

Abstract
Herpes simplex virus‐1 (HSV‐1) is a large enveloped DNA virus that belongs to the family of Herpesviridae. It has been recently shown that the cytoplasmic membranes that wrap the newly assembled capsids are endocytic compartments derived from the plasma membrane. Here, we show that dynamin‐dependent endocytosis plays a major role in this process. Dominant‐negative dynamin and clathrin adaptor AP180 significantly decrease virus production. Moreover, inhibitors targeting dynamin and clathrin lead to a decreased transport of glycoproteins to cytoplasmic capsids, confirming that glycoproteins are delivered to assembly sites via endocytosis. We also show that certain combinations of glycoproteins colocalize with each other and with the components of clathrin‐dependent and ‐independent endocytosis pathways. Importantly, we demonstrate that the uptake of neutralizing antibodies that bind to glycoproteins when they become exposed on the cell surface during virus particle assembly leads to the production of non‐infectious HSV‐1. Our results demonstrate that transport of viral glycoproteins to the plasma membrane prior to endocytosis is the major route by which these proteins are localized to the cytoplasmic virus assembly compartments. This highlights the importance of endocytosis as a major protein‐sorting event during HSV‐1 envelopment.
Keywords: dSTORM, dynamin, endocytosis, glycoprotein, HSV, virus assembly
Herpes simplex virus‐1 (HSV‐1) is a common human pathogen responsible for cold sores. In rare cases it also leads to herpes keratitis and herpes simplex encephalitis, which can progress to blindness and death, respectively 1, 2. Primary HSV‐1 infection usually occurs early in life and results in a latent but lifelong infection with a seroprevalence of 50–80% among adults in Western countries 3. HSV‐1 and 2 can also cause serious disease in transplant recipients that in many cases is prevented by oral acyclovir prophylaxis 3.
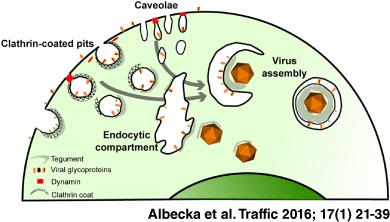
HSV‐1 belongs to the Herpesviridae family of large, enveloped DNA viruses. HSV‐1 establishes latency in sensory neurons, but during primary infection and reactivation it lytically replicates in epithelial cells. The entry of HSV‐1 into permissive cells is dependent on viral glycoproteins and host cell factors. The virus can penetrate a cell via direct fusion at the plasma membrane (PM) or with an endosomal membrane following endocytosis of the virion. The route of entry is dependent on cell type as well as other conditions that are not completely understood (reviewed in 4 5). After membrane fusion, capsids are transported on microtubules to the nucleus and the tegument protein pUL36 has been shown to be crucial for this process 6. Viral DNA enters the nucleus through the nuclear pore and viral gene expression is initiated, with the nucleus being the site of viral genome replication and assembly of new capsids. Newly synthesized DNA‐containing capsids (nucleocapsids) are then transported to the cytoplasm in a process called primary envelopment/de‐envelopment, where nucleocapsids bud at the inner nuclear membrane to form an enveloped particle in the perinuclear space, which then fuses with the outer nuclear membrane 7, 8. Finally, the assembly of infectious virions occurs in the cytoplasm, where the nucleocapsids acquire the full complement of tegument proteins and the viral glycoprotein‐containing lipid envelope through a process of budding/wrapping at intracellular membranes to form an enveloped virus contained within the lumen of a large vesicle/vacuole. Fully assembled particles are secreted out from cells via an exocytic process.
The origin of membrane material for the secondary envelopment of HSV‐1 remains a matter of debate. The presence of viral glycoproteins at the PM has been observed in many studies, but there is a limited understanding of the possible functions of HSV‐1 glycoproteins at the cell surface during normal viral life cycle. Some glycoprotein‐specific functions have been suggested previously for glycoprotein M (gM), which relocalizes both viral and cellular proteins from the PM to intracellular compartments and can antagonize the antiviral function of tetherin 9, 10, 11. Similarly, the gK/pUL20 complex has also been shown to relocalize other viral glycoproteins to intracellular compartments 12. Another glycoprotein complex, gE/gI, functions as an Fc receptor and binds Fc fragments of antibodies, thus decreasing humoral response to infection 13, 14. Furthermore, HSV‐1 glycoproteins have recently been demonstrated to follow Rab6‐dependent post‐Golgi trafficking to the PM. This step was shown to be crucial for the virus production 15, suggesting that transport of glycoproteins to the PM is necessary for the virus assembly process. Previous reports suggested that wrapping membranes might originate from multivesicular bodies (MVBs) 16 or/and from the trans‐Golgi Network (TGN) 17, 18, 19; however, a recent study demonstrated that endocytic tubules are a major source of membrane for secondary envelopment 20. The authors of the latter study used horseradish peroxidase (HRP) as a fluid‐phase marker for endocytosis and showed that HRP‐positive tubules wrapped capsids after 10–30 min of incubation. Furthermore, this process was shown to be Rab5 and Rab11 dependent 20. Overall, these data suggest that HSV‐1 glycoproteins are first trafficked to the PM and then travel to virus assembly sites by endocytosis.
Here, we investigate the mechanism of HSV‐1 glycoprotein endocytosis during the viral assembly process using functional assays. In particular, we investigated the role of dynamin, a member of the GTPase family, which is involved in the scission of endocytic vesicles from the PM. By using dominant‐negative proteins and inhibitors, we show that dynamin function is important for both glycoprotein internalization and production of infectious viral particles. The same methodology to target clathrin assembly resulted in similar but less pronounced effects on glycoprotein endocytosis and production of infectious particles, suggesting that although clathrin‐dependent endocytosis is involved, clathrin‐independent routes also contribute. This is further supported by evidence we obtained from optical super‐resolution microscopy, which shows that glycoproteins colocalize with both the clathrin‐dependent endocytosis marker (AP2) and the clathrin‐independent caveolae marker (Cav‐1), suggesting that both pathways may be important for HSV‐1 assembly. Furthermore, we show that antibodies targeting HSV‐1 glycoproteins added to cells during the timeframe of virus assembly are incorporated into virions and potently neutralize infectivity of newly produced viral particles. Our study clearly demonstrates an important requirement for endocytosis to deliver viral envelope proteins to assembling HSV‐1 particles.
Results
Neutralizing antibodies delivered to assembly sites with endocytosed gD and gH/gL are incorporated into secreted particles
Results obtained by Hollinshead et al. 20 suggest that glycoproteins travel to assembly sites via the PM. We first decided to confirm this hypothesis through the use of neutralizing antibodies. We predicted that glycoprotein‐specific antibodies added to the extracellular media during the HSV‐1 assembly process should be incorporated into virions. In this process, antibodies would bind to their corresponding glycoproteins that have been exposed on the cell surface, and the conjugated complex would then traffic to the intracellular assembly compartments. To verify this hypothesis we used neutralizing antibodies against gD (LP2) and gH/gL (LP11). Non‐neutralizing antibodies (AP7 for gD and BBH‐1 for gH) and an antibody specific to the tegument protein VP16 (LP1) were used as negative controls. We added 5 µg/mL of purified antibodies to HaCaT cells 1 h postinfection and harvested cell‐associated viruses after 16 h incubation. In order to specifically quantify intracellular infectivity cells were harvested using trypsinization to remove or destroy all extracellular infectivity. Trypsin treatment removed on average 65–85% of cell‐associated infectivity in these experiments. As shown in Figure 1A, both neutralizing antibodies (LP2 and LP11) significantly decreased intracellular virus infectivity (down to ∼20 and 40% of the control, respectively), whereas no such effect was observed for control antibodies (LP1, AP7 and BBH‐1). Experiments were also performed in COS7 cells and similar results were observed (Figure S1, Supporting Information). Given the extended period of exposure of infected cells to antibodies in these experiments (16 h), we investigated the localization of internalized antibodies by immunofluorescence microscopy. Antibody signal was detected in numerous puncta throughout the cytoplasm but demonstrated no colocalization with the Golgi, suggesting that internalized HSV‐1 gD or gH/gL remain in the endocytic pathway and do not traffic to the Golgi complex to any detectable levels after internalization from the PM (Figure S1). Furthermore, we also examined the level of intracellular infectivity when neutralizing antibodies to gD (LP2) were added to cells only for the last 30–60 min of infection, and under these conditions intracellular virus infectivity was reduced to 38–46% of the control. These data suggest that the neutralization of intracellular virus in these experiments is not simply due to non‐specific saturation of the cellular secretory and endocytic pathways with antibodies.
Figure 1.
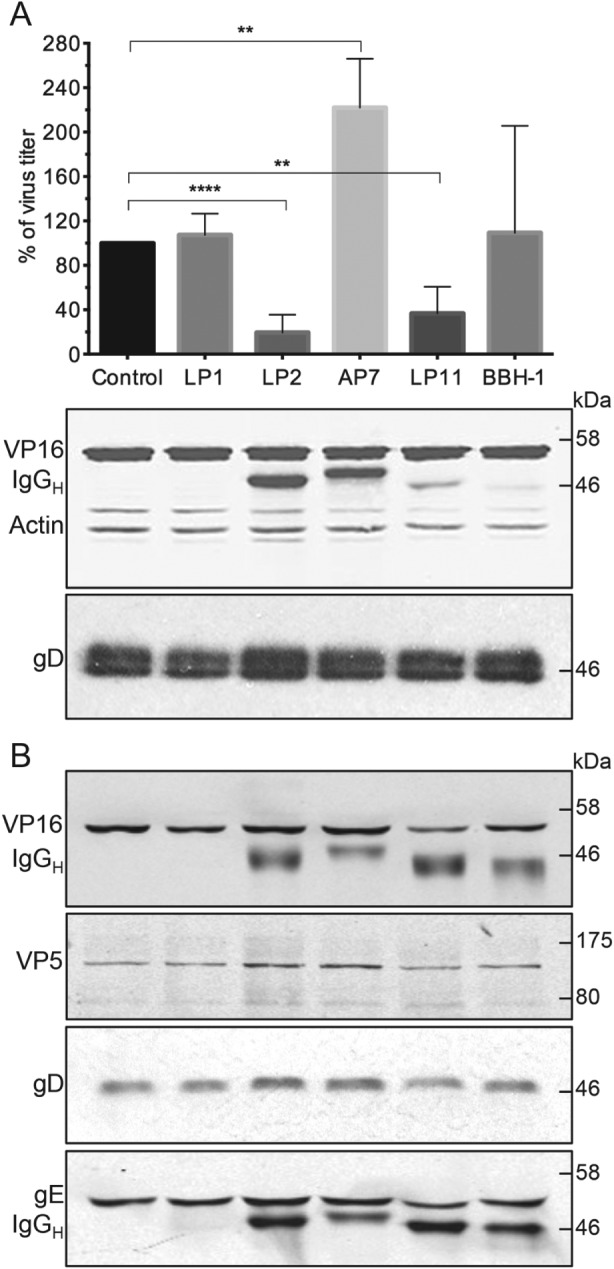
Glycoprotein‐specific neutralizing antibodies incorporated into virions during assembly affect intracellular HSV ‐1 infectivity. A) HaCaT cells were infected with HSV‐1 at MOI = 3 and then incubated with 5 µg/mL of antibodies (LP1 for VP16, LP2 and AP7 for gD and LP11 and BBH‐1 for gH) for 16 h. To remove and inactivate extracellular viruses cells were trypsinized for 10 min. Cell pellets were washed, resuspended in media and intracellular virus released by three freeze–thaw cycles and then titrated using Vero cells. Data are represented as mean ± SD from at least three independent experiments with each experiment containing two biological replicates per condition. T‐tests were performed with Graphpad Prism. ****p value < 0.0001, **p < 0.005. Additional cell pellet samples were lysed in PBS + 1% Triton‐X‐100 + protease inhibitors and analyzed by WB for the presence of VP16, actin and gD. B) HaCaT cells were infected and incubated with antibodies as before. Supernatants were collected and cleared of cellular debris by centrifugation. Secreted viruses were pelleted by ultracentrifugation for 3 h at 75 000× g and analyzed by WB. In the WB analyses, the secondary anti‐mouse antibodies used for the detection of VP16 and actin (A) or VP16 and gE (B) also detected the denatured IgG heavy chains (IgGH) of the antibodies internalized into cells. To avoid detection of internalized antibody heavy chains on gD blots (as both migrate at similar size) Protein A conjugated to HRP and ECL detection was used.
To confirm that these results were due to neutralization of infectivity rather than any indirect effect on virus assembly or release, we collected supernatants from infected HaCaT cells and pelleted viruses for analysis by western blot. Figure 1B shows that particles were released from cells at a similar level regardless of the presence of antibodies, and therefore the observed reduction in infectivity is unlikely to be caused by defects in virion assembly. In fact, a slight increase in particle release was observed where anti‐gD antibodies were used. This could explain the significant increase in infectivity observed with the non‐neutralizing AP7 antibody. Currently, we are not able to explain this phenomenon, although it is conceivable that IgG‐mediated cross‐linking of gD molecules could enhance the stability of tegument interactions with the cytoplasmic domain of gD, thereby increasing the efficiency of assembly.
IgG heavy chains were detected in western blot analysis of cell lysate and/or secreted virus samples for all glycoprotein‐specific antibodies (LP2, AP7, LP11 and BBH‐1) that were added to the media of infected cells (Figure 1A,B), confirming that they are internalized into cells and remain attached to secreted viruses. In contrast, VP16‐specific antibody (LP1) was not present in western blot samples, as predicted for an antibody that recognizes a virion protein that does not become exposed to the extracellular environment. Levels of gD‐specific antibody heavy chain present in cell lysate and secreted virus samples analyzed were comparable. Interestingly, there was a marked difference observed for anti‐gH antibodies as only faint heavy‐chain bands were detected in cellular lysates but strong bands were observed for secreted virion samples. This suggests that of the population of gD and gH molecules that become exposed on the PM and then internalized, a greater proportion of gH is incorporated into virions, perhaps suggesting more efficient targeting of internalized gH to virus assembly sites.
Overall, these results confirm that both gD and the gH/gL complex traffic to the PM and become internalized prior to being targeted to intracellular sites of virus envelopment.
Dominant‐negative dynamin decreases the production of infectious HSV‐1
Next we looked into the mechanism of glycoprotein endocytosis. Dynamin 2 is a protein necessary for pinching off endocytic vesicles from the PM 21. Inhibition of dynamin activity impairs both clathrin‐dependent and ‐independent pathways 22. To investigate the role of dynamin in HSV‐1 assembly, we used dominant‐negative dynamin K44A in a transfection‐based UL36 complementation assay described previously 23. Briefly, this assay is based on the ability to complement the replication defect of a HSV‐1 virus lacking the UL36 gene, which is essential for virus assembly, by expressing pUL36 from a transfected plasmid. COS7 cells were co‐transfected with a pUL36 expression plasmid together with a plasmid encoding the protein of interest (e.g. dominant‐negative dynamin). Subsequently, cells were infected with HSV‐1ΔUL36; productive virus replication of a UL36 deletion virus only occurs in pUL36‐expressing (transfected) cells and thus infected but untransfected cells do not influence the results. The production of infectious virus progeny was determined by plaque assay using a stable pUL36‐complementing cell line. We also used a dominant‐negative form of AP180 (AP180‐C), which is an adaptor necessary for clathrin‐dependent endocytosis 24. We have previously shown that endosomal sorting complex required for transport (ESCRT) proteins plays an important role in HSV‐1 assembly; therefore, dominant‐negative VPS4 (VPS4‐EQ), which potently blocks ESCRT function, was used as a control 25. As shown in Figure 2A, expression of dynamin K44A decreased the production of infectious virus particles by ∼90%. In comparison, expression of dominant‐negative AP180‐C reduced the production of infectious virus by ∼50%. The uptake of transferrin, which is clathrin‐dependent, into cells expressing AP180‐C or dynamin K44A‐transfected cells was strongly inhibited as expected, confirming the activity of these dominant‐negative constructs (Figure 2B). The greater effect of dynamin K44A compared with AP180‐C on production of infectious HSV‐1 suggests that clathrin‐independent pathways may also participate in HSV‐1 glycoprotein endocytosis. The discrepancy between AP180‐C and dynamin results could also be explained by the fact that dynamin 2 is necessary for trafficking of vesicles from the Golgi to the PM 26. However, immunofluorescence experiments clearly show accumulation of gD at the PM (Figure 2C) in dynamin K44A‐transfected cells, supporting the notion that the effect is primarily due to inhibition of clathrin‐independent endocytosis and not Golgi‐to‐PM trafficking. Similar results were also observed for other HSV‐1 envelope proteins gB and gE (Figure S2). To verify whether glycoproteins traffic to intracellular virus assembly sites via the PM, we performed antibody‐uptake experiments using gD‐specific antibodies in cells infected with HSV‐1 expressing the capsid protein VP26 fused to a fluorescent protein mTurquoise (Figure 2C). These experiments and all subsequent fluorescence microscopy experiments were performed at 8 h postinfection, a midpoint of the HSV‐1 replication cycle when virions are being assembled. This allows more reliable identification of enveloped and non‐enveloped capsids in the cytoplasm by fluorescence microscopy because newly assembling virions are better separated and cells are less saturated with viral proteins compared to later times of infection. In cells expressing wild‐type (WT) dynamin, antibodies bound to gD were relocalized into capsid‐positive puncta, whereas in cells expressing dynamin K44A very few puncta where gD colocalized with capsids were present. In addition, accumulation of gD/capsid‐positive particles (enveloped virions) near the cell periphery was clearly observed in WT dynamin‐expressing cells but not in dynamin K44A‐expressing cells (Figure 2C), suggesting that the inhibition of dynamin‐dependent endocytosis prevents gD reaching virus assembly compartments. Taken together, these results suggest that dynamin‐dependent endocytosis is important for the assembly of infectious HSV‐1 particles by mediating the delivery of glycoproteins such as gD to the assembly sites.
Figure 2.
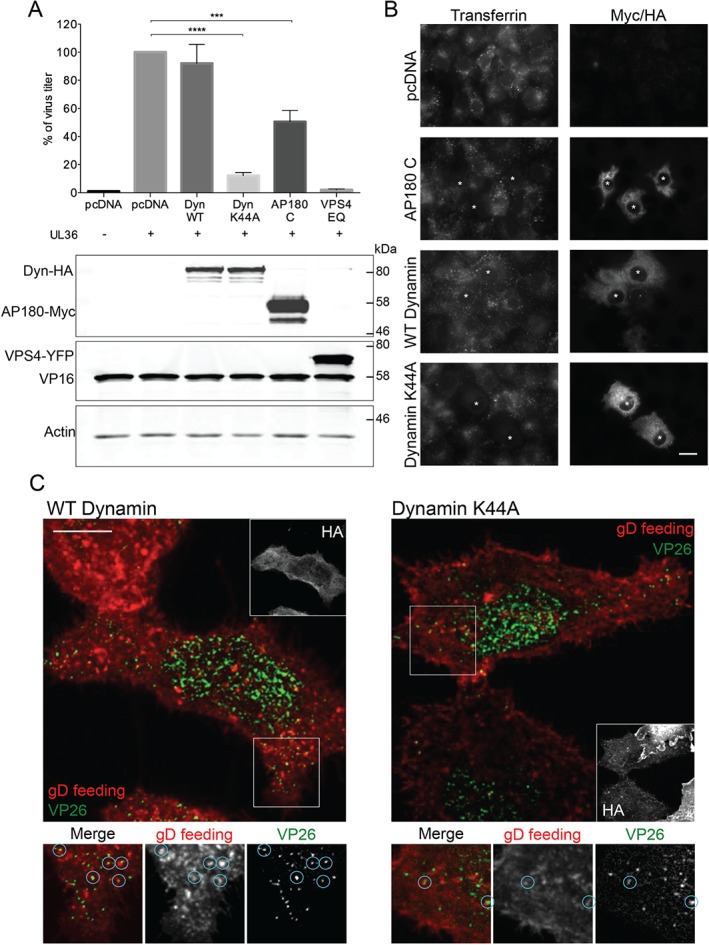
Dominant‐negative dynamin and AP180 impair the production of infectious HSV ‐1. A) COS7 cells were co‐transfected with plasmid expressing UL36 protein and plasmids containing dominant‐negative (K44A) or WT dynamin, dominant‐negative AP180‐C and VPS4‐EQ or empty pcDNA vector control. Cells were then infected with HSV‐1ΔUL36 and after 16 h processed for titration in HS30 cells or for WB analysis of cell lysates. Data are represented as mean ± SD from at least three independent experiments with each experiment containing two biological replicates per condition. T‐tests were performed with Graphpad Prism. ****p value < 0.0001, ***p < 0.0005. B) Transferrin uptake into COS7 cells transfected with pcDNA and WT dynamin controls and dominant/negative dynamin and AP180 was followed by immunodetection of transfected cells by anti‐Myc or anti‐HA antibodies. Asterisks indicate transfected cells. Scale bar = 20 µm. C) WT or K44A dynamin‐transfected COS7 cells were infected with VP26‐mTurquoise/gM‐EYFP for 8 h and then fed with anti‐gD antibody (LP2) for 15 min before fixing. Cells were then permeabilized and immunostained with anti‐HA followed by secondary antibody labeling. Images were acquired using a confocal microscope. Blue circles in the zoom‐in section represent virus particles containing both gD and VP26. Scale bar = 10 µm.
Endocytosis inhibitors prevent glycoproteins from incorporation into mature virions
Our results from dominant‐negative dynamin and AP180 experiments show decrease in virus titer when endocytosis and subsequent trafficking of glycoproteins from the PM to assembly sites is impaired. However, as mentioned above, it is possible that these effects might be partially due to reduced glycoprotein trafficking through the secretory pathway between the Golgi and the PM due to inhibition of dynamin and/or clathrin function at the TGN. In order to more directly investigate the role of endocytosis, we performed experiments to examine the trafficking of HSV glycoproteins in the presence or absence of endocytosis inhibitors. To inhibit dynamin activity, we used Dynole‐34‐2 (Dynole), which allosterically inhibits the GTPase activity of dynamin, and the inactive form, Dynole‐31‐2, as a control (Dynole Negative). Inhibition of clathrin assembly was achieved using PitStop 2 and the inactive form, PitStop 2 Negative, as a control. We used antibody‐uptake experiments in cells infected with HSV‐1 for 8 h and incubating cells with inhibitors or control compounds for the final 30 min prior to fixation to analyze how these inhibitors affect glycoprotein trafficking to assembly sites while avoiding non‐specific effects that may occur due to prolonged endocytosis inhibition. We assessed the effect of the inhibitors on uptake of transferrin and glycoprotein‐specific antibodies into HFF‐Tert (Figure 3) and COS7 cells (Figure S3). Active compounds significantly decreased transferrin uptake into both cell lines, confirming their activity. In infected cells, both Dynole and PitStop 2 changed the localization of HSV‐1 glycoproteins in the cells, and in all cases increased the accumulation of antibodies at the PM and decreased the amount of intracellular puncta; these puncta are likely to be either endosomes or assembled viral particles (Figures 3B and S3B).
Figure 3.

Effect of endocytosis inhibitors on uptake of transferrin and glycoproteins into HFF ‐Tert cells. A) Cells were incubated with DMSO, 15 µm Dynole or Dynole negative control and 30 µm PitStop2 or its negative control for 15 min. Next transferrin conjugated to Alexa Fluor 568 was added for 5 min. Cells were then washed on ice, fixed and imaged using an epifluorescence microscope. Scale bar = 20 µm. B) Cells were infected with HSV‐1 for 8 h. For the last 30 min cells were incubated with DMSO, 15 µm Dynole or Dynole negative control and 30 µm PitStop2 or its negative control. After 15‐min incubation antibodies specific to viral glycoproteins (LP2 for gD, 3063 for gE, CB24 for gB and LP11 for gH) were added and incubated for a further 15 min. After fixation, cells were permeabilized and stained with secondary antibodies and imaged by epifluorescence microscopy. Scale bar = 20 µm.
The uptake experiments also showed that the inhibition of glycoprotein endocytosis is less effective with PitStop 2 (which blocked transferrin uptake at comparable levels to Dynole) because greater numbers of intracellular puncta were still visible compared to Dynole treatment, especially in COS7 cells (Figures 3 and S3). These data also support the notion that clathrin‐independent mechanisms contribute to glycoprotein internalization.
To examine glycoprotein endocytosis inhibition in more detail, we focused on gD trafficking to intracellular capsids. For these experiments, we infected HFF‐Tert cells for 8 h with a doubly fluorescent virus expressing VP26‐mTurquoise (capsid) and gM‐EYFP (envelope) to allow simple identification, and discrimination of enveloped and non‐enveloped capsids in the cytoplasm, and treated them with inhibitors for 30 min, with gD antibody added to the extracellular media for the final 15 min. As shown in Figures 4 and S4A, within 15 min of antibody uptake, the control cells (Dynole Negative and PitStop 2 negative‐treated) accumulated several gD‐positive puncta, many of which were capsid‐positive, thus likely enveloped particles. These data confirm that glycoproteins (gD in this instance) are delivered from the PM to cytoplasmic virion assembly sites. However, in cells treated with active endocytosis inhibitors gD was mostly present close to the PM and very few gD‐positive capsids were observed. We confirmed these data by quantification of gD‐positive capsids, which revealed that only 30 and 39% of VP26‐mTurquoise puncta colocalized with gD staining after the treatment with Dynole and PitStop 2, respectively. In contrast, in control cells (Dynole Negative and PitStop 2 negative‐treated), >70% of capsids were gD‐positive, clearly showing that the treatment with active endocytosis inhibitors decreases gD transport to assembling virions.
Figure 4.
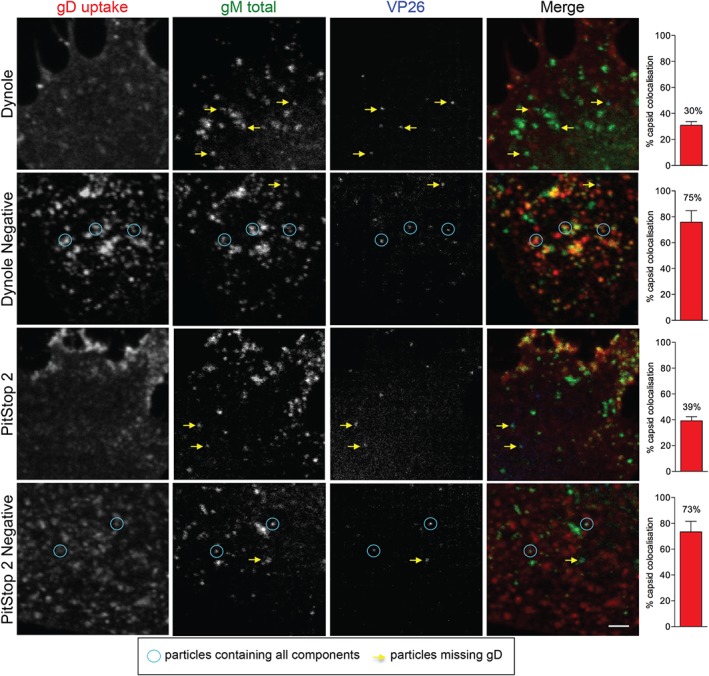
Endocytosis inhibitors impair transport of glycoproteins to cytoplasmic capsids. HFF‐Tert cells were infected with VP26‐mTurquoise/gM‐EYFP recombinant virus for 8 h. Thirty minutes before the end of incubation cells were treated with 15 µm Dynole or Dynole negative control and 30 µm PitStop2 or its negative control. After 15 min antibodies specific to gD (LP2) were added and incubated for a further 15 min. Cells were then fixed, permeabilized and labeled with secondary antibodies. Images were acquired using a confocal microscope. Blue circles indicate complete viral particles while yellow arrows point to particles missing gD staining. Scale bar = 2 µm. Right panels show percentage of intracellular capsids (VP26‐mTurquoise) colocalizing with gD. At least 100 capsids from multiple cells and several fields of view were counted for each condition. Graphs represent the percentage of capsids positive for gD labeling as a mean from two independent experiments ± SEM.
Localization of the TGN
Previous studies suggested that the TGN might provide membranes for HSV‐1 assembly 17, 18, 19. Here, we show that glycoproteins traffic to intracellular capsid‐positive assembly sites from the PM. To show glycoprotein delivery to intracellular capsids in relation to TGN localization, we infected HFF‐Tert cells with a fluorescent virus expressing VP26‐EYFP (capsid) that has been deleted for the gE gene (VP26‐YFP/ΔgE) for 8 h. We used a virus lacking gE to avoid any non‐specific reactivity of the widely used sheep polyclonal anti‐TGN46 antibody with the gE/gI complex of HSV‐1, which is a potent Fc receptor 27. Before fixing the cells, gH‐specific antibodies were added to media for uptake for 15 or 30 min. VP26‐ and gH‐positive puncta were observed in the cytoplasm at both time points, demonstrating endocytic delivery of gH to assembling virions (Figure 5). In contrast, the TGN was clearly distinct from these assembly sites, with the vast majority of TGN46 signal being localized to the perinuclear region, whereas internalized gH and VP26‐positive puncta were more dispersed through the cytoplasm. We also observed occasional puncta positive for both internalized gH‐specific antibodies and TGN46, suggesting that internalized viral glycoproteins can reach TGN46‐containing compartments. The presence of TGN46 in vesicles containing endocytosed gH could arise from the endocytosis of TGN46 following its transport to the PM, or fusion of TGN46‐bearing vesicles that originate from the TGN with endocytic compartments containing internalized gH or a combination of both. However, we did not observe detectable colocalization of TGN46 to assembled virions in the cytoplasm (internalized gH and VP26‐positive puncta). While we cannot rule out the presence of small amounts of TGN46 at HSV‐1 assembly sites, our data suggest that virion envelopment occurs at compartments more characteristic of endosomes than the bulk TGN.
Figure 5.
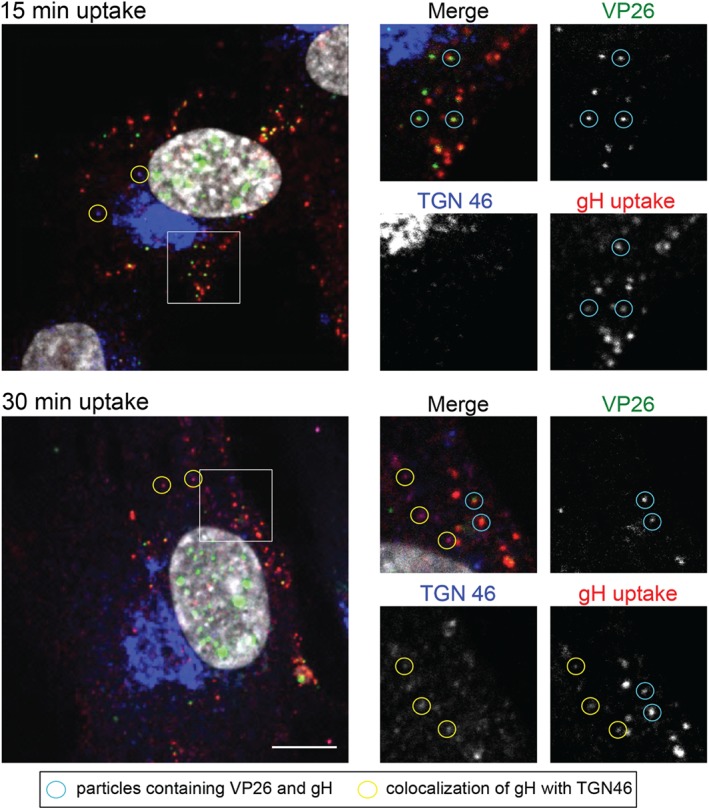
Localization of assembly sites in relation to TGN compartment. HFF‐Tert cells were infected with VP26‐YFP/ΔgE virus for 8 h. gH‐specific antibodies were added to cells for uptake for 15 or 30 min before fixation and permeabilization. Sheep anti‐TGN‐46 antibodies were used to image TGN, followed by secondary Alexa Fluor antibodies. Scale = 10 µm.
Glycoproteins undergo endocytosis in distinct complexes and reach assembling virions at different rates
Many HSV‐1 envelope proteins such as gB, gE, gM, as well as several others, contain potential endocytosis motifs (e.g. tyrosine‐based motifs), which can enable their interactions with clathrin adaptors, some of which have been shown to be important for glycoprotein endocytosis 28, 29. In contrast, gD and gH do not have any predicted trafficking motifs and their endocytosis is dependent on interactions with other viral glycoproteins 9, 10, 12. For these reasons, we investigated potential glycoprotein complexes during endocytosis. By using the dual‐fluorescent VP26‐mTurquoise/gM‐EYFP virus, we were able to visualize capsids and the total amount of gM. Cells were infected for 8 h and antibodies against gB, gE, gD and gH/gL were added to the extracellular media to allow for uptake in various combinations 15 min before fixing the cells. Figures 6A and S4B show some of the combinations of tested glycoprotein antibodies. As predicted, gD and gH/gL did not appear able to enter cells alone and were found to colocalize with tyrosine motif‐containing glycoproteins (either gB, gE or gM). In the case of gH/gL, substantial colocalization was observed with gM. In case of gD, it frequently colocalized with gM; however, some gD–gB puncta (without gM) were also visible. While we cannot conclude that gD or gH/gL are physically interacting with these other glycoproteins, these data suggest that either the colocalizing glycoproteins are endocytosed together in the same vesicles, or if they are in separate vesicles these fuse rapidly after endocytosis (within the 15‐min incubation period). Interestingly, internalized gE and gB as well as gM could be detected in separate puncta (Figure 6A, bottom panels), suggesting that these tyrosine motif‐containing glycoproteins can mediate their own endocytosis independently of each other.
Figure 6.
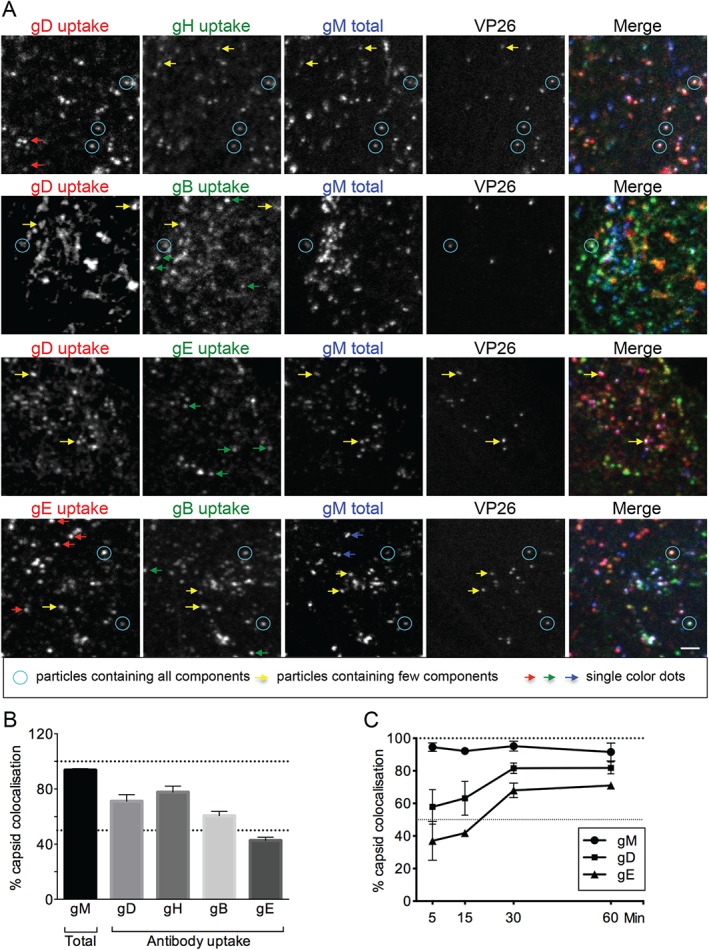
Glycoproteins undergo endocytosis in various complexes. A) HFF‐Tert cells were infected with VP26‐mTurquoise/gM‐EYFP recombinant virus for 8 h. Antibodies specific to HSV‐1 gD, gB, gH and gE were added in various combinations for the last 15 min of incubation. After fixing and permeabilization isotype‐specific secondary Alexa Fluor antibodies were used and images were acquired using a confocal microscope. Blue circles point to particles containing all labeled components. Yellow arrows point to dots containing only some components, while red, green and blue arrows point to dots containing only one type of glycoprotein. Scale bar = 2 µm. B) Quantification of images obtained as for Figure 5A. At least 100 capsids from multiple cells and several fields of view were counted for each combination of glycoproteins. Graph represents the percentage of capsids positive for each glycoprotein labeling as mean from six combinations of antibodies obtained in two independent experiments ± SEM. gM was imaged as fusion to EYFP while other glycoproteins were imaged after primary antibody uptake into cells for 15 min before fixation. C) Kinetics of gD and gE transport to intracellular capsids. Cells infected with VP26‐mTurquoise/gM‐EYFP virus were incubated for 8 h and then fixed with 4% formaldehyde. At 60, 30, 15 and 5 min before fixing gD (LP2) and gE (3063) antibodies were added to cells for uptake. At least 100 capsids from multiple cells and several fields of view were counted for each condition. Data are represented as mean from two independent experiments ± SEM.
In Figure 6A, blue circles highlight the examples of viral particles containing all labeled components. Yellow arrows point to examples of incomplete labeling, indicating that different combinations of glycoproteins can be endocytosed in the same vesicles. Finally, arrows in red, green and blue indicate examples of the presence of only one glycoprotein, confirming that gB, gE and gM do not require each other for endocytosis. Observed capsid‐free puncta might represent the endocytic vesicles but also light particles, which are essentially capsid‐less virions.
In addition, we observed that not all glycoproteins appear to be transported to virion assembly compartments at the same pace because fewer capsids were labeled with internalized gE antibodies compared with gD or gH/gL. To quantify this effect, we counted intracellular capsids from several cells and fields of view for each sample and scored them as positive or negative for each internalized glycoprotein in two independent experiments (Figure 6B). Almost all capsids contained gM‐EYFP (93%), demonstrating that the majority of the cytoplasmic capsids that are detected at this time point are associated with the viral envelope. gD and gH appear to be transported to assembly compartments quite rapidly; within 15 min of antibody uptake 71 and 77% of capsids colocalized with gD and gH, respectively. Sixty percent of capsids were positive for gB but only 42% for gE at this time point. In order to look at this phenomenon in more detail, we performed a time course where gD‐ and gE‐specific antibodies were added to cells for 5, 15, 30 and 60 min (Figure 6C). gD was very rapidly translocated to capsids and within 5 min of uptake 60% of capsids were gD positive. gE transport to capsids was less efficient and even after 60 min of antibody uptake only 70% of capsids appeared gE positive (in contrast to >80% with gD). It is important to note that for the quantification of capsids that colocalize with internalized glycoproteins, intact virions attached to the extracellular surface of cells would also be scored in these assays. For all these analyses, we quantified virions that were clearly within the boundary of the cell membrane and not at the edges of cells to reduce the potential impact of extracellular virions on our data. In addition, to investigate the contribution of extracellular virions to our quantification, we performed antibody labeling of live cells at 4°C prior to fixation, as well as labeling of fixed but non‐permeabilized cells with antibodies to both gD or gE in two independent experiments. Fluorescent capsids were then scored as either positive or negative for glycoprotein antibody. These data consistently demonstrated that 25–30% of capsids were positive for glycoprotein antibody, suggesting that this is the average proportion of capsids that are extracellular in these assays. Overall, these results show that glycoproteins can travel to assembly sites in various complexes or potentially alone, and importantly they are transported at different rates.
Clathrin and caveolae both contribute to endocytosis of HSV‐1 glycoproteins
Our experiments with dominant‐negative dynamin and AP180 as well as endocytosis inhibitors showed that inhibition of dynamin function had a stronger effect on targeting glycoproteins to HSV‐1 envelopment sites than inhibition of clathrin assembly. This suggested that clathrin‐independent endocytosis could also contribute to HSV‐1 envelope protein delivery to assembly sites. To address this question, we used super‐resolution microscopy, specifically direct stochastic optical reconstruction microscopy (dSTORM) 30. Conventional microscopy techniques are limited to a resolution of ca. 250 nm due to optical diffraction, and therefore small vesicles located in close proximity to one another cannot be distinguished. Single‐molecule localization methods like dSTORM are not limited in this way by diffraction and can distinguish objects as close as 20 nm apart. We have previously used dSTORM to analyze the structure of HSV‐1 particles 31. Here, we used dSTORM to investigate the colocalization of glycoproteins with the endocytic machinery. We labeled infected cells with antibodies to HSV‐1 glycoproteins together with antibodies to the clathrin adaptor protein AP2 or the caveolae marker Caveolin 1 (Cav‐1). Here, we also used the VP26‐mTurquoise/gM‐EYFP virus to identify viral capsids and envelope. Cells were infected for 8 h and glycoprotein‐specific antibodies were incubated with the cells for uptake for the last 15 min before fixation. Figure 7 shows AP2 and Cav‐1 labeling with antibody uptake for gD. Images clearly show the presence of gD in AP2‐positive clusters, with dSTORM analysis demonstrating the colocalization of gD and AP2 within the same structures (Figure 7A). Furthermore, localization of gD in Cav‐1‐positive clusters was also observed (Figure 7B). Caveolae are small invaginations of the PM found in many mammalian cells that are often observed associated with lipid rafts and thought to mediate clathrin‐independent endocytosis of certain cargo 32. In these images they appear as aggregates in contrast to often single, round‐shaped AP2‐positive clusters. Similar results demonstrating colocalization of gE with both AP2 and Cav‐1 were also observed (Figure S5). Although further research is necessary to understand the mechanism of HSV‐1 glycoprotein endocytosis, our study suggests a role for both clathrin‐dependent and ‐independent pathways in endocytic delivery of envelope proteins to virus assembly compartments.
Figure 7.
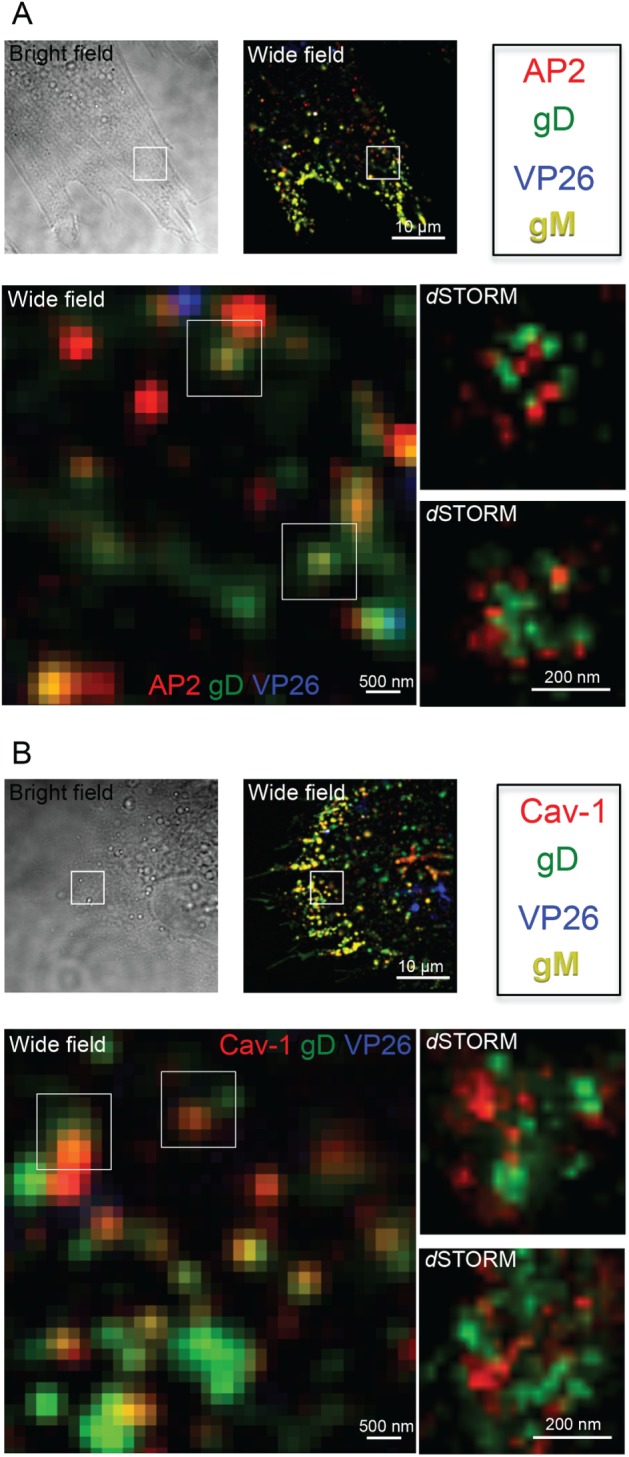
d STORM images of clathrin and caveolae‐dependent endocytosis of gD. HFF‐Tert cells were infected with VP26‐mTurquoise/gM‐EYFP recombinant virus for 8 h. gD‐specific antibodies were added to cells for uptake 15 min before fixation and permeabilization. Clathrin‐coated pits or caveolae were detected with anti‐AP‐2 (A) or Cav‐1 (B) monoclonal antibodies and subtype‐specific secondary Alexa Fluor antibodies. Wide‐field images were acquired for all channels. dSTORM images of gD (Alexa Fluor 568 channel) and AP2/Cav1 (Alexa Fluor 647 channel) are shown. Scale = 10 µm, 500 nm and 200 nm.
Discussion
The identity of the cytoplasmic membrane compartment(s) where final HSV‐1 envelopment occurs has been the topic of much recent debate. Our data support the proposal by Hollinshead et al. 20 that the cytoplasmic compartments where HSV‐1 envelopment takes place are endocytic in nature. Previous studies suggested that membranes wrapping cytoplasmic capsids originate from the TGN or MVB‐related organelles 17, 18, 19. The TGN is a major sorting organelle, which directs newly synthesized proteins to various cellular destinations including the PM. It is composed of interconnected vesicles and tubules and its function is not limited to secretion as it also regulates recycling and retrograde transport from the PM. In fact, the TGN has been shown to contain both newly synthesized and internalized proteins 33 and this recycling route is regulated by both SNARE and Rab proteins 34, 35. The MVBs are a heterogeneous class of endocytic compartments that are formed owing to inward budding of the intralumenal vesicles (ILV) from the limiting membrane of endosomes during their maturation 36. Therefore, the observations of Hollinshead et al. showing that HSV‐1 nucleocapsids are wrapped in membranes positive for an endocytic tracer 20 and our data showing the importance of dynamin‐dependent endocytosis for glycoprotein delivery to assembling virions are not necessarily contradictory to previous research suggesting the TGN and/or MVB as the origin of wrapping membranes. Cellular membrane traffic within the secretory and endocytic pathways comprises of a network of highly dynamic and interconnected compartments that shuffle proteins between each other using vesicle carriers and/or direct fusion, and so, not surprisingly, the content of different compartments can mix in various locations. In fact, during later stages of HSV‐1 lytic infection, the membranes of the secretory and endocytic pathways are likely to be packed with viral glycoproteins, thus assessing the origin of these membranes is not straightforward. Therefore, while it is clear that the HSV‐1 assembly compartment(s) receive endocytic cargo, and that endocytic transport routes are important for virus production, it is difficult to assign a precise definition for HSV‐1 assembly sites, which are likely to be quite heterogeneous.
Further to this, it is important to note that the requirement for the activity of the ESCRT machinery during HSV‐1 envelopment does not necessarily indicate MVBs as budding/wrapping sites for HSV‐1 16, 23, 25. The ESCRT machinery (which is normally responsible for ILV budding during MVB formation) is relatively mobile and can be recruited to multiple cellular membranes under different conditions, and so the involvement of the ESCRT machinery in itself does not allow any firm conclusions to be drawn on sites of HSV‐1 envelopment. Indeed, the ESCRT machinery is involved in the budding of several other enveloped virus families at the PM and abscission at the end of cytokinesis, demonstrating ESCRT activity at multiple different subcellular locations 37, 38.
Our results clearly show that dynamin‐dependent endocytosis plays a major role in glycoprotein delivery to cytoplasmic virus assembly sites. Experiments with dominant‐negative dynamin and with the dynamin inhibitor Dynole show significant decrease in infectious virus production and glycoprotein trafficking to cytoplasmic capsids. It should be noted that dynamin has been shown to be important for the entry of HSV‐1 in some cell lines, such as keratinocytes 39, which could complicate the interpretation of data on HSV‐1 replication in dominant‐negative dynamin‐expressing cells. However, the entry of HSV‐1 has recently been shown to be independent of dynamin in several epithelial cell lines 40, and we do not detect any difference in viral gene expression between cells expressing WT or dominant‐negative dynamin in our experiments with the epithelial cell line COS7. We therefore consider it unlikely that our results are due to effects of dominant‐negative dynamin on virus entry.
Interestingly, we observed that some infectious viruses are still produced under conditions of dynamin inhibition. This could be explained by incomplete inhibition of dynamin activity in our experimental setting. Alternatively, dynamin‐independent endocytosis pathways might also contribute to glycoprotein delivery, or glycoproteins could be trafficked directly to assembly sites without trafficking via the PM. Interestingly, Rab6‐dependent exocytosis of glycoproteins has been shown to be crucial for the secondary envelopment of HSV‐1 15, suggesting that viral envelope proteins must exit the Golgi/TGN to gain access to assembly sites. In agreement with this, our experiments demonstrate that the majority of assembled viruses are wrapped in membranes that are at least partially derived from the PM via dynamin‐dependent endocytosis. However, we cannot exclude that in some conditions, especially when other pathways are blocked, glycoproteins can traffic directly to the assembly sites bypassing the PM. It is also possible that during the inhibition of dynamin activity, virus glycoproteins are able to engage dynamin‐independent endocytosis routes, which results in limited production of infectious virions.
Many HSV‐1 glycoproteins, including gE, gB and gM, contain motifs allowing them to interact with clathrin adaptors 28, 29, and thus should be able to enter cells via endocytosis independently of other viral proteins. In our experiments, these glycoproteins appear as single puncta in antibody‐uptake experiments. In contrast, we have previously shown that gD and gH/gL do not encode their own internalization motifs and require other endocytosis motif‐containing viral glycoproteins, such as gM, for their internalization 9, 10, 12. Our data here also support this notion as we only observe internalized gD or gH/gL when they are colocalized with other viral glycoproteins (namely gM or gB for gD and only gM for gH/gL).
Our observation that inhibition of clathrin assembly has a less pronounced effect on infectious virus production or glycoprotein internalization than the inhibition of dynamin suggests that clathrin‐independent endocytosis also contributes to glycoprotein delivery from the PM to assembly sites. Interestingly, we observed that both gD and gE colocalize not only with AP2 but also with caveolae marker Cav‐1. The role of caveolae in HSV‐1 assembly is not within the scope of this study. However, it will be interesting to assess in the future whether caveolae or other clathrin‐independent endocytosis pathways contribute significantly to glycoprotein delivery. Furthermore, investigating the mechanisms involved in these processes during HSV‐1 replication may provide important information on these cellular pathways, as the role of caveolae in endocytosis is still much debated 41, 42.
Our study clearly confirms the importance of dynamin‐dependent endocytosis for targeting HSV‐1 envelope proteins to sites of virion assembly. However, it is intriguing to speculate why HSV‐1 glycoproteins have to travel via the PM for their efficient targeting to assembly sites and why they do not use more direct routes from the secretory pathway (i.e. the Golgi/TGN) to sites of secondary envelopment that would avoid exposure of viral glycoproteins to the extracellular environment. One tempting hypothesis is that transporting viral glycoproteins via the PM into endocytic organelles is necessary to gather the large number of different viral membrane proteins that are incorporated into mature virions in the same membrane compartment. Given that the different viral envelope proteins (up to 16 for HSV‐1) have varying expression kinetics, using a subcellular compartment that receives large quantities of endocytosed cargo, such as the recycling endosome, could serve as an efficient mechanism to bring all these membrane proteins together for secondary envelopment. As such, the PM could be considered a principal sorting organelle for HSV‐1 proteins, with the process of packaging viral envelope proteins into endocytic vesicles having the effect of concentrating them within the receiving endocytic compartment. Furthermore, interactions between various glycoproteins as well as underlying tegument proteins could be acquired during this transport to and from the PM.
In summary, our data demonstrate that dynamin‐dependent endocytosis is a major pathway for transporting HSV‐1 envelope proteins to intracellular sites of virus assembly. In the future, it will be important to investigate the transport of the other viral membrane proteins as well as membrane‐associated tegument proteins, and how all the structural components come together to form mature viruses. A key question yet to be answered is whether HSV‐1 envelope proteins are transported as discrete subcomplexes, perhaps with associated tegument proteins, as this will shed further insights into the assembly of these large and complex viruses.
Experimental Procedures
Cell lines and viruses
Vero cells (ATCC), COS7 cells (ATCC) and telomerase‐immortalized human foreskin fibroblasts (HFF‐Tert) were grown in DMEM supplemented with 10% FBS, 2 mm glutamine, 100 U/mL penicillin and 100 µg/mL streptomycin. HaCaT cells 43 were maintained in Glasgow minimal essential medium supplemented with 10% FBS, 2 mm glutamine, 100 U/mL penicillin and 100 µg/mL streptomycin. The pUL36 complementing cell line (HS30; grown with addition of 200 mg/mL of G418) and HSV‐1ΔUL36 virus were a gift from P. Desai, John Hopkins University 44. Strain KOS of HSV‐1 genome cloned as a bacterial artificial chromosome was used in this study 45. Recombinant VP26‐mTurquoise/gM‐EYFP virus was described previously 31. Recombinant VP26‐YFP/ΔgE virus was generated by coinfecting cells with HSV‐1ΔgE‐lacZ (described in 46) and HSV‐1‐VP26‐YFP (described in 47), followed by selection of plaque‐purified recombinant virus both expressing VP26‐YFP and lacking gE expression.
Antibodies and reagents
Monoclonal antibodies against viral proteins were all described previously: gD (LP2, AP7, AP12, LP14) 48, gH [LP11 49, BBH‐1; Abcam, ab110227], gB (CB24) 50, gE (3063, 3114) 51, VP16 (LP1; Abcam, ab110226) 52 and UL36 (CB4) 53. VP5 (ab6508), anti‐alpha Adaptin antibody (AP2) (AC1‐M11), anti‐actin antibody (AC‐40), anti‐HA tag antibody (16B12) and Protein A conjugate to HRP (ab7456) were from Abcam. Anti‐Caveolin‐1 (610406) and anti‐GM130 (610822) were from BD Bioscience. Anti‐Myc tag (9E10) was from Sigma‐Aldrich. Anti‐GFP antibody JL8 was from Clontech. TGN46 antibody was from Dr S. Ponnambalam (University of Leeds). LI‐COR antibodies for WB detection were from LI‐COR Biosciences. All secondary Alexa Fluor antibodies and Transferrin AF‐568 were from Molecular Probes. Dynamin inhibitors: Dynole™ Series Kit (ab120474) including Dynole‐34‐2 (Dynole) and Dynole‐31‐2 (Dynole Negative) were used at 15 µm, while PitStop 2™ (ab120687) and PitStop2 negative control (ab120688) at 30 µm. Dilutions were prepared in serum‐free medium. All inhibitors were purchased from Abcam.
Dominant‐negative protein assays
WT HA‐dynamin 2 pcDNA3.1 (34684) and K44A HA‐dynamin 2 pcDNA3.1 (34685) were obtained from Addgene. AP180‐C myc and VPS4‐EQ YFP were described previously 24, 54. Plasmids of interest or empty pcDNA3.1 were co‐transfected with pcDNA‐UL36 25 or empty control plasmid into COS7 cells. Next day cells were infected at multiplicity of infection (MOI) of 5 PFU/mL with HSV‐1ΔUL36 virus. After 16‐h infection cells and supernatants were harvested together and prepared for titration by three freeze–thaw cycles. Virus titers were assessed using the pUL36 complementing cell line HS30. Cells for WB analysis were lysed with 1% Triton‐X‐100 in PBS with protease inhibitors cocktail (Roche) and run on SDS–PAGE followed by detection of VP16 and actin. Cells for immunostaining were fixed with 4% ultra‐pure formaldehyde (Polysciences, cat # 04018‐1) 10 h after infection. Antibodies specific to gD (LP2) were added 15 min before fixing. For transferrin uptake cells were transfected with pcDNA3.1, dominant negative AP180, dynamin WT or dynamin K44A for 24 h. Cells were incubated with Transferrin Alexa Fluor 568 for 5 min in serum‐free medium. After fixing and permeabilization immunodetection of HA‐tag and Myc‐tag was performed.
Neutralization assay
HaCaT and COS7 cells were seeded on 24‐well plates at 105 cells per well 1 day prior to infection with HSV‐1 at MOI = 3. After 1 h cells were incubated with acid wash (40 mm citric acid, 135 mm NaCl, 10 mm KCl, pH 3.0) for 1 min to inactivate residual virus particles that had not entered cells. After three washes with PBS, fresh medium containing 5 µg/mL of purified monoclonal antibodies directed to VP16, gD and gH was added to the cells. VP16 tegument protein antibody was used as negative control. For each glycoprotein, neutralizing (LP2 and LP11) and non‐neutralizing (AP7 and BBH‐1) antibodies were used. Cells were incubated with antibodies for 16 h, then washed with PBS and trypsinized for 10 min to remove all extracellular infectivity. After pelleting cells and washing with PBS, cells were resuspended in complete media, subjected to freeze–thaw three times and intracellular viruses were titrated using Vero cells. Wells used for WB analysis were washed with PBS and lysed with 1% Triton‐X‐100 in PBS with protease inhibitors cocktail (Roche). Supernatants for WB analysis of secreted particles were collected and cellular debris was removed by two subsequent centrifugations at 6200× g for 10 min. Viruses were pelleted by ultracentrifugation at 75 000× g for 3 h at 4°C using a Beckman Coulter TLA‐55 rotor. Pellets were lysed and run on SDS–PAGE. All proteins were detected with specific primary antibodies and secondary LI‐COR antibodies. gD was detected using Protein‐A HRP and enhanced chemiluminescence (ECL) to avoid detection of antibodies used in uptake assay, which migrate on SDS–PAGE gels at a similar position as gD.
Immunofluorescence assay
All samples were fixed for 10 min using 4% ultra‐pure formaldehyde. Permeabilization was performed with 0.1% Triton‐X‐100 in PBS for 5 min. Cells were then blocked with 2% FBS for 30 min and incubated with primary antibodies for 1 h. For some experiments, primary antibodies specific to viral glycoproteins were added for uptake before cells were fixed. Isotype‐specific secondary antibodies conjugated to Alexa Fluor dyes were used for detection. Cells were imaged using epifluorescence microscope (Olympus IX81) or confocal microscope (Zeiss LSM780).
Wide‐field fluorescence, bright field and dSTORM imaging
Imaging was performed on an inverted TIRF microscope (Nikon TE‐300) custom‐built for dSTORM acquisition, as previously described 31, 55, 56, using highly inclined illumination 57. The microscope was equipped with an iXon3 897 EM‐CCD camera (Andor). The following laser and filter combinations were used: 640 nm diode laser (either iBeam Smart, Toptica) and filter 676/37 (Semrock) for AF647, 561 nm laser (DPSS laser, Oxxius) and 607/70 (Semrock) for AF568, 491 nm (DPSS laser, Cobolt) and 530/55 (Semrock) for EYFP and 405 nm diode laser with 530/55 (Semrock) for mTurquoise. The wide‐field fluorescence images were taken before the dSTORM acquisition using low‐power illumination (∼1 mW), acquiring a 50‐frames stack (100‐millisecond exposure). The bright‐field images were taken after the dSTORM imaging to minimize photobleaching from the transmitted light illumination, acquiring a 50‐frames stack (100‐millisecond exposure). For the two‐color dSTORM acquisition, 16 000–20 000 frames were sequentially acquired for AF647 and AF568 at a frame rate of ∼65 Hz (15‐millisecond exposure) with irradiation intensities at 1–5 kW/cm2 in switching buffer [100 mm mercaptoethylamine (Sigma) in PBS, pH 7.2].
Image analysis
The wide‐field fluorescence and bright‐field images were obtained by averaging the 50‐frames stack. Additionally, the continuous background was removed from the wide‐field fluorescence images by applying a rolling‐ball algorithm 58. All dSTORM datasets were analyzed using rapidSTORM 3.3 59. For the two‐color dSTORM, the chromatic offset between the two channels was corrected by using localization‐based correction using a custom‐written MATLAB (The MathWorks) routine 60. The chromatic transformations were obtained from the combination of multiple fields of view containing randomly distributed white fluorescent microspheres (100‐nm TetraSpeck microsphere, Invitrogen).
Supporting information
Figure S1 : Antibodies incorporated into virions during assembly affect HSV ‐1 infectivity. A) COS7 cells were infected with HSV‐1 at MOI = 3 and then incubated with 5 µg/mL of antibodies (LP1 for VP16, LP2 and AP7 for gD and LP11 and BBH‐1 for gH) for 16 h. To remove and inactivate extracellular viruses, cells were trypsinized for 10 min. Pellets were washed and harvested for titration in Vero cells by three freeze–thaw cycles. Additional cell pellet samples were lysed in PBS + 1% Triton‐X‐100 + protease inhibitors and analyzed by WB for the presence of VP16 and actin. Secondary anti‐mouse antibodies used for detection were also able to detect the heavy chains of antibodies used for uptake (IgGH). Data are represented as mean ± SD from at least three independent experiments with each experiment containing two biological replicates per condition. B) COS7 cells were infected with VP26‐mTurquoise/gM‐EYFP virus and incubated with 5 µg/mL of gD‐specific antibodies (LP2) for 16 h. Cells were then fixed, permeabilized and labeled with primary antibodies to GM130 followed by isotope‐specific secondary Alexa Fluor antibodies. Cells were imaged by confocal microscope. Scale bar = 10 µm.
Figure S2 : Dominant‐negative dynamin decreases endocytosis of gB and gE. WT or K44A dynamin‐transfected COS7 cells were infected with VP26‐mTurquoise/gM‐EYFP for 8 h and then fed with anti‐gB (CB24) or gE (3114) antibodies for 15 min before fixing. Cells were then permeabilized, immunostained with anti‐HA antibodies and then labeled with secondary antibodies. Images were obtained using epifluorescence microscope. Scale bar = 10 µm.
Figure S3 : Effect of endocytosis inhibitors on uptake of transferrin and glycoproteins into COS7 cells. A) Cells were incubated with DMSO, 15 µm Dynole or Dynole negative control and 30 µm PitStop2 or its negative control for 15 min. Next, transferrin conjugated to Alexa Fluor 568 was added for 5 min. Cells were then washed on ice, fixed and imaged using an epifluorescence microscope. Scale bar = 20 µm. B) Cells were infected with HSV‐1 for 8 h. For the last 30 min, cells were incubated with DMSO, 15 µm Dynole or Dynole negative control and 30 µm PitStop2 or its negative control. After 15‐min incubation, antibodies specific to viral glycoproteins (LP2 for gD, 3063 for gE, CB24 for gB and LP11 for gH) were added and incubated for a further 15 min. After fixing and permeabilizing, cells were stained with secondary antibodies and imaged by epifluorescence microscopy. Scale bar = 20 µm.
Figure S4 : Low‐power images corresponding to Figures 4 and 6. A) HFF‐Tert cells were infected with VP26‐mTurquoise/gM‐EYFP recombinant virus for 8 h. Thirty minutes before the end of incubation cells were treated with 15 µm Dynole or Dynole negative control and 30 µm PitStop2 or its negative control. After 15 min antibodies specific to gD (LP2) were added and incubated for a further 15 min. Cells were then fixed, permeabilized and labeled with secondary antibodies. Images were acquired using a confocal microscope. Scale bar = 10 µm. B) HFF‐Tert cells were infected with VP26‐mTurquoise/gM‐EYFP recombinant virus for 8 h. Antibodies specific to HSV‐1 gD, gB, gH and gE were added in various combinations for the last 15 min of incubation. After fixing and permeabilization, isotype‐specific secondary Alexa Fluor antibodies were used and images were acquired using a confocal microscope. Scale bar = 5 µm.
Figure S5 : dSTORM images of clathrin and caveolae‐dependent endocytosis of gE. HFF‐Tert cells were infected with VP26‐mTurquoise/gM‐EYFP recombinant virus for 8 h. gE‐specific antibodies were added to the cells for uptake 15 min before fixation and permeabilization. Clathrin‐coated pits or caveolae were detected with anti AP‐2 (A) or Cav‐1 (B) monoclonal antibodies and subtype‐specific secondary Alexa Fluor antibodies. Wide‐field images were acquired for VP26 (blue) and gM (yellow), and merged with dSTORM images of gE (Alexa Fluor 568 channel) and AP2/Cav1 (Alexa Fluor 647 channel). Scale bars = 10 µm, 500 nm and 200 nm.
Acknowledgments
The authors would like to thank Michael Hollinshead and Kevin Moreau for helpful discussions, and Christopher M. Smith for generating the YFP‐VP26/ΔgE virus. This work was supported by grants from the Leverhulme Trust (grant RPG‐2012‐793), the Royal Society (University Research Fellowship UF090010), the Engineering and Physical Sciences Research Council, UK (grant EP/H018301/1, EP/L015889/1) and by the Medical Research Council (grant MR/K015850/1). Raw numerical data, raw microscopy image data used for quantification and sample dSTORM data are archived at https://www.repository.cam.ac.uk/handle/1810/251273. The authors declare no conflicts of interest. A. A. and C. M. C. designed the study. A. A., R. F. L. and A. F. J. J. performed experiments. All authors contributed to data interpretation and to the writing of the manuscript.
References
- 1. Farooq AV, Shukla D. Herpes simplex epithelial and stromal keratitis: an epidemiologic update. Surv Ophthalmol 2012;57:448–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Whitley RJ. Herpes simplex encephalitis: adolescents and adults. Antiviral Res 2006;71:141–148. [DOI] [PubMed] [Google Scholar]
- 3. Jenkins FJ, Rowe DT, Rinaldo CR. Herpesvirus infections in organ transplant recipients. Clin Diagn Lab Immunol 2003;10:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Akhtar J, Shukla D. Viral entry mechanisms: cellular and viral mediators of herpes simplex virus entry. FEBS J 2009;276:7228–7236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Campadelli‐Fiume G, Menotti L, Avitabile E, Gianni T. Viral and cellular contributions to herpes simplex virus entry into the cell. Curr Opin Virol 2012;2:28–36. [DOI] [PubMed] [Google Scholar]
- 6. Abaitua F, Hollinshead M, Bolstad M, Crump CM, O'Hare P. A nuclear localization signal in herpesvirus protein VP1‐2 is essential for infection via capsid routing to the nuclear pore. J Virol 2012;86:8998–9014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Skepper JN, Whiteley A, Browne H, Minson A. Herpes simplex virus nucleocapsids mature to progeny virions by an envelopment → deenvelopment → reenvelopment pathway. J Virol 2001;75:5697–5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mettenleiter TC. Budding events in herpesvirus morphogenesis. Virus Res 2004;106:167–180. [DOI] [PubMed] [Google Scholar]
- 9. Crump CM, Bruun B, Bell S, Pomeranz LE, Minson T, Browne HM. Alphaherpesvirus glycoprotein M causes the relocalization of plasma membrane proteins. J Gen Virol 2004;85:3517–3527. [DOI] [PubMed] [Google Scholar]
- 10. Ren Y, Bell S, Zenner HL, Kathy Lau SY, Crump CM. Glycoprotein M is important for the efficient incorporation of glycoprotein H‐L into herpes simplex virus type 1 particles. J Gen Virol 2012;93:319–329. [DOI] [PubMed] [Google Scholar]
- 11. Blondeau C, Pelchen‐Matthews A, Mlcochova P, Marsh M, Milne RSB, Towers GJ. Tetherin restricts herpes simplex virus 1 and is antagonized by glycoprotein M. J Virol 2013;87:13124–13133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lau S, Crump C. HSV‐1 gM and the gK/pUL20 complex are important for the localization of gD and gH/L to viral assembly sites. Viruses 2015;7:915–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lubinski JM, Lazear HM, Awasthi S, Wang F, Friedman HM. The herpes simplex virus 1 IgG fc receptor blocks antibody‐mediated complement activation and antibody‐dependent cellular cytotoxicity in vivo. J Virol 2011;85:3239–3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ndjamen B, Farley AH, Lee T, Fraser SE, Bjorkman PJ. The herpes virus Fc receptor gE‐gI mediates antibody bipolar bridging to clear viral antigens from the cell surface. PLoS Pathog 2014;10:e1003961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Johns HL, Gonzalez‐Lopez C, Sayers CL, Hollinshead M, Elliott G. Rab6 dependent post‐golgi trafficking of HSV1 envelope proteins to sites of virus envelopment. Traffic 2014;15:157–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Calistri A, Sette P, Salata C, Cancellotti E, Forghieri C, Comin A, Göttlinger H, Campadelli‐Fiume G, Palù G, Parolin C. Intracellular trafficking and maturation of herpes simplex virus type 1 gB and virus egress require functional biogenesis of multivesicular bodies. J Virol 2007;81:11468–11478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Johnson DC, Baines JD. Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol 2011;9:382–394. [DOI] [PubMed] [Google Scholar]
- 18. Sugimoto K, Uema M, Sagara H, Tanaka M, Sata T, Hashimoto Y, Kawaguchi Y. Simultaneous tracking of capsid, tegument, and envelope protein localization in living cells infected with triply fluorescent herpes simplex virus 1. J Virol 2008;82:5198–5211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Henaff D, Radtke K, Lippé R. Herpesviruses exploit several host compartments for envelopment. Traffic 2012;13:1443–1449. [DOI] [PubMed] [Google Scholar]
- 20. Hollinshead M, Johns HL, Sayers CL, Gonzalez‐Lopez C, Smith GL, Elliott G. Endocytic tubules regulated by Rab GTPases 5 and 11 are used for envelopment of herpes simplex virus. EMBO J 2012;31:4204–4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Marks B, Stowell MH, Vallis Y, Mills IG, Gibson A, Hopkins CR, McMahon HT. GTPase activity of dynamin and resulting conformation change are essential for endocytosis. Nature 2001;410:231–235. [DOI] [PubMed] [Google Scholar]
- 22. Henley JR, Krueger EWA, Oswald BJ, McNiven MA. Dynamin‐mediated internalization of caveolae. J Cell Biol 1998;141:85–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pawliczek T, Crump CM. Herpes simplex virus type 1 production requires a functional ESCRT‐III complex but is independent of TSG101 and ALIX expression. J Virol 2009;83:11254–11264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhao X, Greener T, Al‐Hasani H, Cushman SW, Eisenberg E, Greene LE. Expression of auxilin or AP180 inhibits endocytosis by mislocalizing clathrin: evidence for formation of nascent pits containing AP1 or AP2 but not clathrin. J Cell Sci 2001;114:353–365. [DOI] [PubMed] [Google Scholar]
- 25. Crump CM, Yates C, Minson T. Herpes simplex virus type 1 cytoplasmic envelopment requires functional Vps4. J Virol 2007;81:7380–7387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jones SM, Howell KE, Henley JR, Cao H, McNiven MA. Role of dynamin in the formation of transport vesicles from the trans‐Golgi network. Science 1998;279:573–577. [DOI] [PubMed] [Google Scholar]
- 27. Chapman TL, You I, Joseph IM, Bjorkman PJ, Morrison SL, Raghavan M. Characterization of the interaction between the herpes simplex virus type I Fc receptor and immunoglobulin G. J Biol Chem 1999;274:6911–6919. [DOI] [PubMed] [Google Scholar]
- 28. Ortiz Beitia, de Zarate I, Cantero‐Aguilar L, Longo M, Berlioz‐Torrent C, Rozenberg F. Contribution of endocytic motifs in the cytoplasmic tail of herpes simplex virus type 1 glycoprotein B to virus replication and cell‐cell fusion. J Virol 2007;81:13889–13903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nixdorf R, Klupp BG, Mettenleiter TC. Role of the cytoplasmic tails of pseudorabies virus glycoproteins B, E and M intracellular localization and virion incorporation. J Gen Virol 2001;82:215–226. [DOI] [PubMed] [Google Scholar]
- 30. Van de Linde S, Löschberger A, Klein T, Heidbreder M, Wolter S, Heilemann M, Sauer M. Direct stochastic optical reconstruction microscopy with standard fluorescent probes. Nat Protoc 2011;6:991–1009. [DOI] [PubMed] [Google Scholar]
- 31. Laine RF, Albecka A, van de Linde S, Rees EJ, Crump CM, Kaminski CF. Structural analysis of herpes simplex virus by optical super‐resolution imaging. Nat Commun 2015;6:5980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nichols B. Caveosomes and endocytosis of lipid rafts. J Cell Sci 2003;116:4707–4714. [DOI] [PubMed] [Google Scholar]
- 33. Fishman JB, Fine RE. A trans Golgi‐derived exocytic coated vesicle can contain both newly synthesized cholinesterase and internalized transferrin. Cell 1987;48:157–164. [DOI] [PubMed] [Google Scholar]
- 34. Mallard F, Tang BL, Galli T, Tenza D, Saint‐Pol A, Yue X, Antony C, Hong W, Goud B, Johannes L. Early/recycling endosomes‐to‐TGN transport involves two SNARE complexes and a Rab6 isoform. J Cell Biol 2002;156:653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Monier S, Jollivet F, Janoueix‐Lerosey I, Johannes L, Goud B. Characterization of novel Rab6‐interacting proteins involved in endosome‐to‐TGN transport. Traffic 2002;3:289–297. [DOI] [PubMed] [Google Scholar]
- 36. Hanson PI, Cashikar A. Multivesicular body morphogenesis. Annu Rev Cell Dev Biol 2012;28:337–362. [DOI] [PubMed] [Google Scholar]
- 37. Votteler J, Sundquist WI. Virus budding and the ESCRT pathway. Cell Host Microbe 2013;14:232–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Agromayor M, Martin‐Serrano J. Knowing when to cut and run: mechanisms that control cytokinetic abscission. Trends Cell Biol 2013;23:433–441. [DOI] [PubMed] [Google Scholar]
- 39. Rahn E, Petermann P, Hsu M‐J, Rixon FJ, Knebel‐Mörsdorf D. Entry pathways of herpes simplex virus type 1 into human keratinocytes are dynamin‐ and cholesterol‐dependent. PLoS One 2011;6:e25464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Devadas D, Koithan T, Diestel R, Prank U, Sodeik B, Döhner K. Herpes simplex virus internalization into epithelial cells requires Na(+)/H(+) exchangers and p21‐activated kinases but neither clathrin‐ nor caveolin‐mediated endocytosis. J Virol 2014;88:13378–13395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sinha B, Köster D, Ruez R, Gonnord P, Bastiani M, Abankwa D, Stan RV, Butler‐Browne G, Vedie B, Johannes L, Morone N, Parton RG, Raposo G, Sens P, Lamaze C, et al. Cells respond to mechanical stress by rapid disassembly of caveolae. Cell 2011;144:402–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Parton RG, del Pozo MA. Caveolae as plasma membrane sensors, protectors and organizers. Nat Rev Mol Cell Biol 2013;14:98–112. [DOI] [PubMed] [Google Scholar]
- 43. Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol 1988;106:761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Desai PJ. A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA‐filled capsids in the cytoplasm of infected cells. J Virol 2000;74:11608–11618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gierasch WW, Zimmerman DL, Ward SL, VanHeyningen TK, Romine JD, Leib DA. Construction and characterization of bacterial artificial chromosomes containing HSV‐1 strains 17 and KOS. J Virol Methods 2006;135:197–206. [DOI] [PubMed] [Google Scholar]
- 46. Browne H, Bell S, Minson T. Analysis of the requirement for glycoprotein m in herpes simplex virus type 1 morphogenesis. J Virol 2004;78:1039–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hutchinson I, Whiteley A, Browne H, Elliott G. Sequential localization of two herpes simplex virus tegument proteins to punctate nuclear dots adjacent to ICP0 domains. J Virol 2002;76:10365–10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Minson AC, Hodgman TC, Digard P, Hancock DC, Bell SE, Buckmaster EA. An analysis of the biological properties of monoclonal antibodies against glycoprotein D of herpes simplex virus and identification of amino acid substitutions that confer resistance to neutralization. J Gen Virol 1986;67:1001–1013. [DOI] [PubMed] [Google Scholar]
- 49. Buckmaster EA, Gompels U, Minson A. Characterization and physical mapping of an HSV-1 glycoprotein of approximately 115 × 103 molecular weight. Virology 1984;139:408–413. [DOI] [PubMed] [Google Scholar]
- 50. Zenner HL, Yoshimura S‐I, Barr FA, Crump CM. Analysis of Rab GTPase‐activating proteins indicates that Rab1a/b and Rab43 are important for herpes simplex virus 1 secondary envelopment. J Virol 2011;85:8012–8021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Collins WJ, Johnson DC. Herpes simplex virus gE/gI expressed in epithelial cells interferes with cell‐to‐cell spread. J Virol 2003;77:2686–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McLean C, Buckmaster A, Hancock D, Buchan A, Fuller A, Minson A. Monoclonal antibodies to three non‐glycosylated antigens of herpes simplex virus type 2. J Gen Virol 1982;63:297–305. [DOI] [PubMed] [Google Scholar]
- 53. Svobodova S, Bell S, Crump CM. Analysis of the interaction between the essential herpes simplex virus 1 tegument proteins VP16 and VP1/2. J Virol 2012;86:473–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bishop N, Woodman P. ATPase‐defective mammalian VPS4 localizes to aberrant endosomes and impairs cholesterol trafficking. Mol Biol Cell 2000;11:227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Erdelyi M, Rees E, Metcalf D. Correcting chromatic offset in multicolor super‐resolution localization microscopy. Opt Express 2013;21:12177–12183. [DOI] [PubMed] [Google Scholar]
- 56. Schierle GK. In situ measurements of the formation and morphology of intracellular β‐amyloid fibrils by super‐resolution fluorescence imaging. J Am Chem Soc 2011;133:12902–12905. [DOI] [PubMed] [Google Scholar]
- 57. Tokunaga M, Imamoto N, Sakata‐Sogawa K. Highly inclined thin illumination enables clear single‐molecule imaging in cells. Nat Methods 2008;5:159–161. [DOI] [PubMed] [Google Scholar]
- 58. Sternberg S. Biomedical image processing. IEEE Comput 1983;16:22–34. [Google Scholar]
- 59. Wolter S, Löschberger A, Holm T. rapidSTORM: accurate, fast open‐source software for localization microscopy. Nat Methods 2012;9:1040–1042. [DOI] [PubMed] [Google Scholar]
- 60. Annibale P, Scarselli M, Greco M, Radenovic A. Identification of the factors affecting co‐localization precision for quantitative multicolor localization microscopy. Opt Nanoscopy 2012;1:9. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 : Antibodies incorporated into virions during assembly affect HSV ‐1 infectivity. A) COS7 cells were infected with HSV‐1 at MOI = 3 and then incubated with 5 µg/mL of antibodies (LP1 for VP16, LP2 and AP7 for gD and LP11 and BBH‐1 for gH) for 16 h. To remove and inactivate extracellular viruses, cells were trypsinized for 10 min. Pellets were washed and harvested for titration in Vero cells by three freeze–thaw cycles. Additional cell pellet samples were lysed in PBS + 1% Triton‐X‐100 + protease inhibitors and analyzed by WB for the presence of VP16 and actin. Secondary anti‐mouse antibodies used for detection were also able to detect the heavy chains of antibodies used for uptake (IgGH). Data are represented as mean ± SD from at least three independent experiments with each experiment containing two biological replicates per condition. B) COS7 cells were infected with VP26‐mTurquoise/gM‐EYFP virus and incubated with 5 µg/mL of gD‐specific antibodies (LP2) for 16 h. Cells were then fixed, permeabilized and labeled with primary antibodies to GM130 followed by isotope‐specific secondary Alexa Fluor antibodies. Cells were imaged by confocal microscope. Scale bar = 10 µm.
Figure S2 : Dominant‐negative dynamin decreases endocytosis of gB and gE. WT or K44A dynamin‐transfected COS7 cells were infected with VP26‐mTurquoise/gM‐EYFP for 8 h and then fed with anti‐gB (CB24) or gE (3114) antibodies for 15 min before fixing. Cells were then permeabilized, immunostained with anti‐HA antibodies and then labeled with secondary antibodies. Images were obtained using epifluorescence microscope. Scale bar = 10 µm.
Figure S3 : Effect of endocytosis inhibitors on uptake of transferrin and glycoproteins into COS7 cells. A) Cells were incubated with DMSO, 15 µm Dynole or Dynole negative control and 30 µm PitStop2 or its negative control for 15 min. Next, transferrin conjugated to Alexa Fluor 568 was added for 5 min. Cells were then washed on ice, fixed and imaged using an epifluorescence microscope. Scale bar = 20 µm. B) Cells were infected with HSV‐1 for 8 h. For the last 30 min, cells were incubated with DMSO, 15 µm Dynole or Dynole negative control and 30 µm PitStop2 or its negative control. After 15‐min incubation, antibodies specific to viral glycoproteins (LP2 for gD, 3063 for gE, CB24 for gB and LP11 for gH) were added and incubated for a further 15 min. After fixing and permeabilizing, cells were stained with secondary antibodies and imaged by epifluorescence microscopy. Scale bar = 20 µm.
Figure S4 : Low‐power images corresponding to Figures 4 and 6. A) HFF‐Tert cells were infected with VP26‐mTurquoise/gM‐EYFP recombinant virus for 8 h. Thirty minutes before the end of incubation cells were treated with 15 µm Dynole or Dynole negative control and 30 µm PitStop2 or its negative control. After 15 min antibodies specific to gD (LP2) were added and incubated for a further 15 min. Cells were then fixed, permeabilized and labeled with secondary antibodies. Images were acquired using a confocal microscope. Scale bar = 10 µm. B) HFF‐Tert cells were infected with VP26‐mTurquoise/gM‐EYFP recombinant virus for 8 h. Antibodies specific to HSV‐1 gD, gB, gH and gE were added in various combinations for the last 15 min of incubation. After fixing and permeabilization, isotype‐specific secondary Alexa Fluor antibodies were used and images were acquired using a confocal microscope. Scale bar = 5 µm.
Figure S5 : dSTORM images of clathrin and caveolae‐dependent endocytosis of gE. HFF‐Tert cells were infected with VP26‐mTurquoise/gM‐EYFP recombinant virus for 8 h. gE‐specific antibodies were added to the cells for uptake 15 min before fixation and permeabilization. Clathrin‐coated pits or caveolae were detected with anti AP‐2 (A) or Cav‐1 (B) monoclonal antibodies and subtype‐specific secondary Alexa Fluor antibodies. Wide‐field images were acquired for VP26 (blue) and gM (yellow), and merged with dSTORM images of gE (Alexa Fluor 568 channel) and AP2/Cav1 (Alexa Fluor 647 channel). Scale bars = 10 µm, 500 nm and 200 nm.