) IL‐6 stimulation of STAT3 decreased Cten protein expression.
Discussion
Cten is gaining prominence as an oncogene in a variety of different cancers. We have sought to dissect the pathways regulating Cten expression in CRC. Published data have suggested that the EGFR signalling pathway may be a major regulator of Cten (Katz et al. 2007). Herein we have shown that EGF stimulation of CRC cell lines can cause upregulation of Cten. In the cell lines expressing moderate levels of Cten, the upregulation is dose‐dependent, while in cell lines with high levels of Cten, there may be some responsiveness, but high doses of EGF may result in the eventual downregulation of Cten. This phenomenon of inhibition of signalling pathways with high levels of stimulation is not uncommon in biological experiments. The exact mechanism through which it is occurring with regard to Cten is uncertain although there may be a negative feedback loop once cytoplasmic levels go beyond a certain level. Interestingly, cell lines which do not endogenously produce Cten did not respond to EGF stimulation. It is likely that regulation of Cten will be through the balance of activating and inhibitory factors. We have previously described cell lines (both CRC and pancreatic cancer) which are mutant for Kras and which yet do not express Cten (Al‐Ghamdi et al. 2011). This indicates that there are some inhibitory mechanisms which may be stronger than the EGFR/Kras pathway in regulating Cten.
As Kras is the main signal transducer downstream of the EGFR, we had assumed that signalling through the EGFR would become irrelevant in the presence of a Kras‐activating mutation. Perhaps the best known example of this is the lack of efficacy of anti‐EGFR biological therapies (such as cetuximab) when the tumour contains a Kras mutation (Misale et al. 2012). However, a study has shown that EGFR activation (through stimulation with EGF) led to an upregulation of Cten protein and mRNA in the Kras mutant cell line SW480, thus suggesting a Kras‐independent mechanism of Cten regulation by EGF (Hung et al. 2013). We have partly confirmed this observation although our data suggest that EGF‐mediated regulation of Cten is mostly at a post‐transcriptional level. We are uncertain of the reason for the discrepancy between our results and those of Hung et al., but our study has used multiple cell lines, whereas others only tested a single colorectal cell line (Hung et al. 2013). The downstream signalling pathways of EGFR and Kras will overlap but will not be identical, and it would be interesting to ascertain which specific pathway is responsible for the activation of Cten transcription.
In addition to showing that EGFR/Kras signalling positively regulates Cten expression and function, we have identified pathways which are negative regulators of Cten. Calpains catalyse the proteolysis of many substrates localized to focal adhesions including Src, paxillin, talin, vinculin and focal adhesion kinase (FAK) to regulate focal adhesion dynamics and cell motility (Franco et al. 2004; Serrano & Devine 2004; Chan et al. 2010; Cortesio et al. 2011; Hossain et al. 2013). We have now demonstrated that treatment with calpeptin, a calpain inhibitor, causes a small increase in Cten protein expression, suggesting that calpain might negatively regulate Cten expression. The cleavage of paxillin by calpain has been shown to negatively regulate cell motility, and as Cten is known to promote cell migration, it is plausible that calpain could inhibit cell motility through modulation of Cten expression (Cortesio et al. 2011). We have shown that STAT3 negatively regulates Cten in colorectal cell lines using 2 different methodologies. Firstly, knockdown of STAT3 resulted in upregulation of Cten, and secondly, activation of STAT3 through stimulation with IL‐6 resulted in downregulation of Cten. This is in contrast to published data which have shown that Cten is positively regulated by STAT3 in breast tumour cells (Barbieri et al. 2010). Others, however, have shown that mutations in Cten SH2 domain increase phosphorylated STAT3, suggesting that Cten is a negative regulator of STAT3 (Kwon et al. 2011). IL‐6 has been shown to positively regulate Cten in untransformed breast and prostate cell lines as well as a colon tumour cell line (Barbieri et al. 2010; Hung et al. 2013). The biological activity of STAT3 in cancer is complex. It is often described as an oncogene; however, some studies have shown that STAT3 can act as a tumour suppressor in colorectal cancer (Ecker et al. 2009; Musteanu et al. 2010). There may be a difference in the effect in the cell lines used, but we have no explanation for the discrepancy between our results and those of Hung et al. (2013) and Barbieri et al. (2010).
A Tensin switch has been described in a normal breast cell line MCF10, and this was investigated in CRC cell lines and shown not to occur (Katz et al. 2007). We found that across all of our experiments, the changes in Tensin 3 protein generally reflected the changes in Cten irrespective of the method used to modulate Cten. This prompted us to investigate the relationship between Cten and Tensin 3. We found that overexpression of Cten resulted in an increase in Tensin 3 levels, suggesting that Cten is a positive regulator of Tensin 3. Previous reports have suggested that Tensin 3 can have oncogenic or tumour suppressive activity. In breast, lung and melanoma, Tensin 3 has been shown to increase cell migration, anchorage‐independent growth and tumourigenicity (Katz et al. 2007; Qian et al. 2009). A role for Tensin 3 has not been demonstrated in CRC. A recent evaluation of microarray expression data has shown that Tensin 3 is downregulated in colorectal tumours; however, others have shown through microarray data (GSE1323) that Tensin 3 is upregulated in SW620 in comparison with SW480, suggesting that Tensin 3 may play a role in metastasis (Provenzani et al. 2006; Muharram et al. 2014). However, our data suggest that Tensin 3 protein is higher in SW480 than in SW620, and as Tensin 3 (as well as Cten) can be regulated at both transcriptional and post‐transcriptional levels, mRNA expression profiling data cannot be taken entirely at face value. Tensin 3 is expressed in cell lines not expressing Cten and therefore may also be regulated by Cten‐independent mechanisms.
In summary, our results show that Cten is upregulated by both EGF and Kras and that EGF‐induced upregulation may be both Kras‐dependent and Kras‐independent. In addition, we have identified STAT3 as a negative regulator and calpain as a putative negative regulator of Cten expression in CRC (Figure 7). We have shown that whatever the mechanism of upregulating Cten, there is no Tensin switch in CRC and, in contrast, Cten may serve to stabilize Tensin 3. Our data are derived in models which are simplistic representations of CRCs, and these data need to be validated in models which are more reflective of the CRCs which occur in patients.
Figure 7.
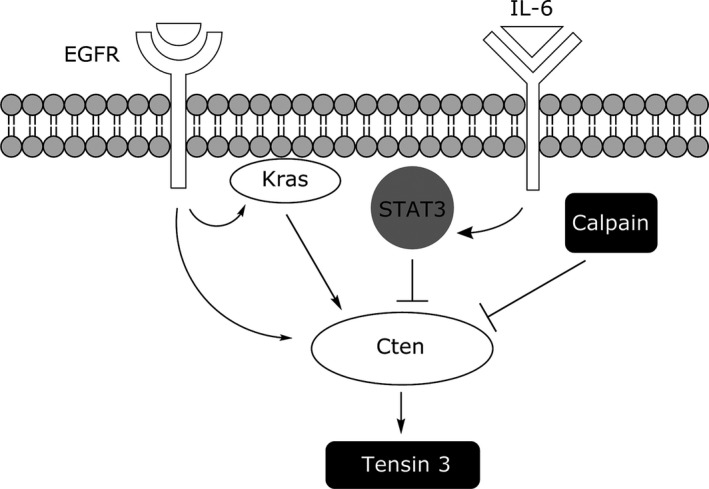
The regulation of Cten in colorectal cancer. In colorectal cell lines, Cten is positively regulated by EGF and Kras but negatively regulated by STAT3 and calpain.
Conflict of Interests
The authors declare that there are no conflicts of interest.
Supporting information
Table S1. Densitometric analysis of western blots.
Acknowledgements
This work was funded by The Pathological Society of Great Britain and Ireland and the Cancer and Polio Research Fund.
References
- Albasri A., Al‐Ghamdi S., Fadhil W. et al (2011a) Cten signals through integrin‐linked kinase (ILK) and may promote metastasis in colorectal cancer. Oncogene 30, 2997–3002. [DOI] [PubMed] [Google Scholar]
- Albasri A., Aleskandarany M., Benhasouna A. et al (2011b) CTEN (C‐terminal tensin‐like), a novel oncogene overexpressed in invasive breast carcinoma of poor prognosis. Breast Cancer Res. Treat. 126, 47–54. [DOI] [PubMed] [Google Scholar]
- Al‐Ghamdi S., Albasri A., Cachat J. et al (2011) Cten is targeted by Kras signalling to regulate cell motility in the colon and pancreas. PLoS ONE 6, e20919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Ghamdi S., Cachat J., Albasri A. et al (2013) C‐terminal tensin‐like gene functions as an oncogene and promotes cell motility in pancreatic cancer. Pancreas 42, 135–140. [DOI] [PubMed] [Google Scholar]
- Barbieri I., Pensa S., Pannellini T. et al (2010) Constitutively active Stat3 enhances Neu‐mediated migration and metastasis in mammary tumors via upregulation of Cten. Cancer Res. 70, 2558–2567. [DOI] [PubMed] [Google Scholar]
- Bromberg J.F. (2001) Activation of STAT proteins and growth control. BioEssays 23, 161–169. [DOI] [PubMed] [Google Scholar]
- Cao X., Voss C., Zhao B., Kaneko T. & Li S.S. (2012) Differential regulation of the activity of deleted in liver cancer 1 (DLC1) by tensins controls cell migration and transformation. Proc. Natl. Acad. Sci. U. S. A. 109, 1455–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K.T., Bennin D.A. & Huttenlocher A. (2010) Regulation of adhesion dynamics by calpain‐mediated proteolysis of focal adhesion kinase (FAK). J. Biol. Chem. 285, 11418–11426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N.T., Kuwabara Y., Conley C. et al (2013) Phylogenetic analysis, expression patterns, and transcriptional regulation of human CTEN gene. Gene 520, 90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortesio C.L., Boateng L.R., Piazza T.M., Bennin D.A. & Huttenlocher A. (2011) Calpain‐mediated proteolysis of paxillin negatively regulates focal adhesion dynamics and cell migration. J. Biol. Chem. 286, 9998–10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecker A., Simma O., Hoelbl A. et al (2009) The dark and the bright side of Stat3: proto‐oncogene and tumor‐suppressor. Front. Biosci. (Landmark Ed) 14, 2944–2958. [DOI] [PubMed] [Google Scholar]
- Franco S.J., Rodgers M.A., Perrin B.J. et al (2004) Calpain‐mediated proteolysis of talin regulates adhesion dynamics. Nat. Cell Biol. 6, 977–983. [DOI] [PubMed] [Google Scholar]
- Hossain M.I., Roulston C.L., Kamaruddin M.A. et al (2013) A truncated fragment of Src protein kinase generated by calpain‐mediated cleavage is a mediator of neuronal death in excitotoxicity. J. Biol. Chem. 288, 9696–9709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung S.‐Y., Shih Y.‐P., Chen M., Lo S.H. (2013) Up‐regulated Cten by FGF2 contributes to FGF2‐mediated cell migration. Mol. Carcinog. 53, 787–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz M., Amit I., Citri A. et al (2007) A reciprocal tensin‐3‐cten switch mediates EGF‐driven mammary cell migration. Nat. Cell Biol. 9, 961–U124. [DOI] [PubMed] [Google Scholar]
- Kwon S.H., Nedvetsky P.I. & Mostov K.E. (2011) Transcriptional profiling identifies TNS4 function in epithelial tubulogenesis. Curr. Biol. 21, 161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y.C., Chen N.T., Shih Y.P., Dong Y. & Lo S.H. (2009) Up‐regulation of C‐terminal tensin‐like molecule promotes the tumorigenicity of colon cancer through beta‐catenin. Cancer Res. 69, 4563–4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo S.H. & Lo T.B. (2002) Cten, a COOH‐terminal tensin‐like protein with prostate restricted expression, is down‐regulated in prostate cancer. Cancer Res. 62, 4217–4221. [PubMed] [Google Scholar]
- Misale S., Yaeger R., Hobor S. et al (2012) Emergence of KRAS mutations and acquired resistance to anti‐EGFR therapy in colorectal cancer. Nature 486, 532–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muharram G., Sahgal P., Korpela T. et al (2014) Tensin‐4‐dependent MET stabilization is essential for survival and proliferation in carcinoma cells. Dev. Cell 29, 421–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musteanu M., Blaas L., Mair M. et al (2010) Stat3 is a negative regulator of intestinal tumor progression in Apc(Min) mice. Gastroenterology 138, 1003–1011 e1‐5. [DOI] [PubMed] [Google Scholar]
- Provenzani A., Fronza R., Loreni F., Pascale A., Amadio M. & Quattrone A. (2006) Global alterations in mRNA polysomal recruitment in a cell model of colorectal cancer progression to metastasis. Carcinogenesis 27, 1323–1333. [DOI] [PubMed] [Google Scholar]
- Qian X.L., Li G.R., Vass W.C. et al (2009) The Tensin‐3 protein, including its SH2 domain, is phosphorylated by Src and contributes to tumorigenesis and metastasis. Cancer Cell 16, 246–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki H., Moriyama S., Mizuno K. et al (2003) Cten mRNA expression was correlated with tumor progression in lung cancers. Lung Cancer 40, 151–155. [DOI] [PubMed] [Google Scholar]
- Selliah N., Brooks W.H. & Roszman T.L. (1996) Proteolytic cleavage of alpha‐actinin by calpain in T cells stimulated with anti‐CD3 monoclonal antibody. J. Immunol. 156, 3215–3221. [PubMed] [Google Scholar]
- Serrano K. & Devine D.V. (2004) Vinculin is proteolyzed by calpain during platelet aggregation: 95 kDa cleavage fragment associates with the platelet cytoskeleton. Cell Motil. Cytoskeleton 58, 242–252. [DOI] [PubMed] [Google Scholar]
- Seth R., Crook S., Ibrahem S., Fadhil W., Jackson D. & Ilyas M. (2009) Concomitant mutations and splice variants in KRAS and BRAF demonstrate complex perturbation of the Ras/Raf signalling pathway in advanced colorectal cancer. Gut 58, 1234–1241. [DOI] [PubMed] [Google Scholar]
- Storr S.J., Carragher N.O., Frame M.C., Parr T. & Martin S.G. (2011) The calpain system and cancer. Nat. Rev. Cancer 11, 364–374. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Densitometric analysis of western blots.