

Abstract
Objective
How hexanucleotide (GGGGCC) repeat expansions in C9ORF72 cause amyotrophic lateral sclerosis (ALS) remains poorly understood. Both gain‐ and loss‐of‐function mechanisms have been proposed. Evidence supporting these mechanisms in vivo is, however, incomplete. Here we determined the effect of C9orf72 loss‐of‐function in mice.
Methods
We generated and analyzed a conditional C9orf72 knockout mouse model. C9orf72fl/fl mice were crossed with Nestin‐Cre mice to selectively remove C9orf72 from neurons and glial cells. Immunohistochemistry was performed to study motor neurons and neuromuscular integrity, as well as several pathological hallmarks of ALS, such as gliosis and TDP‐43 mislocalization. In addition, motor function and survival were assessed.
Results
Neural‐specific ablation of C9orf72 in conditional C9orf72 knockout mice resulted in significantly reduced body weight but did not induce motor neuron degeneration, defects in motor function, or altered survival.
Interpretation
Our data suggest that C9orf72 loss‐of‐function, by itself, is insufficient to cause motor neuron disease. These results may have important implications for the development of therapeutic strategies for C9orf72‐associated ALS. Ann Neurol 2015;78:426–438
A hexanucleotide repeat expansion (GGGGCC) in a noncoding region of C9ORF72 is the most common genetic cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD; C9ALS/FTD).1, 2, 3 Three possible pathogenic mechanisms have been linked to C9ORF72 repeat expansions: sequestration of RNA‐binding proteins, toxicity mediated by dipeptides formed as a result of repeat‐associated non‐ATG translation, and haploinsufficiency.1, 2, 4, 5, 6 Experimental evidence supporting each of these mechanisms is accumulating.7 However, how these mechanisms cause ALS/FTD and their relevance in vivo remain poorly understood.
The function of C9ORF72 is not known, but it has been suggested to play a role in protein trafficking.8, 9 Several observations identify C9ORF72 haploinsufficiency as a hallmark of C9ALS/FTD. First, different studies report decreased C9ORF72 mRNA expression in brain tissue, lymphoblast cells, and induced pluripotent stem cell–derived neurons of patients.1, 10, 11 Second, one study shows decreased C9ORF72 protein expression in frontal cortex of C9ALS/FTD patients.12 Third, knockdown of C9orf72 in model organisms such as Caenorhabditis elegans and zebrafish embryos causes motor deficits.10, 13 However, knockdown of C9orf72 caused by a single intracerebroventricular injection of antisense oligonucleotides (ASOs) in mice does not affect motor function or anxiety.14
To test the haploinsufficiency model and to determine whether lack of C9orf72 expression leads to motor neuron degeneration or abnormal motor function, we generated and analyzed a conditional C9orf72 knockout mouse model.
Materials and Methods
Mouse Husbandry, Breeding, and Genotyping
All animal use and care were in accordance with local institution guidelines. Mice were kept on a 12‐hour light/dark cycle with food and water available ad libitum. B6;SJL‐Tg(ACTFLPe)9205Dym/J mice and B6.Cg‐Tg(Nes‐cre)1Kln/J mice were obtained from Jackson Laboratory (Bar Harbor, ME; 003800; 003771) and C57Bl/6J mice from Charles River Laboratories (Wilmington, MA). To generate C9orf72loxP mice, a targeting construct was designed to insert an Frt‐flanked neomycin cassette and 1 loxP site upstream of exon 4 and 1 loxP site downstream of exon 5 of C9orf72. Deletion of exons 4 and 5 targets all reported C9orf72 isoforms in the mouse. This construct was electroporated into C57Bl/6 embryonic stem cells. Correctly targeted stem cells, as determined by polymerase chain reaction (PCR) and Southern blot analysis, were injected into blastocysts, and chimeric mice were bred with C57Bl/6J mice. The resulting C9orf72loxP‐neo mice were then bred with mice expressing Flp recombinase in their germline to remove the Frt‐flanked neomycin cassette, generating C9orf72loxP/+ offspring. Female C9orf72loxP/loxP or C9orf72loxP/+ mice were crossed with male Nestin‐Cre+/−;C9orf72loxP mice to generate neural‐specific C9orf72 conditional knockout mice. Mice were genotyped using primers to detect the Cre gene (forward = 5′‐GCGGTCTGGCAGTAAAAACTATC‐3′, reverse = 5‐′GTGAAACAGCATTGCTGTCACTT‐3′) and the genomic region containing the loxP sequences (forward = 5′‐CCACGGAGGGATGTTCTTTA‐3′, reverse = 5′‐GAAACCAGACCCAAACACAGA‐3′).
Antibody Generation
The anti‐C9orf72 rabbit polyclonal antibody C9‐2034 was generated from an N‐terminal thioredoxin fusion of a stretch of 58 amino acids present in all human C9orf72 isoforms comprising MEDQGQSIIPMLTGEVIPVMELLSSMKSHSVPEEIDIADTVLNDDDIGDSCHEGFLLK. This was generated from a C9orf72 short isoform expression construct using the primers 5′‐CCCGAATTCGAGAGAATGGAAGATCAGGGT‐3′ and 5′‐GAAGCGGCCGCATCTGCTTCATCCAGCTTTTATGA‐3′. The PCR product was digested and cloned into the EcoRI/NotI sites of pET32a (Clontech Laboratories, Mountain View, CA). Thioredoxin (Thx)‐C9orf72‐tail expression was induced in transformed BL21(DE3) Escherichia coli (Agilent Technologies, Santa Clara, CA) using 1mM Isopropyl β‐D‐1‐thiogalactopyranoside (IPTG) for 3 hours at 37°C. Thx‐C9orf72‐Short was purified using HisPur resin according to the manufacturer's instructions (Thermo Fisher Scientific, Waltham, MA) following solubilization in sonication buffer (20mM Tris pH 8.0, 100mM NaCl) using a Vibra‐Cell Ultrasonic Processor (Sonics & Materials, Newtown, CT). The anti‐C9orf72 rabbit polyclonal antibody C9‐2074 was generated by reimmunization of the previously described N‐terminal thioredoxin fusion of full‐length C9orf72 short isoform.12 The respective purified fusion proteins were used as antigens for custom rabbit polyclonal antibody generation (Covalab, Villeurbanne, France). Custom antisera were immunoaffinity‐purified against their antigen following preabsorption against Thx and glial fibrillary acidic protein (Thx‐GFAP expressed in BL21[DE3] transformed with GFAP pET32a). Both antibodies detected a 50 to 55kDa protein in mouse and human brain lysates, as well as myc‐tagged C9orf72 from transfected HEK293T cells.
The anti‐C9orf72 rabbit polyclonal antibodies were validated for Western blotting using siRNA‐mediated knockdown of endogenous C9orf72 in HEK293T cells using 27‐mer oligonucleotide duplexes as previously described.12 Additional validation in SH‐SY5Y neuroblastoma cells was performed following knockdown of endogenous C9orf72 using Dicer substrate RNAi interference using custom dicer substrate siRNA (DsiRNA; Integrated DNA Technologies, Coralville, IA). Custom DsiRNA oligonucleotide duplex sequences are C9ORF72‐DsiRNA1F 5‐GGAAAGAAUAUGGAUGCAUAAGGAAA‐3′/C9ORF72‐DsiRNA1R 5′‐UUUCCUUAUGCAUCCAUAUUCUUCCUU‐3′ and C9ORF72‐DsiRNA2F 5′‐GUACUCAAUGAUGAUGAUAUUGGTG‐3′/C9ORF72‐DsiRNA2R 5′‐CACCAAUAUCAUCAUCAUUGAGUACUG‐3′. The inventoried FLuc‐S1 exogenous reporter gene DsiRNA duplex was used as a nontargeting control. DsiRNA was transfected using the same conditions as 27mer oligonucleotide duplexes. Whole cell and tissue lysate preparation and Western blotting were performed as described previously.12 Western blotting using the anti‐C9orf72 rabbit polyclonal antibodies detected no C9orf72 expression in knockout mice (Fig 1) or reduced expression following knockdown of C9orf72 (in HEK293 or SHSY5Y cells; data not shown).
Figure 1.

Expression of C9orf72 and generation of C9orf72 conditional knockout mice. (A–G) In situ hybridization shows expression of C9orf72 in coronal sections of the adult brain and spinal cord. (F, G) Double staining combining in situ hybridization for C9orf72 and immunohistochemistry for SMI132, a marker for motor neurons (indicated by arrowheads). (E, M) Sections incubated with sense probes do not display specific staining. (H–O) At embryonic and early postnatal stages, C9orf72 is expressed in spinal cord motor neurons (arrowheads in H–J), the retina, olfactory system, and other brain regions, in addition to non‐neuronal tissues such as kidney. (P) Schematic representation of the C9orf72 targeting vector, targeted allele, floxed allele following Flp recombination, and deleted allele following Cre recombination. Black arrows indicate transcription start site, gray arrows indicate location of loxP primers (P1, P2), and numbers indicate exons. Please note that exons 4 and 5 are shared by all 3 reported mouse C9orf72 isoforms. (Q) Genotyping using loxP primers on DNA isolated from ear and brain tissue of Nestin‐Cre+/−;C9orf72+/+ and Nestin‐Cre+/−;C9orf72fl/fl mice shows brain‐specific recombination. (R) Quantitative real‐time polymerase chain reaction shows tissue‐specific reduction of C9orf72 levels in Nestin‐Cre+/−;C9orf72fl/+ and/or Nestin‐Cre+/−;C9orf72fl/fl in brain (0.493, p = 0.0004 and 0.044, p = 0.00007) and spinal cord (0.684, p = 0.083 and 0.145, p = 0.002), but not in muscle (0.789, p = 0.594 and 0.585, p = 0.169) or liver (1.94, p = 0.219 and 1.74, p = 0.473). Results are shown with Tbp as a housekeeping gene. Data are presented as mean ± standard deviation, n = 3 for each genotype. (S) In situ hybridization for C9orf72 shows loss of C9orf72 expression in spinal cord of Nestin‐Cre+/−;C9orf72fl/fl mice. **p < 0.01, ***p < 0.001. CA1 = cornu ammonis 1; Cx = cortex; DG = dentate gyrus; DRG = dorsal root ganglia; F = Frt site; GL = granular cell layer; GM = gray matter; L = loxP site; Mol = molecular layer; N = neomycin cassette; OE = olfactory epithelium; P1 = forward loxP primer; P2 = reverse loxP primer; Ret = retina; RN = red nucleus; VNO = vomeronasal organ; WM = white matter; WT = wild type. Scale bars: A, 200μm; H, 100μm; K, 100μm, S, 150μm.
In Situ Hybridization
Fresh frozen mouse embryos, or spinal cord and brain tissue from adult mice were cut at 16 to 20µm on a cryostat. Nonradioactive in situ hybridization was performed as described previously.15 A digoxigenin‐labeled C9orf72 probe was transcribed from mouse cDNA (a 736bp fragment corresponding to nucleotides 298–1,034 of 3110043O21Rik) using a DIG RNA labeling SP6/T7 kit (Roche, Basel, Switzerland). Sense probes were used to confirm specificity. Images were taken on a Zeiss (Oberkochen, Germany) AxioSkop2 microscope.
Quantitative Real‐Time PCR
Fresh frozen tissue was homogenized in TRIzol reagent and total RNA was isolated. After DNAse treatment, cDNA was synthesized using superscript II reverse transcriptase (Life Technologies, Carlsbad, CA) and oligo‐dT primers. Quantitative real‐time PCR was performed using SYBR green reagent (Roche) on a QuantStudio 6 Flex real‐time PCR system (Applied Biosystems, Foster City, CA) using primers against 3110043O21Rik (Q1 [exon 2]: forward = 5′‐GACCTGGAATGCAGTGAGAC‐3′, reverse = 5′‐TCCCAGTAAGCAAAGGTAGC; Q2 [exons 3–6]: forward = 5′‐GGCACAGAGAGGATGGAAGA‐3′, reverse = 5′‐CTGCCAACTACAACGGAACA‐3′; Q3 [exons 6–8]: forward = 5′‐TGGCTGTTCCGTTGTAGTTG‐3′, reverse = 5′‐AACTGCCTGTTGCATCCTTT‐3′; Q4 [exons 7–8]: FW = 5′‐GTACGAATCGGGACTCTTTG‐3′, reverse = 5′‐GCTTGACAGTGTTGACATCC‐3′; Q5 [exons 8–9]: forward = 5′‐CAACGCAGATACATGAGGTC‐3′, reverse = 5′‐GGAAGGCTTTCACTAGAGTGTC‐3′; Q6 [exons 10–11]: forward = 5′‐CCCTTTAAGTCTCTTCGGAACC‐3′, reverse = 5′‐AGACAGACACAGCAAACCAC‐3′) and mRNA levels were normalized to Gapdh (forward = 5′‐GAGACGGCCGCATCTTCTTGT‐3′, reverse = 5‐′ CACACCGACCTTCACCATTTT‐3′) and Tbp (QT00198443; Qiagen, Valencia, CA) housekeeping genes. Samples from 3 mice per genotype were amplified in triplicate, and relative quantification using comparative ΔΔCT values was statically analyzed using Student t test.
Western Blotting
Western blotting was performed as described previously.16 In brief, fresh frozen tissue was homogenized in radioimmunoprecipitation assay buffer (50mM Tris pH 7.6, 150mM NaCl, 0.5mM ethylenediaminetetraacetic acid, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 1% Triton‐X‐100, and protease inhibitor cocktail), centrifuged twice at 10,000 × g, and the supernatant was used as total protein. Western blotting was performed using 50µg of total protein per lane as measured by bicinchoninic acid assay (Pierce Biotechnology, Rockford, IL), and blots were probed with the following antibodies: anti‐C9orf72 (2034; 1:250), anti‐C9orf72 (2074; 1:250), and anti–β‐actin (Sigma, St Louis, MO; 1:2,000). After overnight incubation, blots were probed with horseradish peroxidase–conjugated secondary antibodies and developed with SuperSignal West Femto chemiluminescent substrate (Pierce Biotechnology).
Motor Function Testing
Mice were weighed and tested for their hindlimb extension reflex weekly. Every 3 weeks, mice performed 5 trials on an accelerating rotarod (Ugo Basile, Varese, Italy) at 4rpm with an acceleration of 0.2rpm/s for 180 seconds and the latency to fall was recorded. Mice that rotated passively were scored as fallen. Grip strength of front paws and of front and hind paws together was measured every 3 months using a grip strength meter (Bioseb, Vitrolles, France). Strength was measured 5 times for each mouse both for front and for front and hind paws. All behavioral tests were performed on ≥11 mice per genotype for each time point. Statistical analysis was carried out using R software (http://www.r‐project.org). (Non‐)linear mixed models were used to account for repeated measurements and other sources of correlation within the longitudinal data and to provide a correct model in case of missing data. For statistical analysis of body weight, a nonlinear mixed effects model was used with gender as a covariate. Interactions between time and behavioral tests were analyzed to study possible changes during follow‐up. For rotarod and grip strength statistical analysis, a nonlinear mixed effects model was used with gender and body weight as covariates. Survival analysis was carried out using a Cox regression analysis with mean body weight as covariate. Cox proportional hazard assumptions were tested.
Immunohistochemistry
Immunohistochemistry was performed as described previously.17 Tissues were removed and cryoprotected in 30% sucrose after transcardial perfusion with 4% paraformaldehyde. Spinal cord and brain were cut at 20µm and mounted on SuperFrost Plus slides (Thermo Fisher Scientific). For immunofluorescent stainings, sections were blocked and incubated overnight at 4°C with the following antibodies: anti‐GFAP (Dako, Carpinteria, CA), anti‐Iba1 (Wako Chemicals, Richmond, VA), anti–choline acetyltransferase (ChAT; Millipore, Billerica, MA), and anti–TDP‐43 (Proteintech, Chicago, IL). Alexa Fluor–conjugated secondary antibodies were incubated for 2 hours at room temperature (RT) and 4′,6‐diamidino‐2‐phenylindole was used to stain nuclei. For diaminobenzidine (DAB) stainings, sections were incubated overnight at 4°C with anti‐ubiquitin antibodies (Millipore) and for 1 h at RT with goat anti‐mouse biotin. Slides were incubated with ABC reagent (Vector Laboratories, Burlingame, CA) for 1 h and developed with DAB reagent (Sigma). For motor neuron counting, at least 10 spinal cord sections (L1–L6) per mouse using 3 mice per genotype were analyzed and only ChAT‐positive neurons with a clear nucleus were counted and measured. For GFAP and Iba1 quantification, at least 10 spinal cord sections (L1–L6) from 3 mice per genotype were analyzed for positive staining area using ImageJ (NIH, Bethesda, MD). For neuromuscular junction staining, gastrocnemius muscle was cut longitudinally at 40µm and stained with anti–α‐bungarotoxin‐488 (Life Technologies), anti‐sv2 (Developmental Studies Hybridoma Bank [DSHB], Iowa City, IA), and anti‐neurofilament (DSHB) antibodies. At least 150 neuromuscular junctions were counted per mouse. Images were taken on an Olympus (Tokyo, Japan) FluoView FV1000 confocal microscope or a Zeiss AxioScope microscope followed by image analysis using ImageJ. Statistical analysis was performed using Student t test (SPSS Statistics; IBM, Armonk, NY).
Results
Genetic Ablation of C9orf72 in Mice
Previous work analyzing C9orf72‐LacZ knockin mice reported C9orf72 expression in neurons known to degenerate in ALS and FTD, such as cortical and spinal cord motor neurons.18 To confirm and extend these findings, we performed in situ hybridization for the mouse C9orf72 orthologue, 3110043O21Rik (here referred to as C9orf72), at embryonic, postnatal, and adult stages. As reported previously, neuronal C9orf72 expression was detected in several regions of the adult brain and spinal cord (see Fig 1; Table1).18 In contrast to previous work, however, prominent expression of C9orf72 was also observed in embryonic and early postnatal neurons. These included retinal ganglion cells, sensory neurons in the olfactory epithelium and in dorsal root ganglia, and spinal motor neurons. In addition, expression in non‐neuronal tissues such as kidney and tooth was detected. These results indicate that C9orf72 may also function during embryonic and postnatal development.
Table 1.
Overview of C9orf72Expression in the Developing Mouse Embryo and Adult Central Nervous System
| Location | E13.5 | E15.5 | E17.5 | P1 | P15 | Adult |
|---|---|---|---|---|---|---|
| Brain | ||||||
| Cortex | + | + | + | + | ++ | + |
| Hippocampal formation | + | + | ++ | ++ | ++ | ++ |
| Dentate gyrus | N/D | N/D | ++ | ++ | ++ | ++ |
| Cornu ammonis | N/D | N/D | ++ | ++ | ++ | ++ |
| Thalamus | N/D | N/D | N/D | + | ++ | + |
| Midbrain/pons | N/D | + | + | + | ++ | ++ |
| Ventral tegmental area | N/D | N/D | N/D | − | − | − |
| Pontine nuclei | N/D | N/D | N/D | + | ++ | ++ |
| Red nucleus | N/D | N/D | N/D | + | ++ | ++ |
| Facial motor nucleus | N/D | N/D | N/D | + | ++ | ++ |
| Cerebellum | + | + | + | + | + | + |
| Molecular layer | N/D | N/D | N/D | + | + | + |
| Granule cell layer | N/D | N/D | N/D | + | + | + |
| Spinal cord | ||||||
| Gray matter | ++ | ++ | ++ | ++ | ++ | ++ |
| White matter | − | − | − | − | − | − |
| Dorsal root ganglia | ++ | ++ | ++ | ++ | ++ | ++ |
| Other | ||||||
| Eye | ++ | ++ | ++ | ++ | N/D | N/D |
| Olfactory epithelium | ++ | ++ | ++ | ++ | N/D | N/D |
| Tooth | ++ | ++ | ++ | ++ | N/D | N/D |
| Kidney | + | + | + | + | N/D | N/D |
N/D = not determined, − = no expression, + = weak expression, ++ = moderate/strong expression.
To circumvent problems such as embryonic lethality, we generated conditional C9orf72 knockout mice using the Cre‐loxP system and bred the resulting C9orf72fl/fl mice to Nestin‐Cre mice (see Fig 1). In Nestin‐Cre mice, Cre recombination occurs in neuronal and glial precursors from E10.5 onward.19 The specific ablation of C9orf72 mRNA and protein in Nestin‐Cre+/−; C9orf72fl/fl mice was confirmed using different approaches. First, DNA genotyping on ear clips or brain tissue showed excision of exons 4 and 5 only in brain tissue. Second, quantitative real‐time PCR detected a reduction in C9orf72 mRNA levels in brain and spinal cord tissue but not in other tissues of Nestin‐Cre+/−;C9orf72fl/fl mice. Third, in situ hybridization detected C9orf72 in the spinal cord of Nestin‐Cre+/−; C9orf72+/+ but not in Nestin‐Cre+/−;C9orf72fl/fl mice.
Three C9orf72 isoforms have been reported in mice. These isoforms all contain exons 4 and 5 and are targeted in the C9orf72 conditional knockout mouse (Fig 2). To confirm that the expression of all isoforms is affected by the excision of exons 4 and 5, we performed quantitative real‐time PCR to detect expression of different exonic regions present in 1 or multiple isoforms. As expected, a reduction of C9orf72 mRNA levels was found for all exonic regions analyzed. In addition, we generated a new anti‐C9orf72 antibody. This antibody was raised against a 58 amino acid stretch shared by all isoforms (C9‐2034). The other antibody used was reported previously and was raised using a large part of the C9orf72 protein. The region used is present in 2 of the 3 isoforms (C9‐2074).12 Western blotting using these antibodies showed a predicted ∼55kDa band, corresponding to isoform 1, that was lost in brain tissue of Nestin‐Cre+/−;C9orf72fl/fl mice. The other 2 C9orf72 isoforms (isoforms 2 and 3) were not detected in wild‐type mouse brain tissue (data not shown). Together, these results confirm the neural‐specific and successful ablation of C9orf72 in Nestin‐Cre+/−;C9orf72fl/fl mice.
Figure 2.
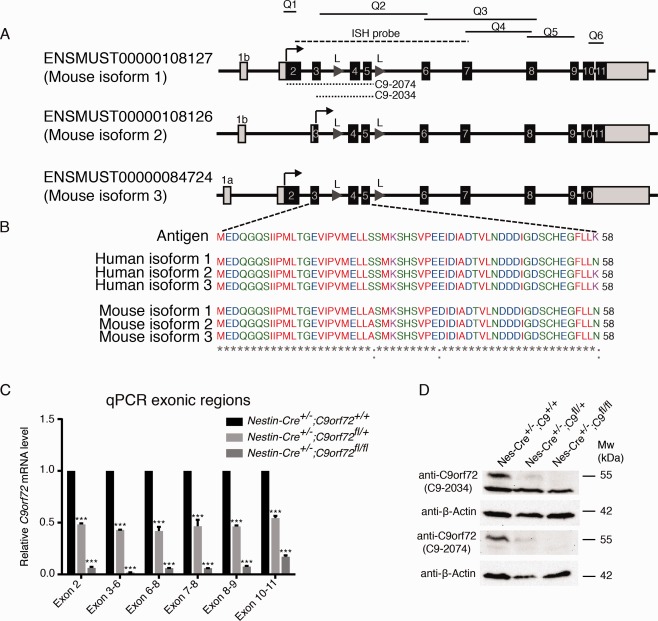
Cre‐mediated excision of C9orf72 affects all C9orf72 isoforms. (A) Schematic overview of the 3 reported mouse C9orf72 isoforms. Q1 to Q6 indicate regions amplified by quantitative real‐time polymerase chain reaction (qPCR). The in situ probes used are indicated. Dotted lines indicate antigen regions used to generate the 2 anti‐C9orf72 antibodies (C9‐2034 and C9‐2074). (B) Amino acid sequence of the antigen used to generate the novel anti‐C9orf72 antibody C9‐2034, and its comparison to human C9ORF72 isoforms and mouse C9orf72 isoforms. (C) qPCR using primers that target different exonic regions shared by 1 or multiple mouse isoforms shows a significant reduction of brain C9orf72 mRNA levels in Nestin‐Cre+/−;C9orf72fl/+ and/or Nestin‐Cre+/−;C9orf72fl/fl mice. Results are shown using Tbp as a housekeeping gene. Data are presented as mean ± standard deviation, n = 2 for each genotype. (D) Western blot analysis of whole brain lysates using 2 different anti‐C9orf72 antibodies shows a ∼55kDa band in control mice that is reduced or absent in Nestin‐Cre+/−;C9orf72fl/+ or Nestin‐Cre+/−;C9orf72fl/fl mice, respectively. Note that only isoform 1 is detected at the protein level in mouse brain. β‐Actin was used as a loading control. ***p < 0.001. L = loxP sites.
Mice Lacking C9orf72 in Neurons and Glial Cells Do Not Display Motor Neuron Degeneration or Defects in Motor Function
ALS is characterized by progressive motor neuron degeneration and a decline in motor performance. To examine a possible role for C9orf72 haploinsufficiency in ALS pathogenesis, we therefore assessed motor function, motor neuron number, and neuromuscular integrity in 12‐month‐old (data not shown) and 18‐month‐old Nestin‐Cre+/−;C9orf72fl/fl mice and littermate controls. Nestin‐Cre+/−;C9orf72fl/fl mice were born at the expected Mendelian ratio and were indistinguishable from Nestin‐Cre+/−;C9orf72+/+ control littermates in viability, appearance, fertility, and the gross anatomical and histological appearance of the brain. Immunohistochemistry for ChAT, to visualize spinal motor neurons, did not reveal changes in neuron number or size (Fig 3A, B). Furthermore, innervation of the gastrocnemius muscle by motor axons, as assessed by overlap between the pre‐ and postsynaptic compartment of the neuromuscular junction, was similar in Nestin‐Cre+/−;C9orf72fl/fl mice and controls (see Fig 3C, D). Astro‐ and microgliosis are hallmarks of ALS. However, no differences in immunostaining for GFAP or Iba1, to label astrocytes or microglia, respectively, were detected between spinal cord or brain tissue from Nestin‐Cre+/−;C9orf72fl/fl mice and littermate controls (Figs 4A–D, 5A–F″, 6A–F″). Similarly, other hallmarks of ALS pathogenesis, such as TDP‐43 mislocalization or changes in ubiquitin staining, were absent in Nestin‐Cre+/−;C9orf72fl/fl mice (see Figs 4E–H, 7). Similar results were obtained in 12‐month‐old Nestin‐Cre+/−;C9orf72fl/fl mice, that is, absence of defects in motor neuron number, size, and connectivity and no signs of gliosis or other pathological hallmarks of ALS (data not shown). Thus, C9orf72 deficiency in mice fails to cause several of the pathological hallmarks reported in ALS patients or mouse models.
Figure 3.
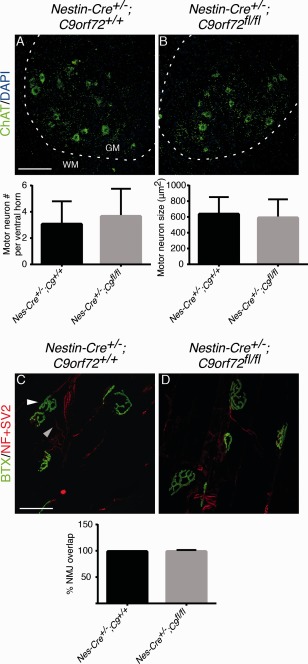
Neural‐specific ablation of C9orf72 does not cause changes in motor neuron number, size, or neuromuscular junction (NMJ) connectivity in 18‐month‐old Nestin‐Cre+/−;C9orf72fl/fl mice. (A, B) Immunofluorescent staining of adult spinal cord using anti–choline acetyltransferase (ChAT) antibodies. Graphs show quantification of motor neuron number and size. Dotted lines indicate border between gray matter (GM) and white matter (WM). (C, D) Representative images of NMJs in gastrocnemius muscle visualized by immunohistochemistry for neurofilament (NF) and synaptic vesicle protein 2 (SV2) in combination with Alexa Fluor 488–conjugated bungarotoxin (BTX) binding. Graph shows overlap between presynaptic (gray arrowhead) and postsynaptic staining (white arrowhead). Motor neurons and their NMJs are intact in Nestin‐Cre+/−;C9orf72fl/fl mice. Data in graphs represent mean ± standard deviation, n = 3 per genotype. Scale bars: A, 100μm; C, 30μm. DAPI = 4′,6‐diamidino‐2‐phenylindole.
Figure 4.
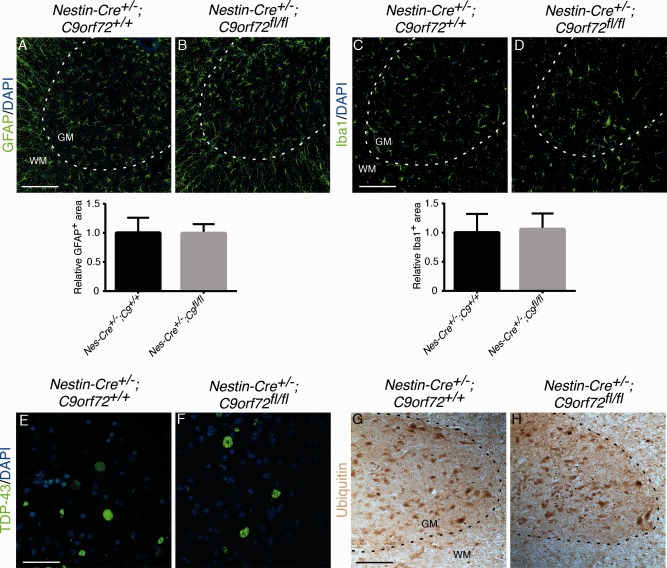
Neural‐specific ablation of C9orf72 does not cause signs of neuroinflammation or other pathological hallmarks of amyotrophic lateral sclerosis in 18‐month‐old Nestin‐Cre+/−;C9orf72fl/fl mice. (A–D) Immunofluorescent staining of adult spinal cord using anti–glial fibrillary acidic protein (GFAP) or anti‐Iba1 antibodies. Graphs show quantification of GFAP‐ or Iba1‐positive areas. No signs of gliosis are observed in Nestin‐Cre+/−;C9orf72fl/fl mice. (E, F) Immunofluorescent staining of adult spinal cord using anti–TDP‐43 antibodies. Nuclear TDP‐43 staining is detected in Nestin‐Cre+/−;C9orf72fl/fl mice and control littermates. (G, H) Representative images showing immunostaining for ubiquitin in adult spinal cord. Ubiquitin expression is not affected by loss of C9orf72. Data in graphs represent mean ± standard deviation, n = 3 per genotype. Scale bars: A, C, G, 100μm; E, 30μm. DAPI = 4′,6‐diamidino‐2‐phenylindole; GM = gray matter; WM = white matter.
Figure 5.
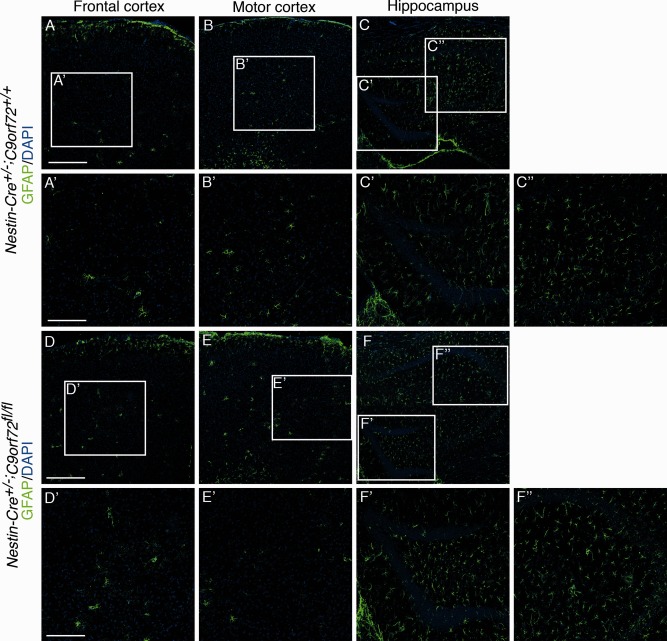
Immunostaining for glial fibrillary acidic protein (GFAP) in the brain of 18‐month‐old mice. Immunofluorescent staining is shown for GFAP in frontal cortex, motor cortex, and hippocampus of Nestin‐Cre+/−;C9orf72+/+ mice (A–C″) and Nestin‐Cre+/−;C9orf72fl/fl mice (D–F″). Boxes in A–C and D–F are shown at higher magnification in A′–C″ and D′–F″, respectively. No changes in the distribution, number, or appearance of GFAP‐positive astrocytes were detected in Nestin‐Cre+/−;C9orf72fl/fl mice. Scale bars: A, D, 200μm; A′, D′, 100μm. DAPI = 4′,6‐diamidino‐2‐phenylindole.
Figure 7.

Immunostaining for TDP‐43 and ubiquitin in several brain regions of 18‐month‐old mice. (A–C, G–I) Immunofluorescent staining of adult spinal cord using anti–TDP‐43 antibodies is shown. Nuclear TDP‐43 staining is detected in Nestin‐Cre+/−; C9orf72fl/fl mice and control littermates. (D–F, J–L) Representative images show immunostaining for ubiquitin in adult spinal cord. Ubiquitin expression is not affected by loss of C9orf72. DAPI = 4′,6‐diamidino‐2‐phenylindole; GCL = granule cell layer; H = hilus. Scale bars = 60μm.
Figure 6.

Immunostaining for Iba1 in the brain of 18‐month‐old mice. Immunofluorescent staining is shown for Iba1 in frontal cortex, motor cortex, and hippocampus of Nestin‐Cre+/−;C9orf72+/+ mice (A–C″) and Nestin‐Cre+/−;C9orf72fl/fl mice (D–F″). Boxes in A–C and D–F are shown at higher magnification in A′–C″ and D′–F″, respectively. No changes in the distribution, number, or appearance of Iba1‐positive microglia are detected in Nestin‐Cre+/−;C9orf72fl/fl mice. Scale bars: A,D, 200μm; A′, D′, 100μm. DAPI = 4′,6‐diamidino‐2‐phenylindole.
A reduction in body weight was detected in all Nestin‐Cre–positive mice, as has been previously reported for other studies using this Cre driver.20 However, even when considering this effect, Nestin‐Cre+/−;C9orf72fl/fl mice displayed a small but significant decrease in body weight, as compared to Nestin‐Cre+/−;C9orf72+/+ control mice (−6.02%, p = 0.019). This difference was detected at the first time point of measurements and did not significantly change in time (Fig 8). To assess motor performance, the hindlimb extension reflex was tested (data not shown) and mice were assessed using an accelerating rotarod and a grip strength meter for a period of 18 months after birth. No differences in motor performance or grip strength were found between Nestin‐Cre+/−; C9orf72fl/fl mice and various control mice. A similar result was obtained in a smaller cohort of 2‐year‐old mice (n ≥ 4; data not shown). Finally, survival was not significantly altered in Nestin‐Cre+/−;C9orf72fl/fl mice, the oldest mice living >24 months. Thus, Nestin‐Cre+/−;C9orf72fl/fl mice do not display overt defects in motor function or decreased survival.
Figure 8.
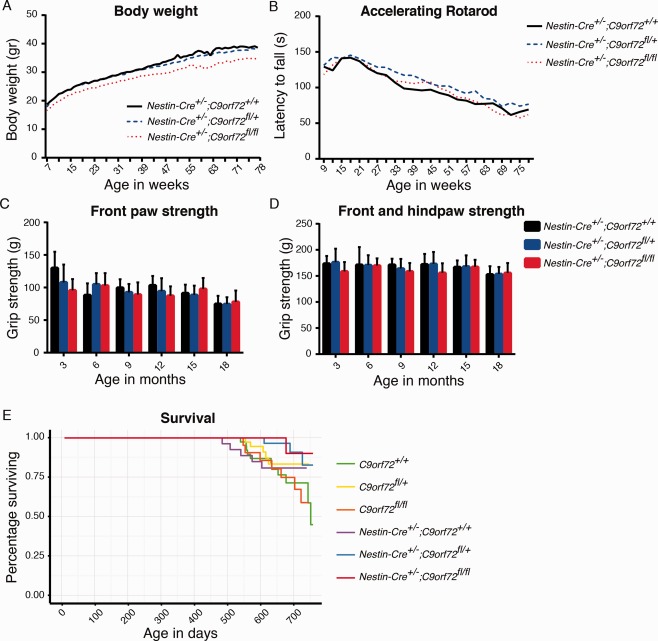
Loss of C9orf72 in mice does not affect motor function and survival. (A) A small decrease in body weight is detected in Nestin‐Cre+/−;C9orf72fl/fl mice as compared to Nestin‐Cre+/−;C9orf72+/+ controls (−6.02%, p = 0.019, linear mixed‐effect model for repeated measurements [LME]; n ≥ 11 for each genotype at each time point). (B–D) Rotarod performance and grip strength is similar in Nestin‐Cre+/−;C9orf72+/+, Nestin‐Cre+/−;C9orf72fl/+, and Nestin‐Cre+/−;C9orf72fl/fl mice (rotarod, p = 0.084 and p = 0.899; grip strength front, p = 0.346 and p = 0.295; front and hind, p = 0.731 and p = 0.305, LME). Data are shown as mean ± standard deviation, n ≥ 11 for each genotype at each time point. (E) Survival rates as shown by a Kaplan–Meier curve. Survival of Nestin‐Cre+/−;C9orf72fl/fl mice is similar to control mice.
Discussion
C9orf72 haploinsufficiency may contribute to the pathogenic mechanism in C9ALS/FTD patients. To test this hypothesis, we generated a conditional C9orf72 knockout mouse model. Two previous studies have reported the generation of C9orf72 knockout mice, but neither study provided a histological or behavioral characterization.18, 21 In one of these studies, using C9orf72‐LacZ knockin reporter mice, C9orf72 expression was not detected at embryonic and early postnatal stages in the mouse.18 In contrast, we show neuronal C9orf72 expression at several sites in the embryonic nervous system, as well as in non‐neuronal tissues. This observation is in line with a recent study showing expression of C9orf72 both inside and outside the nervous system of zebrafish embryos.10 The cause of this apparent discrepancy is unknown but may include the inability of heterozygous LacZ reporter mice to detect low levels of gene expression.22 Together, our data suggest that C9orf72 plays a role during embryonic and postnatal development.
The expression of C9orf72 at embryonic stages and in several tissues outside the nervous system prompted us to selectively ablate C9orf72 in neurons and glial cells by crossing C9orf72fl/fl mice with Nestin‐Cre mice. The resulting Nestin‐Cre+/−;C9orf72fl/fl mice did not show overt defects in motor function or pathological hallmarks of ALS, such as reduced motor neuron number, gliosis, TDP‐43 mislocalization, or enhanced ubiquitination. A small but significant decrease in body weight was found in Nestin‐Cre+/−;C9orf72fl/fl mice. Our results are in apparent contrast with recent work in model organisms such as zebrafish and C. elegans. Knockdown or knockout, respectively, of C9orf72 orthologues in zebrafish and C. elegans causes motor deficits. These include age‐dependent motility defects and γ‐aminobutyric acidergic motor neuron degeneration in C. elegans, and motor axon and behavior deficits in zebrafish.10, 13 The discrepancy with our results may be caused by the low homology of C9orf72 orthologues in humans and in zebrafish and C. elegans (76% and 23% compared to human C9ORF72, respectively) as compared to the high homology between human and mouse C9orf72 (98%). Another possible explanation could be redundancy of the C9orf72 gene in mammalian species. In line with our results, a single treatment of wild‐type mice with ASOs directed against C9orf72 did not lead to motor abnormalities.14
Together, our data suggest that loss of C9orf72 on its own is insufficient to cause ALS. Our results, however, do not rule out the possibility that C9ORF72 loss‐of‐function modulates the disease process and influences disease onset, severity, and duration in C9ALS/FTD patients. The disease mechanisms caused by repeat expansions in C9ORF72 are likely to be complex and may ultimately result from interplay between C9ORF72 loss‐ and gain‐of‐function mechanisms.23, 24 In support of a gain‐of‐function mechanism, recent work shows that AAV‐mediated expression of 66 GGGGCC repeats in the adult mouse brain induces key features of ALS/FTD, such as TDP‐43 pathology and behavioral deficits.6 A different study, using transgenic mice with inducible expression of hexanucleotide (GGGGCC) repeats, found ubiquitinated inclusions, but these inclusions were not detected in neuronal populations relevant for ALS/FTD and no motor phenotype was found.25 Our finding that loss of C9orf72 function does not lead to motor neuron disease in mice has consequences for therapeutic strategies, because it indicates that ASOs directed at the repeat expansion that also lower C9ORF72 expression are unlikely to have negative side effects due to reduced C9ORF72 expression.
Authorship
M.K., A.M.B., H.‐J.W., M.L.T., C.A.C.Z., R.V.d.S., and R.D.S. performed experiments and data analysis. A.J.W. and D.J.B. provided unpublished reagents. M.K., A.M.B., H.‐J.W., J.H.V., L.H.v.d.B., and R.J.P. designed and coordinated the study. M.K. and R.J.P. prepared the manuscript with input from all coauthors. L.H.v.d.B. and R.J.P. are joint senior authors.
Potential Conflicts of Interest
L.H.v.d.B.: scientific advisory board, Biogen Idec, Cytokinetics, Baxter; grant, travel expenses, Baxter.
Acknowledgment
This study was supported by the Netherlands Organization for Health Research and Development (L.H.v.d.B.), Thierry Latran Foundation (J.H.V., R.J.P.), Prinses Beatrix Spierfonds (R.J.P., L.H.v.d.B.), Van Meer Stichting, Netherlands ALS Foundation (TOTALS; R.J.P., L.H.v.d.B.) European Community Health Seventh Framework Program (259867; L.H.v.d.B.), and Motor Neurone Disease Association (grant core Blake/Mar12/6088; A.J.W., D.J.B).
We thank Y. Adolfs for help with mouse experiments an L. Olsthoorn for immunohistochemical studies. Anti‐SV2 (developed by K. M. Buckley) and anti‐neurofilament (2H3, developed at Howard Hughes Medical Institute/Columbia University) antibodies were obtained from the DSHB, created by the NIH National Institute of Child Health and Human Development and maintained at the Department of Biology, University of Iowa, Iowa City, Iowa.
The copyright line for this article was changed on 21 August 2015 after original online publication.
References
- 1. DeJesus‐Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p‐linked FTD and ALS. Neuron 2011;72:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p12‐linked ALS‐FTD. Neuron 2011;72:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Majounie E, Renton AE, Mok K, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross‐sectional study. Lancet Neurol 2012;11:323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ash PE, Bieniek KF, Gendron TF, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 2013;77:639–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mori K, Weng SM, Arzberger T, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide‐repeat proteins in FTLD/ALS. Science 2013;339:1335–1338. [DOI] [PubMed] [Google Scholar]
- 6. Chew J, Gendron TF, Prudencio M, et al. C9ORF72 repeat expansions in mice cause TDP‐43 pathology, neuronal loss, and behavioral deficits. Science 2015;348:1151–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mizielinska S, Isaacs AM. C9orf72 amyotrophic lateral sclerosis and frontotemporal dementia: gain or loss of function? Curr Opin Neurol 2014;27:515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Levine TP, Daniels RD, Gatta AT, et al. The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab‐GEFs. Bioinformatics 2013;29:499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Farg MA, Sundaramoorthy V, Sultana JM, et al. C9ORF72, implicated in amyotrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet 2014;23:3579–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ciura S, Lattante S, Le Ber I, et al. Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann Neurol 2013;74:180–187. [DOI] [PubMed] [Google Scholar]
- 11. Donnelly CJ, Zhang PW, Pham JT, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 2013;80:415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Waite AJ, Baumer D, East S, et al. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol Aging 2014;35:1779.e5–1779.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Therrien M, Rouleau GA, Dion PA, Parker JA. Deletion of C9ORF72 results in motor neuron degeneration and stress sensitivity in C. elegans. PLoS One 2013;8:e83450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lagier‐Tourenne C, Baughn M, Rigo F, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A 2013;110:E4530–E4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schmidt ER, Brignani S, Adolfs Y, et al. Subdomain‐mediated axon‐axon signaling and chemoattraction cooperate to regulate afferent innervation of the lateral habenula. Neuron 2014;83:372–387. [DOI] [PubMed] [Google Scholar]
- 16. Groen EJ, Fumoto K, Blokhuis AM, et al. ALS‐associated mutations in FUS disrupt the axonal distribution and function of SMN. Hum Mol Genet 2014;22:3690–3704. [DOI] [PubMed] [Google Scholar]
- 17. Kolk SM, Gunput RA, Tran TS, et al. Semaphorin 3F is a bifunctional guidance cue for dopaminergic axons and controls their fasciculation, channeling, rostral growth, and intracortical targeting. J Neurosci 2009;29:12542–12557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Suzuki N, Maroof AM, Merkle FT, et al. The mouse C9ORF72 ortholog is enriched in neurons known to degenerate in ALS and FTD. Nat Neurosci 2013;16:1725–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tronche F, Kellendonk C, Kretz O, et al. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet 1999;23:99–103. [DOI] [PubMed] [Google Scholar]
- 20. Braincon N, McNay DE, Maratos‐Flier JE, Flier JS. Combined neural inactivation of suppressor of cytokine signaling‐3 and protein‐tyrosine phosphatase‐1B reveals additive, synergistic, and factor‐specific roles in the regulation of body energy balance. Diabetes 2010;59:3074–3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Panda SK, Wefers B, Ortiz O, et al. Highly efficient targeted mutagenesis in mice using TALENs. Genetics 2013;195:703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sundararajan S, Wakamiya M, Behringer RR, Rivera‐Peréz JA. A fast and sensitive alternative for β‐galactosidase detection in mouse embryos. Development 2012;139:4484–4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mizielinska S, Gronke S, Niccoli T, et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine‐rich proteins. Science 2014;345:1192–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Haeusler AR, Donnelly CJ, Periz G, et al. C9orf72 nucleotide repeat structues initiate molecular cascades of disease. Nature 2014;507:195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hukema RK, Riemslagh FW, Melhem S, et al. A new inducible transgenic mouse model for C9orf72‐associated GGGGCC repeat expansion supports a gain‐of‐function mechanism in C9orf72 associated ALS and FTD. Acta Neuropathol Commun 2014;2:166. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]