

Abstract
Mycobacterium tuberculosis HN878 represents a virulent clinical strain from the W-Beijing family, which has been tested in small animal models in order to study its virulence and its induction of host immune responses following infection. This isolate causes death and extensive lung pathology in infected C57BL/6 mice, whereas lab-adapted strains, such as M. tuberculosis H37Rv, do not. The use of this clinically relevant isolate of M. tuberculosis increases the possibilities of assessing the long-lived efficacy of tuberculosis vaccines in a relatively inexpensive small animal model. This model will also allow for the use of knockout mouse strains to critically examine key immunological factors responsible for long-lived, vaccine-induced immunity in addition to vaccine-mediated prevention of pulmonary immunopathology. In this study, we show that the ID93/glucopyranosyl lipid adjuvant (GLA)-stable emulsion (SE) tuberculosis vaccine candidate, currently in human clinical trials, is able to elicit protection against M. tuberculosis HN878 by reducing the bacterial burden in the lung and spleen and by preventing the extensive lung pathology induced by this pathogen in C57BL/6 mice.
INTRODUCTION
The development of an effective Mycobacterium tuberculosis vaccine, and increased availability of novel drugs targeting drug-resistant strains, would significantly contribute to decreasing the continuing spread of this global disease. Nine million people were estimated to have contracted tuberculosis (TB) in 2013, with 1.5 million deaths occurring annually (1). Overall, 3.5% of new cases and 20.5% of previously treated cases are multidrug-resistant (MDR) TB (1), although this can be as high as 35% and 75%, respectively, in central Asian countries. The Beijing lineage of TB has been associated with a number of MDR TB outbreaks (2–6) and accounts for approximately 13% of isolates worldwide (7). The Beijing lineage itself has expanded more rapidly than other lineages (8); this may be due to the higher virulence of modern strains compared with phylogenetically older sublineages (9–11). The clinical isolate, HN878, is a representative strain from the W-Beijing lineage. The HN878 isolate has been studied in mice to characterize the infectivity and pathogenicity of this virulent clinical strain of M. tuberculosis (12–14). These same studies have provided insight into the role of the immune response generated to this pathogen in animals and humans and how the immune response to HN878 contributes to the pathology induced following infection with this isolate. Mice infected with the hypervirulent HN878 strain have increased type I interferon (IFN) expression in the lung and decreased Th1 immunity (12), and HN878 infection in IFN-α/βR-deficient mice results in decreased bacterial growth (13). In humans, a type I IFN-α molecular signature, expressed by a population of isolated neutrophils, was observed in patients with active TB (15). More recently, analysis using a whole-genome microarray approach showed a similar type I interferon molecular signature in neutrophil-depleted blood from TB-infected patients (16). Ordway et al. have shown a transient increase in IFN-γ from CD4+ T cells in the lung following HN878 infection in mice, but these IFN-γ-secreting cells in the lung were decreased by 30 days, while CD4+ Foxp3+ regulatory T (Treg) cells continued to increase to greater than 30% in the lungs 60 days later (14). Vaccination with Mycobacterium bovis bacille Calmette-Guérin (BCG), while effective at 30 days, was shown in the same study to only delay the expansion of the Treg response; Tregs were observed at 60 days at high percentages in the lung at this time point following infection with HN878 (14). Extensive pathology was also observed in the lungs of untreated HN878-infected mice, which survived less than 90 days following infection, whereas pathology was delayed in BCG-vaccinated mice. While survival was prolonged in BCG-vaccinated mice, mice ultimately succumbed to infection (14). This has led Ordway et al. (14) to hypothesize that the inflammation induced by the virulent M. tuberculosis strains, including HN878, leads to the induction of CD4+ Foxp3+ Treg cells, followed by suppression or interference with protective host immunity.
Currently, the only TB vaccine approved for humans is Mycobacterium bovis bacille Calmette-Guérin (BCG), a live attenuated vaccine dating to the 1920s. BCG has proved effective for preventing severe disseminated disease in children but does not protect against pulmonary TB in adults (17). Additionally, the live attenuated BCG vaccine is unsafe for administration to HIV-positive or other immunocompromised individuals due to the possibility of developing regional BCG infection (BCG-itis) (18) or disseminated BCG (BCG-osis) (19-22). Development of an effective TB vaccine will be critical for reducing the worldwide burden of this pathogen. Ideally, a new vaccine would provide immunity against growing as well as dormant phases of the bacterium (23) and would also be safe for use in immunocompromised recipients, providing an alternative to the live attenuated BCG vaccine. We have developed a vaccine candidate fusion of 4 M. tuberculosis proteins (Rv1813, Rv2608, Rv3619, and Rv3620, belonging to the PE/PPE [proteins containing Pro-Glu/Pro-Pro-Glu motifs, respectively], EsX, and latency protein categories) (24), designated ID93. Protein antigens used in vaccines frequently result in low immunogenicity (25, 26); thus, we elected to combine ID93 with a Toll-like receptor 4 (TLR4) agonist as an adjuvant.
The TLR4 agonist in our vaccine adjuvant, glucopyranosyl lipid adjuvant (GLA), is a synthetic hexaacylated lipid A analog. When formulated in an oil-in-water stable emulsion (SE), GLA induces in vivo innate immune responses as well as Th1-skewed cellular immunity to coadministered vaccine antigens (27, 28). GLA adjuvant formulations have been used with experimental vaccines for influenza, HIV (29), respiratory syncytial virus (RSV) (30), leishmaniasis (31), malaria (32), and leprosy (33) in addition to TB (34, 35). Many of these vaccines using GLA formulations have also resulted in protection in preclinical animal models, including mice (34–36), ferrets (37, 38), guinea pigs (34, 35, 39), cotton rats (30), and hamsters (40), against infectious challenge.
Prior studies have shown that mice immunized with the ID93/GLA-SE candidate vaccine generate a Th1-biased response, with antigen-specific polyfunctional CD4+ T cells. ID93/GLA-SE prophylactic immunization has led to protection in mice against challenge with H37Rv (34, 35) and a multidrug-resistant TN5904 strain of M. tuberculosis (35). When ID93/GLA-SE is used alone as a vaccine, or as a booster to BCG, postinfection survival of guinea pigs is increased (35). Additionally, this vaccine is safe and immunogenic in nonhuman primates (35). Therapeutic vaccination with ID93/GLA-SE has also proved to be an effective adjunct to antibiotic therapy in infected mice and nonhuman primates (41).
Most preclinical efficacy studies of candidate TB vaccines use the M. tuberculosis laboratory strain H37Rv or Erdman. M. tuberculosis H37Rv was isolated in 1905 and was maintained as in vitro cultures until at least 1923 (42); the Erdman strain was isolated in 1945 (43). These strains have not been observed in humans for 70 years or more and, thus, may not be relevant to the M. tuberculosis strains circulating today. Use of clinically relevant M. tuberculosis isolates for preclinical vaccine efficacy and basic research has been recommended (44). M. tuberculosis HN878 is a recent clinical isolate of the Beijing lineage. In this study, we tested the efficacy of ID93/GLA-SE against the W-Beijing strain, M. tuberculosis HN878, using a homologous vaccination strategy; however, for clinical use, this vaccine candidate, and other new TB vaccine candidates, will likely be used as a booster in BCG-vaccinated populations (45, 46).
Here, we tested our hypothesis that ID93/GLA-SE can elicit protection against M. tuberculosis HN878 through reduction of bacterial burden, decreased lung pathology, and increased survival by induction of long-lived Th1 immunity.
MATERIALS AND METHODS
Antigen and adjuvant.
ID93 is a recombinant fusion of the M. tuberculosis proteins Rv2608, Rv3620, Rv1813, and Rv3619. Glucopyranosyl lipid adjuvant (GLA) formulated in SE was prepared as previously described (35).
Animals.
Female C57BL/6 mice (5 to 7 weeks old) were purchased from Charles River Laboratories (Wilmington, MA). Mice were housed in the Infectious Disease Research Institute (IDRI) animal facility under specific pathogen-free conditions. Animals were treated according to the regulations and guidelines of the IDRI animal care and use committee.
Immunizations.
Mice were immunized intramuscularly (i.m.) with saline, 5 μg GLA-SE alone, or 0.5 μg of ID93 admixed with 5 μg of GLA-SE, three times at 3-week intervals. Control mice were immunized intradermally (i.d.) with one dose of the bacille Calmette-Guérin Pasteur strain (Sanofi Pasteur, Swiftwater, PA) at 5 × 104 CFU.
Flow cytometry.
Four weeks after the final immunization, splenocytes were isolated from 4 mice per group, and red blood cells were lysed with red blood cell lysis buffer (eBioscience). Cells were resuspended in RPMI 1640 (Life Technologies)-10% fetal bovine serum (FBS) (BioWhittaker) with penicillin-streptomycin (Life Technologies) and glutamine (Gemini) (i.e., complete RPMI [cRPMI]) and were dispensed at 2 × 106 cells/well in 96-well round-bottom plates. Cells were stimulated with medium alone, 10 μg/ml of ID93, or phorbol myristate acetate (PMA) (Calbiochem, San Diego, CA)-ionomycin (Sigma-Aldrich) (1 μg/ml of each) control for 2 h at 37°C. Subsequently, brefeldin A at 1 μg/μl (GolgiPlug; BD Biosciences, San Jose, CA) was added, and samples were incubated an additional 8 h at 37°C. Plates were held at 4°C overnight before staining with antibodies.
Splenocytes were stained with fluorochrome-conjugated monoclonal antibodies to CD4 (clone RM4-5; eBioscience, San Diego, CA), CD8 (clone 53-67; Biolegend, San Diego, CA), and CD44 (clone IM7; eBioscience) in 1% bovine serum albumin (BSA) in phosphate-buffered saline (PBS) with 1 μg/ml of Fc block (CD16/CD32, clone 93; eBioscience) for 15 min at room temperature. Following surface staining, cells were fixed and permeabilized with Cytofix/Cytoperm (BD Biosciences) for 10 min at room temperature. Intracellular staining was done with fluorochrome-conjugated monoclonal antibodies to tumor necrosis factor (TNF) (clone MP6-XT22), interleukin 2 (IL-2) (clone JES6-5H4), IFN-γ (clone XMG1.2), IL-5 (clone TRFK5), and IL-17 (clone tc11-18h10.1) (purchased from BioLegend), plus IL-10 (clone JES5-16E3,) and CD154 (clone mr1) (purchased from eBioscience) in Perm/Wash buffer (BD Biosciences). All antibodies were used at a 1:100 dilution. Stained cells were washed and resuspended in 1% BSA in PBS and filtered before analysis on a modified 4-laser Fortessa with FACSDiva software (BD Biosciences). Lymphocytes were gated by forward and side scatter, and a minimum of 20,000 CD4+ events were acquired for each sample. Data were analyzed with FlowJo version 9.7.5 (Treestar, Ashland, OR) and SPICE (National Institutes of Health, http://exon.niaid.nih.gov/spice).
Postinfection lung samples taken 4 weeks after challenge were stimulated as described above for splenocytes. Cells were washed with PBS (Life Sciences) before staining with live/dead fixable stain (Invitrogen). Samples were then washed, stained with antibodies, and analyzed, as detailed above for splenocytes. Lung tissue was harvested, processed, and stimulated in a manner similar to that described for the spleen.
Culture of HN878.
M. tuberculosis HN878 (a gift from Ian Orme, Colorado State University, Fort Collins, CO) was thawed and cultured in 10 ml of Middlebrook 7H9 liquid medium with 10% (vol/vol) oleic acid-albumin-dextrose-catalase (OADC) supplement (Becton Dickinson) and 0.05% (wt/vol) Tween 80. The culture was expanded to roller bottles and grown to an optical density at 600 nm (OD600) of approximately 0.5 to 0.6. Sterile glycerol (10%) was added to the bacterial culture, and aliquots were frozen at −80°C until needed for infection.
Establishment of the bacterial dose for aerosol delivery of 50 to 100 CFU in the lungs of mice.
Four C57BL/6 mice per group were infected with 1 × 106, 3 × 106, 1 × 107, or 2 × 107 CFU/ml of HN878 according to the procedure described in the supplemental material, with the intent to deliver 50 to 100 CFU per mouse. Twenty-four hours postinfection, the entire lung was harvested, homogenized, and plated on 7H10 agar plates. Plates were incubated for 3 weeks at 37°C, 5% CO2. Colonies were counted, and the aerosol dose yielding approximately 100 CFU per mouse was selected.
Aerosol challenge.
Four weeks after the final immunization, mice were challenged with a low-dose aerosol of M. tuberculosis HN878. An aerosol exposure chamber (University of Wisconsin, Madison, WI) was calibrated to deliver 50 to 100 viable M. tuberculosis HN878 CFU into the lungs (see supplemental material). An aliquot of frozen stock was thawed and diluted in PBS with 0.05% Tween 80 (Sigma-Aldrich). Twenty-four hours after infection, lungs of 3 mice were homogenized and plated onto Middlebrook 7H10 agar (Molecular Toxicology, Inc., Boone, NC) to ensure bacterial delivery of 50 to 100 CFU. Plates were incubated for 2 to 3 weeks at 37°C, 5% CO2, and then examined for colonies. Infection and all subsequent procedures were performed under biosafety level 3 conditions.
Bacterial burden.
Four weeks after infection with M. tuberculosis HN878, 7 mice per group were euthanized with CO2. The accessory lobe of the lung was reserved for histology. Lung and spleen were harvested and homogenized in PBS with 0.05% Tween 80 (Sigma-Aldrich), using an Omni tissue homogenizer (Omni International, Kennesaw, GA). Serial 5-fold dilutions of homogenates were made in PBS with 0.05% Tween 80, and aliquots of the dilutions were plated on Middlebrook 7H10 agar plates. Plates were incubated for 2 to 3 weeks at 37°C, 5% CO2, before counting colonies. Bacterial burden in CFU per organ was calculated and expressed as log10. The reduction in bacterial burden was calculated as mean log10 CFUsaline − mean log10 CFUvaccine.
Survival.
Ten mice from each group were monitored for survival after infection. Moribund animals, and those losing 20% of body weight, were euthanized with CO2. Bacterial burdens in lungs and spleens were determined as described above. The accessory lobe of the lung was reserved for histology. At experiment termination, all remaining mice were euthanized for sample processing in the same manner.
Histology.
The accessory lobe of the lung was fixed in 10% normal buffered formalin for at least 7 days. Fixed tissues were embedded in paraffin, cut, and stained with hematoxylin and eosin (H&E) by the Benaroya Research Institute Histology Core Facility (Seattle, WA). Histological analysis was done in a blind manner by group and performed by a board-certified veterinary anatomic pathologist (Brendan Podell, CSU, Fort Collins). Disease severity, based on area affected, cellular composition, and presence of inflammatory tissue injury, was quantitatively and subjectively evaluated in three tissue sections each, from 7 and 10 mice per group at after 4 weeks of infection and at survival endpoints of the study, respectively. Area morphometry for quantification of lesion-to-tissue area proportions was performed on an Eclipse 80i microscope (Nikon Instruments, Melville, NY) equipped with an automated, computer-controlled stage and Stereo investigator software version 11.01 (MBF Bioscience, Williston, VT), with tissue and lesion area estimated using the fraction-fractionator method. Fifteen to 26 counting frames for lungs of mice sampled at week 4 of infection and 22 to 42 counting frames for lungs of mice sampled at study termination were assigned randomly by the software depending on tissue size, and a counting frame of 1,195 μm2 with a grid spacing of 100 μm was used to define the areas of interest. Data were expressed as a percentage ratio of lesion to total tissue area.
Statistical analysis.
Flow cytometry data were analyzed with FlowJo version 9.7.5 (Treestar, Ashland, OR) and SPICE (National Institutes of Health, http://exon.niaid.nih.gov/spice) using the Wilcoxon signed-rank test. CFU data were analyzed with GraphPad Prism 6 (GraphPad Software, San Diego, CA) using a standard one-way analysis of variance (ANOVA) followed by Tukey's multiple-comparison test versus saline. A P value of <0.05 was considered significant. Survival curves were compared to saline using the Mantel-Cox log rank test with Bonferroni correction for multiple comparisons, based on a significance level of P < 0.05.
RESULTS
ID93 adjuvanted with GLA-SE induces a CD4+ Th1 antigen-specific population.
We first conducted an experiment to characterize the immunogenicity of ID93/GLA-SE. Four weeks after the third immunization, splenocytes were harvested from 4 mice and stimulated with ID93. Lymphocytes were gated on CD44+, and cytokine expression in CD4+ or CD8+ T cells was measured. Background expression in unstimulated (medium only) samples was subtracted. Only the ID93/GLA-SE-immunized animals had significant percent frequency of CD4 T cells expressing CD154 (1.5%), IFN-γ, and TNF (1.5%) (see Fig. S1 in the supplemental material). A large majority (>1% frequency) of the activated ID93-specific CD4+ T cells were multifunctional, expressing double cytokines (CD154+ IFN-γ/TNF). Less than 0.1% induction of CD154, IFN-γ, or TNF-expressing CD8+ T cells was observed (data not shown), and no ID93-specific IL-10, IL-17, or IL-5 was produced from CD4+ T cells (see Fig. S1 in the supplemental material). These results are consistent with our previous reports on ID93/GLA-SE immunization (34, 35).
Antigen-specific Th1 cells infiltrate the lungs of ID93/GLA-SE-immunized mice post-HN878 infection.
Four weeks after challenge with M. tuberculosis HN878, single cell suspensions were prepared from lungs of 3 mice for each group, stimulated with ID93, and analyzed as for preinfection splenocytes for cytokine induction. CD4+ CD44+ T cells of ID93/GLA-SE-immunized mice produced significant amounts of IFN-γ (∼2.3%) and TNF (∼4%) in response to ID93; significant expression of CD154 (an activation marker) was also induced (Fig. 1A). Small but statistically significant IL-17 induction was generated as well (0.35%) (Fig. 1B). Whereas IL-17 has been shown to be dispensable against infection with M. tuberculosis H37Rv, a requirement for IL-17 in protection against HN878 has recently been described (47). CD154+ IFN-γ/TNF double-cytokine-positive cells comprised the majority of the Th1 cytokine-producing CD44+ CD4+ cells, although some CD154+ TNF-positive single positives were seen (Fig. 1C).
FIG 1.

ID93-specific Th1 T cells infiltrate the lungs of ID93/GLA-SE immunized mice postchallenge. C57BL/6 mice were immunized 3 times, at 3-week intervals. Four weeks after the final immunization, mice were challenged with low-dose aerosol M. tuberculosis strain HN878. Four weeks later, single-cell suspensions were made from lungs of 3 mice per group and stimulated with ID93 or medium alone. Medium-only background was subtracted from ID93-stimulated samples. (A) Percentage of single-cytokine-producing CD4+ CD44+ T cells; (B) percentage of IL-17-producing CD44+ CD4+ T cells; (C) Percentage of CD4+ CD44+ polyfunctional Th1-cytokine-producing T cells. Bars are mean plus standard deviation; dots indicate individual values. *, P < 0.05 versus saline immunized, using the Wilcoxon signed-rank test.
ID93/GLA-SE immunization reduces M. tuberculosis HN878 bacterial burden.
ID93/GLA-SE vaccine efficacy was tested in mice challenged with M. tuberculosis HN878. Bacterial titration was performed to determine the dose of the aerosol to include for infection (see Fig. S2 in the supplemental material). BCG-immunized mice were included as a positive control. Four weeks after challenge, organs from 7 mice per group were harvested, and bacterial burdens in lungs and spleens were determined (Fig. 2; see Table S1 in the supplemental material). Compared to saline-injected mice, ID93/GLA-SE immunization reduced CFU in the lungs by 0.54 log10 (P < 0.001) and in the spleen by 0.52 log10 (P < 0.01) (Fig. 2; see Table S1 in the supplemental material). The BCG control was also effective, with protection of 0.96 log10 (P < 0.0001) in the lungs and 0.79 log10 (P < 0.0001) in the spleen (Fig. 2; see Table S1 in the supplemental material). The GLA-SE adjuvant alone did not afford protection in either lung (0.28 log10, P > 0.05) or spleen (0.12 log10, P > 0.05) (Fig. 2; see Table S1 in the supplemental material).
FIG 2.

Bacterial counts within the lungs (A) and spleens (B) of immunized C57BL/6 mice (7 mice per group) 4 weeks after aerosol infection with M. tuberculosis HN878. Results are presented as log10 CFU within each organ. Asterisks represent statistically significant difference from saline using the one-way ANOVA with Tukey's multiple-comparison test: **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
ID93/GLA-SE immunization prolongs survival of mice challenged with HN878.
We next determined whether the protective effect afforded by ID93/GLA-SE was long lived. After a low-dose HN878 aerosol challenge, 10 mice per group were monitored for survival. Saline-treated animals had a median survival time of 231 days, as did those receiving GLA-SE without antigen. Weights were also monitored following infection and were represented as an average for each group over time (Fig. 3A). By study termination on day 248, 80% of mice immunized with ID93/GLA-SE and 90% of BCG-vaccinated controls had survived (Fig. 3B). Both groups were statistically significantly different from saline controls by the Mantel-Cox log rank test with Bonferroni's correction for multiple comparisons, whereas GLA-SE alone did not enhance survival (Fig. 3B).
FIG 3.
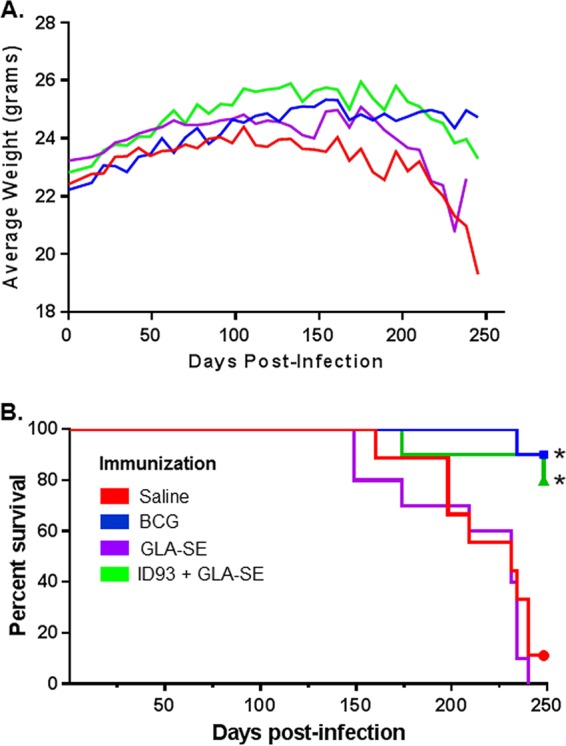
Protection with ID93/GLA-SE. (A) Change in weights of immunized C57BL/6 mice following M. tuberculosis HN878 challenge. (B) Survival of immunized C57BL/6 mice following M. tuberculosis HN878 challenge. Groups of 10 mice were monitored for survival for about 250 days after challenge with M. tuberculosis HN878. The median survival for saline- and GLA-SE-immunized groups was 231 days; median survival for BCG- and ID93/GLA-SE-immunized groups was undefined. Results indicated by an asterisk were considered statistically significantly different from saline: *, P < 0.05, using the Mantel-Cox log-rank test with Bonferroni correction for multiple comparisons.
Longer survival time trends with lower bacterial burden.
Bacterial burden was also monitored throughout the survival study. CFU plotted by day of euthanasia yielded a line with a negative slope for the lung (Fig. 4), whereas a negative slope was less obvious for the spleen (data not shown). Log10 CFU data from individual mice were also organized by treatment and time of euthanasia or death and were included as well (Table 1).
FIG 4.

Early death due to infection trends with higher bacterial burden. Bacterial burden (CFU) in lungs of the survival arm was graphed by euthanasia day. Solid line, nonlinear regression curve of untransformed CFU with 1/Y weighting; dotted lines, 95% confidence interval (CI).
TABLE 1.
Bacterial loads in the lungs of individual long-term survivors
| Group | Time to death in days (bacterial load in lung, log10) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
| Saline | 198 (7.21) | 209 (6.90) | 231 (6.47) | 248 (7.18) | 240 (7.25) | 160 (7.41) | 65a (NDb) | 240 (7.19) | 198 (7.06) | 234 (6.59) |
| BCG | 248 (6.50) | 248 (6.19) | 234 (6.51) | 248 (6.06) | 248 (6.46) | 248 (6.40) | 248 (6.70) | 248 (6.34) | 248 (7.00) | 248 (6.97) |
| GLA-SE | 234 (6.59) | 234 (6.90) | 231 (6.64) | 240 (7.60) | 234 (6.89) | 231 (6.58) | 149 (8.27) | 209 (7.96) | 149 (8.80) | 174 (7.20) |
| ID93/GLA-SE | 248c (7.17) | 248 (6.06) | 174 (7.09) | 248 (5.98) | 248 (6.46) | 248 (6.49) | 248 (6.38) | 248 (6.47) | 248 (6.34) | 248 (6.16) |
Euthanized due to severe ulcerative dermatitis.
ND, not done.
Fulfilled criteria for euthanasia on experiment end date.
Decreased lung immunopathology with ID93/GLA-SE in HN878-infected mice.
Because infection with M. tuberculosis HN878 has been shown to induce severe pathology in the lungs of untreated mice, we wanted to determine whether ID93/GLA-SE could provide protection against pulmonary immunopathology induced by this M. tuberculosis strain. H&E-stained slides of lung sections from the week 4 time point (Fig. 5) and the survival arm of the study (Fig. 6) were blinded and examined by a board-certified veterinary pathologist. At week 4, saline and GLA-SE (adjuvant only) control mice had similar disease profiles, with lesion burdens ranging from 12% to 36% (Fig. 7). Inflammation was mostly composed of macrophages, with few infiltrating neutrophils and lymphocytes (Fig. 5A and C). In contrast, inflammatory cell populations in the lungs of BCG- and ID93/GLA-SE-immunized mice consisted mostly of lymphocytes, with fewer macrophages (Fig. 5B and D). The ID93/GLA-SE and BCG groups showed significantly less lesion involvement and more evidence of protection within the lungs 4 weeks following infection with HN878, ranging from a 7.6% to a 27% lesion-to-tissue burden (Fig. 7). The cellular composition in the lung of BCG-vaccinated mice was similar to that seen in ID93/GLA-SE-immunized mice.
FIG 5.

Lymphocyte infiltration is increased and inflammation is reduced in the lungs 4 weeks after M. tuberculosis HN878 infection in mice vaccinated with ID93/GLA-SE. (A, C) Lungs of mice receiving saline or GLA-SE alone are dominated by innate immune cell infiltrates consisting of a high proportion of macrophages and neutrophils (arrows). (B, D) In contrast, BCG- and ID93/GLA-SE-vaccinated mice display similar inflammatory infiltrates with frequent incorporation of lymphocytes (arrows), fewer macrophages, and nearly absent neutrophils. Magnification, ×400.
FIG 6.

Protection from progressive pulmonary pathology with ID93/GLA-SE vaccination. Representative photomicrographs from H&E-stained lung sections of mice from survival groups are shown. (A, C) Lungs of mice receiving saline or GLA-SE alone demonstrated progressive pulmonary disease and tissue injury, with larger lesions (lesion is delineated by arrowheads) that consisted of frequent neutrophils and foamy macrophages. Areas of necrosis and lipid accumulation were frequently present (arrows). (B, D) In contrast, lesions within the lungs of BCG- and ID93/GLA-SE-vaccinated mice were limited, with high frequency of lymphocytes (arrows) and absence of neutrophils as well as necrosis. Magnification, ×100.
FIG 7.

Decreased lesion involvement within the lungs of mice immunized with ID93/GLA-SE 4 weeks following challenge with M. tuberculosis HN878. The bars represent the percent mean lesion involvement in the lungs, and the dots represent the lesion-to-tissue percentage from each individual mouse lung. Area of morphometry for quantification of the lesion-to-tissue area proportion was determined on an Eclipse 80i microscope using the fraction-fractionator method. Asterisks represent statistically significant differences from the saline group determined using one-way ANOVA: *, P < 0.05; **, P < 0.01.
In the survival arm of the study, the trends were similar to week 4 results, with severe disease burden most evident in the untreated saline control group. Saline and GLA-SE adjuvant-alone groups showed evidence of destructive pulmonary inflammation, with high numbers of neutrophils, foamy macrophages, and areas of necrosis with lipid accumulation (Fig. 6A and C). In contrast, ID93/GLA-SE-immunized mice displayed very little pathology following infection with M. tuberculosis HN878, with generalized absence of destructive inflammation and higher proportions of infiltrating lymphocytes with few to absent neutrophils (Fig. 6D). Similar to the observation 4 weeks after infection with M. tuberculosis HN878, the ID93/GLA-SE group showed significantly less lesion involvement and more evidence of protection within the lungs at the terminal time point (8 months following infection), ranging from a 21.3% to a 53.8% lesion-to-tissue burden (Fig. 8). BCG-vaccinated mice exhibited a cellular composition similar to that of the ID93/GLA-SE group but with an increased area of inflammation, primarily due to large lymphoid follicles, some of which contained high proportions of large lymphocytes with perinuclear clearing of the cytoplasm, morphology that is consistent with plasma cells (Fig. 6B and 8). The lesion involvement in the lungs of BCG-immunized mice ranged between 38.3% and 68.7% and was not significantly different from that of the groups given either saline (44.7% to 84.2%) or GLA-SE (42.7% to 79%) (Fig. 8).
FIG 8.

Decreased lesion involvement within the lungs of mice immunized with ID93/GLA-SE 8 months following challenge with M. tuberculosis HN878. The bars represent the percent mean lesion involvement in the lungs, and the dots represent the lesion-to-tissue percentage from each individual mouse lung. Area of morphometry for quantification of lesion-to-tissue area proportion was determined on an Eclipse 80i microscope using the fraction-fractionator method. Asterisks represent statistically significant differences determined using one-way ANOVA: **, P < 0.01 compared to GLA-SE; ***, P < 0.001 compared to BCG and saline.
In mice euthanized at the end of the experiment, proliferative changes were also present among epithelial cells that line the alveoli. These changes, consistent with squamous metaplasia of pneumocyte epithelial cells, were present in mice injected with saline or adjuvant alone, affecting 7 of 10 animals in each group (Fig. 9). Notably, epithelial hyperplasia and squamous metaplasia were absent from both BCG- and ID93/GLA-SE-vaccinated mice (see representation of epithelial changes within the M. tuberculosis-infected lung tissue; Fig. 9).
FIG 9.

Epithelial hyperplasia and squamous metaplasia in mice with chronic M. tuberculosis HN878 infection. A representative photomicrograph is shown, demonstrating a proliferation and squamous differentiation of epithelial cells (arrows), which surround aggregates of active inflammation consisting of neutrophils and macrophages (arrowheads). Lung tissue is taken from a saline control animal, 34 weeks after infection. Magnification, ×200.
DISCUSSION
In this study, we show that ID93/GLA-SE, which generates T helper 1 (Th1) CD4+ T cells, elicits long-lived immunity and prevents pulmonary pathology both early (4 weeks) and late (8 months) following challenge with M. tuberculosis HN878. We observed both a reduction in the bacterial burden in the lungs and increased survival in 80% of ID93/GLA-SE-immunized animals up to 250 days following low-dose aerosol challenge with HN878. One of the characteristic features of this clinical isolate (HN878) is its hypervirulence; this strain has been shown to grow much faster than other M. tuberculosis strains, such as H37Rv and Erdman-KO1 (13), which are typically used in challenge models to measure vaccine efficacy.
One of the properties associated with vaccine efficacy is the prevention of lung pathology associated with M. tuberculosis infection. An orchestrated balance of host responses against M. tuberculosis is required to induce protection through acquired immunity and granuloma formation in the lung, while preventing pathology that can lead to lung injury and exacerbation of disease (48). Tuberculosis, if untreated, can lead to an accumulation of mycobacterial products in alveoli, followed by increased necrosis and caseation within the lung, bacterial growth, and spread of infection (49). TB lesions involved in healing can result in chronic fibrocaseous disease and extensive pulmonary fibrosis, displacing normal lung tissue (49). We have shown that adjuvant formulation plays a key role in the protective efficacy of a tuberculosis vaccine in the lungs of guinea pigs (34). A main difference which results in C57BL/6 mice following infection with the HN878 clinical isolate compared to the H37Rv strain is the increased lung pathology in mice observed with HN878. We found reduced lung inflammation in ID93/GLA-SE-immunized mice, with lung infiltrates consisting mostly of lymphocytes, in contrast to saline or adjuvant-alone groups, which had extensive neutrophil accumulation at both 4 weeks and 8 months following infection with M. tuberculosis HN878. Other researchers have also observed greater numbers of lung lesions, increased lesion size, and greater pathology scores (representing greater lung pathology) in mice 30 days following challenge with M. tuberculosis HN878 (13). Vaccine efficacy tested in mice against this clinical isolate can therefore be determined based on additional protective parameters besides reduction in bacterial CFU in the lung and decreased dissemination to other organs. Furthermore, since untreated mice die as a result of low-dose infection with HN878, this provides a model for vaccine durability by looking at survival over time. In our experiments, the untreated C57BL/6 mice challenged with M. tuberculosis HN878 lived longer than what has been seen in previously published studies (12, 13), although we did observe similar growth characteristics in the lungs of mice 4 weeks after challenge with the HN878 strain (∼6.5 log10) (14). There are some differences between our study and these other studies, however, including the source of mice used in this study. Our study used C57BL/6 mice purchased from Charles River, whereas Ordway et al. (13, 14) used mice obtained from Jackson Laboratories. Sun et al., who also purchased C57BL/6 mice from Charles River, showed that mice survived longer with the HN878 strain (50). It is unclear if using the same mouse strain purchased from two different vendors could result in such differences in resistance to infection with HN878. Recently, Chang et al. showed significant differences in airway responsiveness by measuring lung resistance in response to different doses of methacholine (a method often used in asthma research) in inbred C57BL/6 mice, depending on where the mice originated (mice purchased from five vendors were tested, including Jackson Laboratories and Charles River Laboratories) (51). The authors suggest that environmental factors (husbandry practices, such as caging systems, food components, type of bedding, etc.), rather than genotypic differences between the B6 substrains, were most likely the cause of observed phenotypic differences, particularly since F1 mice bred and raised in the same environment did not show differences in airway responsiveness among the substrains. Manca et al., who also showed higher death rates in untreated mice with HN878 challenge, used B6D2/F1 mice for their study (12) and also used an aerosol system (Lovelace nebulizer) different from the Madison aerosol chamber used in our laboratory. Culturing of the M. tuberculosis strain, including the number of passages, could also result in differences seen in different laboratories and could have resulted in decreased virulence in our laboratory. We included our method for culturing the bacterial stock and for determining the challenge dose used (see supplemental data). Regardless of these differences, the utility of the model is still apparent for testing vaccine efficacy with proper controls, due to the induction of lung pathology, decreased survival observed in untreated infected animals (unlike that seen with laboratory M. tuberculosis strains), and reduced costs compared to other animal models, such as the guinea pig, for survival studies. Another benefit of using this model would be to enable determination of the immune requirements of durable vaccine protection using this C57BL/6 challenge model, as several knockout models are generated on the C57BL/6 genetic background. To date, our laboratory has shown through adoptive transfer experiments and use of multiple cytokine and cytokine receptor gene knockout mice that the ID93/GLA-SE candidate vaccine does not require IFN-γ, TNF, or inducible nitric oxide synthase (iNOS) for protection against M. tuberculosis H37Rv (52). It is unclear, however, whether IFN-γ, TNF, and iNOS elicited by ID93/GLA-SE immunization are critical to long-lived, durable vaccine protection against this clinical isolate and the pathology induced by HN878; these vaccine-induced efficacy readouts are not feasible using the laboratory-adapted H37Rv strain in C57BL/6 mice, since this laboratory M. tuberculosis strain is not lethal and significant pathology in the lung due to infection is not observed. Other investigators have suggested that preclinical challenge with relevant clinical isolates in mice and other animal models might be better predictors of vaccine efficacy (14, 53, 54). The data in this study showing protection against a W-Beijing clinical isolate (HN878) support our other protective efficacy studies with ID93/GLA-SE, which have been performed in many animal models and against both sensitive and multidrug-resistant strains of M. tuberculosis. Given that the pathogenesis of M. tuberculosis HN878 (and other pathogenic strains) may partially stem from the presence of lipids, such as the phenolic glycolipids that induce type I IFN and block Th1 immunity (55) or may be due to transient Th1 immunity, which is diminished due to an expansion of CD4+ Foxp3 Treg cells in response to infection with virulent strains of M. tuberculosis (13), we hypothesize that the use of GLA as an adjuvant to induce innate cytokines, such as IL-12p40 (28), to drive a vaccine-mediated Th1 response is an effective strategy against virulent isolates such as HN878. Another possible strategy would be to test the potential synergistic effects of a CCR4 antagonist(s), known to block Treg function (56) combined with the ID93/GLA-SE vaccine. One of these CCR4 antagonists, mogamulizumab, is currently in clinical trials for various cancers, including adult T-cell leukemia-lymphoma, non-small-cell lung cancer, and advanced solid tumors (www.clinicaltrials.gov) (57). Addition of a CCR4 antagonist combined with the MVA85A tuberculosis vaccine candidate showed enhanced purified protein derivative (PPD)-specific IFN-γ-expressing splenocytes in mice approximately 1 week following immunization (58), which supports the feasibility of this strategy.
Another interesting observation in our study was that a majority of mice (7 of 10 mice) in the survival study from both the saline and adjuvant control groups displayed epithelial hyperplasia in the lung tissue, suggesting an association between M. tuberculosis infection and the development of neoplasia. The morphology observed histologically among the epithelial cells that line the alveoli is representative of squamous metaplasia of pulmonary pneumocytes, which may represent preneoplastic changes within the lung. Squamous metaplasia was observed only in the lungs of mice chronically infected with M. tuberculosis HN878; no hyperplasia was present 4 weeks after infection in any of the groups. Chronic infection is a known risk factor for pulmonary cancer, and a recent review of the literature by Falagas et al. has identified several reports showing associations between TB and cancer in humans (59). Another paper recently showed the development of squamous cell carcinoma in mice chronically infected with tuberculosis using M. tuberculosis Erdman (60). The authors report evidence for the induction of DNA-damaging reactive oxygen and nitrogen within TB-infected macrophages and upregulation of epiregulin mRNA from interstitial macrophages within the TB lesions in the mice. The authors also suggest that epiregulin, an epidermal growth factor, produced from TB-infected macrophages may be serving as a paracrine growth factor during the preneoplasia stages in their model. In their study, infection with M. tuberculosis Erdman in C57BL/6 mice led to the appearance of squamous cell carcinoma in 50% of mice (2 of 4 mice) 7 months following infection and in 80% of mice (8 of 10 mice) 12 weeks after infection. The presence of cancer in the lungs was accelerated in B6.C3H-sst1 mice, which include the sst1-tuberculosis susceptible allele (61); 100% (4 of 4 mice) had evidence of squamous cell metaplasia in lungs 5 months after infection. In our study, we used HN878, a W-Beijing clinical isolate, for infection in C57BL/6 mice. Similar to others, we show that the HN878 isolate induces a greater amount of inflammation in the untreated lungs of C57BL/6 mice compared to what is normally observed following infection with the laboratory M. tuberculosis strain H37Rv. In addition, C57BL/6 mice also succumb to infection more rapidly with the HN878 strain compared to the rate observed with H37Rv; while C57BL/6 mice will eventually succumb to infection with H37Rv, they are typically considered relatively resistant to infection with laboratory strains of M. tuberculosis (62). This is the first report, to our knowledge, that shows epithelial hyperplasia and squamous metaplasia in the lung with M. tuberculosis HN878 in untreated C57BL/6 mice and protection against epithelial hyperplasia and lung pathology, in addition to a reduction in HN878 bacterial load in the lung and spleen and increased survival with ID93/GLA-SE.
In summary, a model that utilizes M. tuberculosis HN878 infection in C57BL/6 mice will be useful for testing the efficacy of prophylactic TB vaccines (with and without a BCG prime immunization) where bacterial reduction and immunopathology in the lung can be assessed, and in the context of an adjunct therapeutic vaccine combined with drug treatment against clinical M. tuberculosis isolates. Our future plans will include testing ID93/GLA-SE in these scenarios, will investigate whether ID93/GLA-SE inhibits the elicitation of Foxp3+ T regulatory cells in HN878-infected animals and in addition will characterize other important immunological mechanisms of vaccine-induced protection.
Supplementary Material
ACKNOWLEDGMENTS
We thank Mark Orr for helping design the experiment to establish the dose of M. tuberculosis HN878 required for low-dose aerosol infection in C57BL/6 mice, Dave Argilla and Natasha Dubois for their technical help, and David Roberts for culturing the M. tuberculosis HN878 strain.
S.G.R. is a founder of, and holds an equity interest in, Immune Design Corp., a licensee of certain rights associated with GLA.
This project has been funded in whole or in part with federal funds from the National Institute of Allergy and Infectious Diseases National Institutes of Health, Department of Health and Human Services, under contract HHSN272200800045C and grants R01AI044373 and U01AI078054.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, National Institute of Allergy And Infectious Diseases, or the National Institutes of Health, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Funding Statement
HHS | NIH | National Institute of Allergy and Infectious Diseases (NIAID) provided funding to Rhea N. Coler under contract number HHSN272200800045C. HHS | NIH | National Institute of Allergy and Infectious Diseases (NIAID) provided funding to Steven G. Reed under grant number R01AI044373. HHS | NIH | National Institute of Allergy and Infectious Diseases (NIAID) provided funding to Rhea N. Coler under grant number U01AI078054. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, National Institute of Allergy And Infectious Diseases, or the National Institutes of Health nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/CVI.00458-15.
REFERENCES
- 1.World Health Organization. 2014. Global tuberculosis report 2014. World Health Organization, Geneva, Switzerland: http://reliefweb.int/report/world/global-tuberculosis-report-2014. [Google Scholar]
- 2.Almeida D, Rodrigues C, Ashavaid TF, Lalvani A, Udwadia ZF, Mehta A. 2005. High incidence of the Beijing genotype among multidrug-resistant isolates of Mycobacterium tuberculosis in a tertiary care center in Mumbai, India. Clin Infect Dis 40:881–886. doi: 10.1086/427940. [DOI] [PubMed] [Google Scholar]
- 3.Bifani PJ, Mathema B, Liu Z, Moghazeh SL, Shopsin B, Tempalski B, Driscol J, Frothingham R, Musser JM, Alcabes P, Kreiswirth BN. 1999. Identification of a W variant outbreak of Mycobacterium tuberculosis via population-based molecular epidemiology. JAMA 282:2321–2327. doi: 10.1001/jama.282.24.2321. [DOI] [PubMed] [Google Scholar]
- 4.Casali N, Nikolayevskyy V, Balabanova Y, Harris SR, Ignatyeva O, Kontsevaya I, Corander J, Bryant J, Parkhill J, Nejentsev S, Horstmann RD, Brown T, Drobniewski F. 2014. Evolution and transmission of drug-resistant tuberculosis in a Russian population. Nat Genet 46:279–286. doi: 10.1038/ng.2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson R, Warren R, Strauss OJ, Jordaan AM, Falmer AA, Beyers N, Schaaf HS, Murray M, Cloete K, van Helden PD, Victor TC. 2006. An outbreak of drug-resistant tuberculosis caused by a Beijing strain in the western Cape, South Africa. Int J Tuberc Lung Dis 10:1412–1414. [PubMed] [Google Scholar]
- 6.Kubica T, Rusch-Gerdes S, Niemann S. 2004. The Beijing genotype is emerging among multidrug-resistant Mycobacterium tuberculosis strains from Germany. Int J Tuberc Lung Dis 8:1107–1113. [PubMed] [Google Scholar]
- 7.Brudey K, Driscoll JR, Rigouts L, Prodinger WM, Gori A, Al-Hajoj SA, Allix C, Aristimuno L, Arora J, Baumanis V, Binder L, Cafrune P, Cataldi A, Cheong S, Diel R, Ellermeier C, Evans JT, Fauville-Dufaux M, Ferdinand S, Garcia de Viedma D, Garzelli C, Gazzola L, Gomes HM, Guttierez MC, Hawkey PM, van Helden PD, Kadival GV, Kreiswirth BN, Kremer K, Kubin M, Kulkarni SP, Liens B, Lillebaek T, Ho ML, Martin C, Martin C, Mokrousov I, Narvskaia O, Ngeow YF, Naumann L, Niemann S, Parwati I, Rahim Z, Rasolofo-Razanamparany V, Rasolonavalona T, Rossetti ML, Rusch-Gerdes S, Sajduda A, Samper S, Shemyakin IG, Singh UB, Somoskovi A, Skuce RA, van Soolingen D, Streicher EM, Suffys PN, Tortoli E, Tracevska T, Vincent V, Victor TC, Warren RM, Yap SF, Zaman K, Portaels F, Rastogi N, Sola C. 2006. Mycobacterium tuberculosis complex genetic diversity: mining the fourth international spoligotyping database (SpolDB4) for classification, population genetics, and epidemiology. BMC Microbiol 6:23. doi: 10.1186/1471-2180-6-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Merker M, Blin C, Mona S, Duforet-Frebourg N, Lecher S, Willery E, Blum MG, Rusch-Gerdes S, Mokrousov I, Aleksic E, Allix-Beguec C, Antierens A, Augustynowicz-Kopec E, Ballif M, Barletta F, Beck HP, Barry CE III, Bonnet M, Borroni E, Campos-Herrero I, Cirillo D, Cox H, Crowe S, Crudu V, Diel R, Drobniewski F, Fauville-Dufaux M, Gagneux S, Ghebremichael S, Hanekom M, Hoffner S, Jiao WW, Kalon S, Kohl TA, Kontsevaya I, Lillebaek T, Maeda S, Nikolayevskyy V, Rasmussen M, Rastogi N, Samper S, Sanchez-Padilla E, Savic B, Shamputa IC, Shen A, Sng LH, Stakenas P, Toit K, Varaine F, Vukovic D, Wahl C, Warren R, Supply P, Niemann S, Wirth T. 2015. Evolutionary history and global spread of the Mycobacterium tuberculosis Beijing lineage. Nat Genet 47:242–249. doi: 10.1038/ng.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanekom M, Gey van Pittius NC, McEvoy C, Victor TC, Van Helden PD, Warren RM. 2011. Mycobacterium tuberculosis Beijing genotype: a template for success. Tuberculosis (Edinb) 91:510–523. doi: 10.1016/j.tube.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 10.Palanisamy GS, DuTeau N, Eisenach KD, Cave DM, Theus SA, Kreiswirth BN, Basaraba RJ, Orme IM. 2009. Clinical strains of Mycobacterium tuberculosis display a wide range of virulence in guinea pigs. Tuberculosis (Edinb) 89:203–209. doi: 10.1016/j.tube.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 11.Ribeiro SC, Gomes LL, Amaral EP, Andrade MR, Almeida FM, Rezende AL, Lanes VR, Carvalho EC, Suffys PN, Mokrousov I, Lasunskaia EB. 2014. Mycobacterium tuberculosis strains of the modern sublineage of the Beijing family are more likely to display increased virulence than strains of the ancient sublineage. J Clin Microbiol 52:2615–2624. doi: 10.1128/JCM.00498-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Manca C, Tsenova L, Bergtold A, Freeman S, Tovey M, Musser JM, Barry CE III, Freedman VH, Kaplan G. 2001. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-α/β. Proc Natl Acad Sci U S A 98:5752–5757. doi: 10.1073/pnas.091096998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ordway D, Henao-Tamayo M, Harton M, Palanisamy G, Troudt J, Shanley C, Basaraba RJ, Orme IM. 2007. The hypervirulent Mycobacterium tuberculosis strain HN878 induces a potent TH1 response followed by rapid down-regulation. J Immunol 179:522–531. doi: 10.4049/jimmunol.179.1.522. [DOI] [PubMed] [Google Scholar]
- 14.Ordway DJ, Shang S, Henao-Tamayo M, Obregon-Henao A, Nold L, Caraway M, Shanley CA, Basaraba RJ, Duncan CG, Orme IM. 2011. Mycobacterium bovis BCG-mediated protection against W-Beijing strains of Mycobacterium tuberculosis is diminished concomitant with the emergence of regulatory T cells. Clin Vaccine Immunol 18:1527–1535. doi: 10.1128/CVI.05127-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, Wilkinson KA, Banchereau R, Skinner J, Wilkinson RJ, Quinn C, Blankenship D, Dhawan R, Cush JJ, Mejias A, Ramilo O, Kon OM, Pascual V, Banchereau J, Chaussabel D, O'Garra A. 2010. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466:973–977. doi: 10.1038/nature09247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ottenhoff TH, Dass RH, Yang N, Zhang MM, Wong HE, Sahiratmadja E, Khor CC, Alisjahbana B, van Crevel R, Marzuki S, Seielstad M, van de Vosse E, Hibberd ML. 2012. Genome-wide expression profiling identifies type 1 interferon response pathways in active tuberculosis. PLoS One 7:e45839. doi: 10.1371/journal.pone.0045839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andersen P, Doherty TM. 2005. The success and failure of BCG-implications for a novel tuberculosis vaccine. Nat Rev Microbiol 3:656–662. doi: 10.1038/nrmicro1211. [DOI] [PubMed] [Google Scholar]
- 18.de Souza Campos Fernandes RC, Medina-Acosta E. 2010. BCG-itis in two antiretroviral-treated HIV-infected infants. Int J STD AIDS 21:662–663. doi: 10.1258/ijsa.2010.010267. [DOI] [PubMed] [Google Scholar]
- 19.Alangari AA, Al-Zamil F, Al-Mazrou A, Al-Muhsen S, Boisson-Dupuis S, Awadallah S, Kambal A, Casanova JL. 2011. Treatment of disseminated mycobacterial infection with high-dose IFN-γ in a patient with IL-12Rβ1 deficiency. Clin Dev Immunol 2011:691956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jevtovic D, Salemovic D, Ranin J, Brmbolic B, Djurkovic-Djakovic O. 2009. The prognosis of pediatric AIDS in Serbia. Curr HIV Res 7:287–292. doi: 10.2174/157016209788347912. [DOI] [PubMed] [Google Scholar]
- 21.Sadeghi-Shabestari M, Rezaei N. 2009. Disseminated bacille Calmette-Guerin in Iranian children with severe combined immunodeficiency. Int J Infect Dis 13:e420–e423. doi: 10.1016/j.ijid.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 22.Trimble R, Atkins J, Quigg TC, Burns CC, Wallace GS, Thomas M, Mangla AT, Infante AJ, Centers for Disease Control and Prevention . 2014. Vaccine-associated paralytic poliomyelitis and BCG-osis in an immigrant child with severe combined immunodeficiency syndrome: Texas, 2013. MMWR Morb Mortal Wkly Rep 63:721–724. [PMC free article] [PubMed] [Google Scholar]
- 23.Singh S, Saraav I, Sharma S. 2014. Immunogenic potential of latency associated antigens against Mycobacterium tuberculosis. Vaccine 32:712–716. doi: 10.1016/j.vaccine.2013.11.065. [DOI] [PubMed] [Google Scholar]
- 24.Bertholet S, Ireton GC, Kahn M, Guderian J, Mohamath R, Stride N, Laughlin EM, Baldwin SL, Vedvick TS, Coler RN, Reed SG. 2008. Identification of human T cell antigens for the development of vaccines against Mycobacterium tuberculosis. J Immunol 181:7948–7957. doi: 10.4049/jimmunol.181.11.7948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guy B. 2007. The perfect mix: recent progress in adjuvant research. Nat Rev Microbiol 5:505–517. doi: 10.1038/nrmicro1681. [DOI] [PubMed] [Google Scholar]
- 26.Reed SG, Bertholet S, Coler RN, Friede M. 2009. New horizons in adjuvants for vaccine development. Trends Immunol 30:23–32. doi: 10.1016/j.it.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 27.Anderson RC, Fox CB, Dutill TS, Shaverdian N, Evers TL, Poshusta GR, Chesko J, Coler RN, Friede M, Reed SG, Vedvick TS. 2010. Physicochemical characterization and biological activity of synthetic TLR4 agonist formulations. Colloids Surf B Biointerfaces 75:123–132. doi: 10.1016/j.colsurfb.2009.08.022. [DOI] [PubMed] [Google Scholar]
- 28.Coler RN, Bertholet S, Moutaftsi M, Guderian JA, Windish HP, Baldwin SL, Laughlin EM, Duthie MS, Fox CB, Carter D, Friede M, Vedvick TS, Reed SG. 2011. Development and characterization of synthetic glucopyranosyl lipid adjuvant system as a vaccine adjuvant. PLoS One 6:e16333. doi: 10.1371/journal.pone.0016333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arias MA, Van Roey GA, Tregoning JS, Moutaftsi M, Coler RN, Windish HP, Reed SG, Carter D, Shattock RJ. 2012. Glucopyranosyl lipid adjuvant (GLA), a synthetic TLR4 agonist, promotes potent systemic and mucosal responses to intranasal immunization with HIVgp140. PLoS One 7:e41144. doi: 10.1371/journal.pone.0041144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lambert SL, Aslam S, Stillman E, MacPhail M, Nelson C, Ro B, Sweetwood R, Lei YM, Woo JC, Tang RS. 2015. A novel respiratory syncytial virus (RSV) F subunit vaccine adjuvanted with GLA-SE elicits robust protective TH1-type humoral and cellular immunity in rodent models. PLoS One 10:e0119509. doi: 10.1371/journal.pone.0119509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bertholet S, Goto Y, Carter L, Bhatia A, Howard RF, Carter D, Coler RN, Vedvick TS, Reed SG. 2009. Optimized subunit vaccine protects against experimental leishmaniasis. Vaccine 27:7036–7045. doi: 10.1016/j.vaccine.2009.09.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fox CB, Baldwin SL, Vedvick TS, Angov E, Reed SG. 2012. Effects on immunogenicity by formulations of emulsion-based adjuvants for malaria vaccines. Clin Vaccine Immunol 19:1633–1640. doi: 10.1128/CVI.00235-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duthie MS, Sampaio LH, Oliveira RM, Raman VS, O'Donnell J, Bailor HR, Ireton GC, Sousa AL, Stefani MM, Reed SG. 2013. Development and preclinical assessment of a 73 kDa chimeric fusion protein as a defined subunit vaccine for leprosy. Vaccine 31:813–819. doi: 10.1016/j.vaccine.2012.11.073. [DOI] [PubMed] [Google Scholar]
- 34.Baldwin SL, Bertholet S, Reese VA, Ching LK, Reed SG, Coler RN. 2012. The importance of adjuvant formulation in the development of a tuberculosis vaccine. J Immunol 188:2189–2197. doi: 10.4049/jimmunol.1102696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bertholet S, Ireton GC, Ordway DJ, Windish HP, Pine SO, Kahn M, Phan T, Orme IM, Vedvick TS, Baldwin SL, Coler RN, Reed SG. 2010. A defined tuberculosis vaccine candidate boosts BCG and protects against multidrug-resistant Mycobacterium tuberculosis. Sci Transl Med 2:53ra74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baldwin SL, Shaverdian N, Goto Y, Duthie MS, Raman VS, Evers T, Mompoint F, Vedvick TS, Bertholet S, Coler RN, Reed SG. 2009. Enhanced humoral and type 1 cellular immune responses with Fluzone adjuvanted with a synthetic TLR4 agonist formulated in an emulsion. Vaccine 27:5956–5963. doi: 10.1016/j.vaccine.2009.07.081. [DOI] [PubMed] [Google Scholar]
- 37.Clegg CH, Roque R, Perrone LA, Rininger JA, Bowen R, Reed SG. 2014. GLA-AF, an emulsion-free vaccine adjuvant for pandemic influenza. PLoS One 9:e88979. doi: 10.1371/journal.pone.0088979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clegg CH, Roque R, Van Hoeven N, Perrone L, Baldwin SL, Rininger JA, Bowen RA, Reed SG. 2012. Adjuvant solution for pandemic influenza vaccine production. Proc Natl Acad Sci U S A 109:17585–17590. doi: 10.1073/pnas.1207308109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shanley CA, Ireton GC, Baldwin SL, Coler RN, Reed SG, Basaraba RJ, Orme IM. 2014. Therapeutic vaccination against relevant high virulence clinical isolates of Mycobacterium tuberculosis. Tuberculosis (Edinb) 94:140–147. doi: 10.1016/j.tube.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karmakar S, Zhang W, Ahmad G, Torben W, Alam MU, Le L, Damian RT, Wolf RF, White GL, Carey DW, Carter D, Reed SG, Siddiqui AA. 2014. Cross-species protection: Schistosoma mansoni Sm-p80 vaccine confers protection against Schistosoma haematobium in hamsters and baboons. Vaccine 32:1296–1303. doi: 10.1016/j.vaccine.2013.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Coler RN, Bertholet S, Pine SO, Orr MT, Reese V, Windish HP, Davis C, Kahn M, Baldwin SL, Reed SG. 2013. Therapeutic immunization against Mycobacterium tuberculosis is an effective adjunct to antibiotic treatment. J Infect Dis 207:1242–1252. doi: 10.1093/infdis/jis425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Steenken W Jr, Gardner LU. 1946. History of H37 strain of tubercle bacillus. Am Rev Tuberc 54:62–66. [DOI] [PubMed] [Google Scholar]
- 43.Miyoshi-Akiyama T, Matsumura K, Iwai H, Funatogawa K, Kirikae T. 2012. Complete annotated genome sequence of Mycobacterium tuberculosis Erdman. J Bacteriol 194:2770. doi: 10.1128/JB.00353-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Orme IM. 2015. Tuberculosis vaccine types and timings. Clin Vaccine Immunol 22:249–257. doi: 10.1128/CVI.00718-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ho MM, Southern J, Kang HN, Knezevic I. 2010. WHO informal consultation on standardization and evaluation of BCG vaccines, Geneva, Switzerland, 22-23 September 2009. Vaccine 28:6945–6950. doi: 10.1016/j.vaccine.2010.07.086. [DOI] [PubMed] [Google Scholar]
- 46.Fruth U, Young D. 2004. Prospects for new TB vaccines: stop TB working group on TB vaccine development. Int J Tuberc Lung Dis 8:151–155. [PubMed] [Google Scholar]
- 47.Gopal R, Monin L, Slight S, Uche U, Blanchard E, Fallert Junecko BA, Ramos-Payan R, Stallings CL, Reinhart TA, Kolls JK, Kaushal D, Nagarajan U, Rangel-Moreno J, Khader SA. 2014. Unexpected role for IL-17 in protective immunity against hypervirulent Mycobacterium tuberculosis HN878 infection. PLoS Pathog 10:e1004099. doi: 10.1371/journal.ppat.1004099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Orme IM, Robinson RT, Cooper AM. 2015. The balance between protective and pathogenic immune responses in the TB-infected lung. Nat Immunol 16:57–63. [DOI] [PubMed] [Google Scholar]
- 49.Hunter RL. 2011. Pathology of post primary tuberculosis of the lung: an illustrated critical review. Tuberculosis (Edinb) 91:497–509. doi: 10.1016/j.tube.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun R, Skeiky YA, Izzo A, Dheenadhayalan V, Imam Z, Penn E, Stagliano K, Haddock S, Mueller S, Fulkerson J, Scanga C, Grover A, Derrick SC, Morris S, Hone DM, Horwitz MA, Kaufmann SH, Sadoff JC. 2009. Novel recombinant BCG expressing perfringolysin O and the overexpression of key immunodominant antigens: preclinical characterization, safety, and protection against challenge with Mycobacterium tuberculosis. Vaccine 27:4412–4423. doi: 10.1016/j.vaccine.2009.05.048. [DOI] [PubMed] [Google Scholar]
- 51.Chang HY, Mitzner W, Watson J. 2012. Variation in airway responsiveness of male C57BL/6 mice from 5 vendors. J Am Assoc Lab Anim Sci 51:401–406. [PMC free article] [PubMed] [Google Scholar]
- 52.Orr MT, Windish HP, Beebe EA, Argilla D, Huang PW, Reese VA, Reed SG, Coler RN. 2015. Interferon γand tumor necrosis factor are not essential parameters of CD4+ T-cell responses for vaccine control of tuberculosis. J Infect Dis 212:495–504. doi: 10.1093/infdis/jiv055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jeon BY, Derrick SC, Lim J, Kolibab K, Dheenadhayalan V, Yang AL, Kreiswirth B, Morris SL. 2008. Mycobacterium bovis BCG immunization induces protective immunity against nine different Mycobacterium tuberculosis strains in mice. Infect Immun 76:5173–5180. doi: 10.1128/IAI.00019-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McShane H, Williams A. 2014. A review of preclinical animal models utilised for TB vaccine evaluation in the context of recent human efficacy data. Tuberculosis (Edinb) 94:105–110. doi: 10.1016/j.tube.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Manca C, Reed MB, Freeman S, Mathema B, Kreiswirth B, Barry CE III, Kaplan G. 2004. Differential monocyte activation underlies strain-specific Mycobacterium tuberculosis pathogenesis. Infect Immun 72:5511–5514. doi: 10.1128/IAI.72.9.5511-5514.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bayry J, Tartour E, Tough DF. 2014. Targeting CCR4 as an emerging strategy for cancer therapy and vaccines. Trends Pharmacol Sci 35:163–165. doi: 10.1016/j.tips.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 57.Pease JE, Horuk R. 2014. Recent progress in the development of antagonists to the chemokine receptors CCR3 and CCR4. Expert Opin Drug Discov 9:467–483. doi: 10.1517/17460441.2014.897324. [DOI] [PubMed] [Google Scholar]
- 58.Bayry J, Tchilian EZ, Davies MN, Forbes EK, Draper SJ, Kaveri SV, Hill AV, Kazatchkine MD, Beverley PC, Flower DR, Tough DF. 2008. In silico identified CCR4 antagonists target regulatory T cells and exert adjuvant activity in vaccination. Proc Natl Acad Sci U S A 105:10221–10226. doi: 10.1073/pnas.0803453105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Falagas ME, Kouranos VD, Athanassa Z, Kopterides P. 2010. Tuberculosis and malignancy. QJM 103:461–487. doi: 10.1093/qjmed/hcq068. [DOI] [PubMed] [Google Scholar]
- 60.Nalbandian A, Yan BS, Pichugin A, Bronson RT, Kramnik I. 2009. Lung carcinogenesis induced by chronic tuberculosis infection: the experimental model and genetic control. Oncogene 28:1928–1938. doi: 10.1038/onc.2009.32. [DOI] [PubMed] [Google Scholar]
- 61.Pichugin AV, Yan BS, Sloutsky A, Kobzik L, Kramnik I. 2009. Dominant role of the sst1 locus in pathogenesis of necrotizing lung granulomas during chronic tuberculosis infection and reactivation in genetically resistant hosts. Am J Pathol 174:2190–2201. doi: 10.2353/ajpath.2009.081075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Medina E, North RJ. 1998. Resistance ranking of some common inbred mouse strains to Mycobacterium tuberculosis and relationship to major histocompatibility complex haplotype and Nramp1 genotype. Immunology 93:270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.