

Abstract
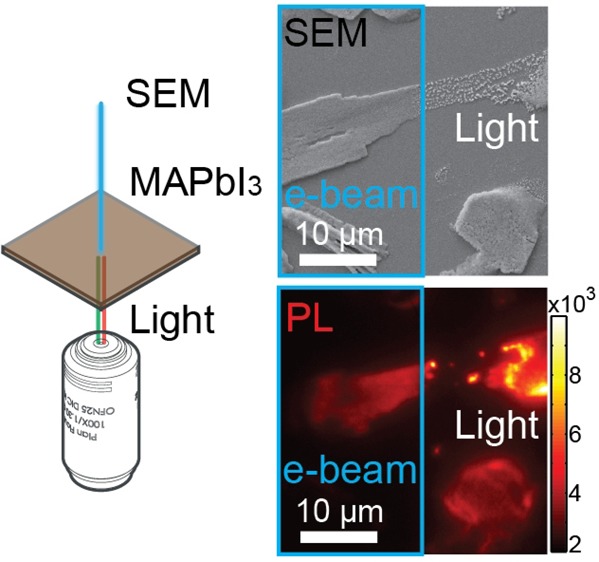
Organometal halide perovskites show promising features for cost-effective application in photovoltaics. The material instability remains a major obstacle to broad application because of the poorly understood degradation pathways. Here, we apply simultaneous luminescence and electron microscopy on perovskites for the first time, allowing us to monitor in situ morphology evolution and optical properties upon perovskite degradation. Interestingly, morphology, photoluminescence (PL), and cathodoluminescence of perovskite samples evolve differently upon degradation driven by electron beam (e-beam) or by light. A transversal electric current generated by a scanning electron beam leads to dramatic changes in PL and tunes the energy band gaps continuously alongside film thinning. In contrast, light-induced degradation results in material decomposition to scattered particles and shows little PL spectral shifts. The differences in degradation can be ascribed to different electric currents that drive ion migration. Moreover, solution-processed perovskite cuboids show heterogeneity in stability which is likely related to crystallinity and morphology. Our results reveal the essential role of ion migration in perovskite degradation and provide potential avenues to rationally enhance the stability of perovskite materials by reducing ion migration while improving morphology and crystallinity. It is worth noting that even moderate e-beam currents (86 pA) and acceleration voltages (10 kV) readily induce significant perovskite degradation and alter their optical properties. Therefore, attention has to be paid while characterizing such materials using scanning electron microscopy or transmission electron microscopy techniques.
In the past few years organometal halide perovskites attracted significant research interest because of their unique physical and chemical properties that make them key components for application in cost-effective photovoltaics,1−3 They generally possess broad absorption spectra, and the composing materials such as CH3NH3PbI3 act as a light harvester to achieve outstanding power conversion efficiencies exceeding 20%.1,3−5 Despite recent advances in the field demonstrating the enormous potential in energy conversion performance, a major obstacle to large-scale application lies within the inherent material instability.6 Under ambient conditions, CH3NH3PbI3 easily undergoes decomposition into PbI2, facilitated by moisture,7 light,8 and electrical voltages,9 severely limiting the device lifetime.10 Upon suppression of the degradation pathway facilitated by moisture, the device lifetime can be improved substantially making use of surface protection layers and encapsulation.11,12 However, the mechanisms underlying the light and electrical degradation pathways are unfortunately poorly understood, making rational strategies to further improve long-term stability of these materials challenging.2,6,13 In addition, deeper insight into the interaction of light and electrons in perovskites is necessary to improve our understanding of many fundamental properties, such as their hysteresis behavior,14 ferroelectric effects,15 photoluminescence (PL),16−18 cathodoluminescence (CL),19 and ion migration.9
In this work, we reveal the impact of ion migration inside the perovskite structure on the material degradation by light and electric currents in methylammonium lead iodide perovskites by means of integrated luminescence and electron microscopy (iLEM) (Figure S1). Morphologies of polycrystalline perovskite films, as well as their PL and CL, evolve differently under light and under the electric current caused by a scanning electron beam (e-beam). The different material responses to light and the e-beam are associated with different electric currents that drive ion migration in the material. Moreover, heterogeneity of material stability is demonstrated in solution-processed perovskite cuboids, indicating potentials to rationally enhance stability of perovskite materials by reducing ion migration while improving crystallinity and morphology control.
Supported CH3NH3PbI3 films were prepared on ITO-coated glass slides using the conventional one-step deposition method.18 The resulting isolated deposits (Figure 1a) have a characteristic perovskite crystal structure, evident from X-ray diffraction analysis (Figure S2). The deposited structures showed uniform PL emission spectra centered at 768 nm ±2 nm with a full width at half-maximum of 38 nm ± 2 nm (Figure S3). The PL intensity from the same structure reduced by a factor of 3 when the sample chamber was altered between ambient conditions and the high vacuum (10–3 Pa) in the iLEM instrument without changes in the emission spectra (Figures S4 and S5). The emission intensity changes are related to the oxygen content in the environment.20
Figure 1.
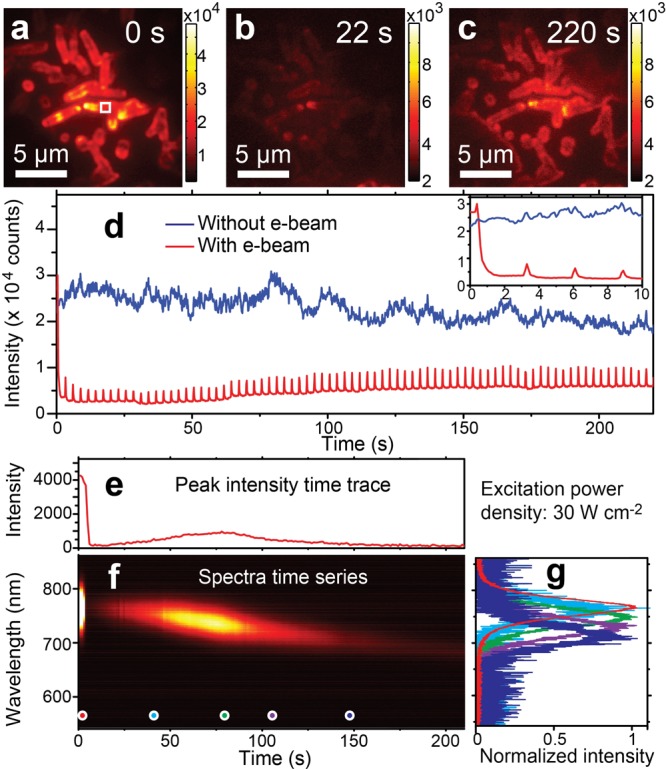
Scanning e-beam altering the perovskite PL. (a–c) PL images taken at different stages of the intensity changes (0, 22, and 220 s). The excitation power density was 14 W/cm2. The scanning e-beam current was 86 pA. (d) PL time traces of regions with (red) and without (blue) the scanning e-beam. The PL intensity trace under e-beam scanning (red) is averaged on a randomly selected region highlighted with a solid white square in panel a. The inset shows both traces in the first 10 s. (e, f) PL intensity time trace and the corresponding spectral evolution under laser illumination (30 W/cm2) and e-beam scanning (86 pA). (g) Emission spectra (normalized) at different time points, which are highlighted with corresponding colors in panel f.
It is worth noting that the samples were kept under the vacuum conditions in the scanning electron microscopy (SEM) chamber throughout the experiments in this study. Without the presence of air and moisture, CH3NH3PbI3 films take several weeks to degrade in vacuum.21 Therefore, the vacuum-induced perovskite degradation of perovskite in the SEM chamber is negligible during the course of our experiments (less than 10 h). However, the vacuum can instantly remove low-boiling degradation products such as CH3NH2 and HI from the surface. Thus, the vacuum eventually promotes light and e-beam-driven degradation of perovskite structures by preventing of recombination or reaction of degradation products at surfaces, leaving PbI2 as residues.
We first examine the influence of the e-beam on the perovskite PL. To reduce photodamages, 532 nm laser illumination of a low power density of 14 W/cm2 was employed. The perovskite PL intensities are spatially heterogeneous in Figure 1a. Unless otherwise stated, a scanning e-beam of a 10 kV acceleration voltage, which resulted in an electric current of 86 pA through the perovskite film, was applied on sample regions of the same area (730 μm2). While scanning over the sample surface, the concentrated e-beam results in a high instantaneous current density at the sample during the dwell time of 100 ns at each pixel. One frame of e-beam scanning took approximately 2.5 s, leading to an e-beam scanning repetition of about 0.4 Hz at each pixel. The scanning parameters were kept constant for all our measurements. Under the scanning e-beam, the PL displayed an immediate decrease in its intensity to become almost indiscernible in Figure 1b, implying formation of quenched species or destruction of emitting sites.18,22 As the e-beam scanning continued, PL at the same region recovered to approximately 20% of the initial intensity (Figure 1c). The altered PL by the scanning e-beam can be clearly recognized in the time trace (in red color) in Figure 1d. The inset shows the first 10 s of the time traces with and without e-beam scanning, demonstrating the abrupt drop in PL upon e-beam scanning. In contrast, other parts of the perovskite sample, where the scanning e-beam was absent, showed only a gradually decreasing PL with time (in blue in Figure 1d).
The electric current induced by the e-beam dominates the PL recovery. We compare the PL changes by e-beams of different acceleration voltages (5, 10, and 20 kV) and currents (21, 86, 106, 208, and 573 pA). In each measurement, the e-beam scanning was applied on a fresh sample area which was not illuminated before. In all cases, PL immediately reduces upon e-beam exposure. After abrupt drops in PL, gradually recovery in PL by extended e-beam scanning was observed under all e-beam parameters. However, the rates of PL recovery by the e-beam are different when different e-beam currents are applied. Under the same acceleration voltage of 10 kV, stronger beam currents lead to faster PL recovery, as shown in Figure S6. At a similar e-beam current of 86 pA, beams with acceleration voltages of 5 kV and 10 kV result in the almost identical PL recovery time trace in Figure S6. In contrast, when the acceleration voltage is increased to 20 kV, PL recovers much more slowly, implying additional material damages by high-energy electrons. To avoid such additional damage on perovskite materials, a 10 kV acceleration voltage was applied in the rest of our experiments.
Besides changes in PL intensities, periodically appearing sharp spikes can also be observed in the time trace under the scanning e-beam in Figure 1d. The periodic spikes that synchronize with the scanning e-beam clearly demonstrate their electron-induced nature. Such electron-induced luminescence is most likely due to CL (Figures S7 and S8). It is noteworthy that the enhanced CL linearly correlates with the recovered PL intensities (Figure S7).
The e-beam did not only alter perovskite luminescence intensity but also induced a continuous spectral shift from 770 to 700 nm in the recovered PL. Figure 1e shows a PL intensity time trace of the perovskite structure under both laser illumination and e-beam scanning. The evolution of emission spectra is given in Figure 1f. After the immediate drop in PL by the e-beam, the initial recovery in PL under the continuously scanning e-beam showed an emission center at 770 nm (cyan curve in Figure 1g), which is identical with that before the e-beam is applied (red curve in Figure 1g). Moreover, the emission center continuously shifted to 750 nm (green curve in Figure 1g) where the PL intensity reached a maximum. Afterward, the emission spectra continued to shift toward shorter wavelengths while the intensity further decreased. During the spectral shift, the shape and width of the emission spectra remained constant, which is different from spectra shifts due to phase transitions of the material.23,24 Similar spectral shifts, however, were absent without the presence of the scanning e-beam (Figure S9). These observations indicate e-beam treatments as a potential method to alter the energy band gaps at room temperature in the range from 1.61 to 1.77 eV. It is worth noting that these e-beam-induced changes in luminescence properties are long-lasting even after keeping the sample in vacuum or at ambient conditions overnight, implying the chemical and structural origin.
Next, we investigate e-beam-driven structural changes in perovskite films. Figure 2a,b shows SEM images over the same sample area before and after patterning the letter “T” using the e-beam (video S1). The drawn pattern can be revealed by contrast of SEM, white-light transmission, and topographical atomic force microscopy (AFM) images in panels b, c, and d of Figure 2, respectively. Deep cracks of about 20 nm widths can be observed in the e-beam-treated area outlined with solid lines in Figure 2b. Due to the material instability at grain boundaries, these cracks occurred along grain boundaries (Figure S10). The very local morphology change clearly reveals the structural defects induced by the e-beam. Accompanied with the morphology change, reduced light absorption after the e-beam treatment (Figure S11) leads to different light transmission intensities at the e-beam-treated region and its nontreated surroundings, illustrated in Figure 2c. Furthermore, the AFM measurement reveals thinning of the perovskite film at the e-beam-treated region in Figure 2d. Figure 2e shows three height profiles along the three white solid lines in Figure 2d. The cyan profile indicates the film thickness to be 200 nm. The black and the red profiles show the heights across the lines drawn using the e-beam, confirming a thinning of about 70 nm in the film area treated with the e-beam. It is noteworthy that the thickness thinning (70 nm) is much more pronounced than the grain boundary broadening (20 nm). These changes in morphology provide evidence for a directional degradation process of the perovskites driven by the e-beam. Moreover, their luminescence properties alter correspondingly upon the structural changes by degradation. In particular, the emission spectral shifted from 767 to 755 nm, which agrees with previous findings observed in the e-beam-degraded region (Figure S12), confirming the structural origin of the changes in luminescence properties by e-beam-driven degradation.
Figure 2.

Structural changes induced by e-beam treatment and the corresponding changes in PL. (a) SEM micrograph with the minimum electron beam dose acquired on the original perovskite film. (b) SEM micrograph acquired on the identical area after drawing the “T” pattern highlighted with solid lines (guide to the eye). (c) White-light transmission image acquired on the same area after the e-beam treatment. (d) AFM image measured on the same area after the e-beam treatment. (e) Height profiles along the three white solid lines in panel d. (f) SEM micrograph acquired before laser illumination. The left half of the structure, highlighted with a cyan square, is subjected to e-beam scanning (86 pA, 10 kV) for 5 min. (g) PL image acquired after the 500 W/cm2 laser illumination is applied for 12 s. (h) SEM micrograph on the same area in panels f and g after laser illumination.
Interestingly, different structural changes were observed in e-beam- or light-driven degradation. The left-hand portion of Figure 2f, highlighted with a cyan box, was subjected to a scanning e-beam for 5 min to trigger the e-beam-driven degradation. The whole structure was then submitted to wide-field laser illumination of 500 W/cm2, which caused rapid photodamages in the structure within several seconds (Figure 2g). The light-driven degradation resulted in structural decomposition and broke the perovskite structure into spatially scattered particles, as shown in Figure 2h. The e-beam degraded half on the left, however, showed little structural deformation by light.
The difference in morphology evolution caused by the e-beam or by light is a consequence of ion migrations in the material. In analogy with the report by Xiao et al.9 in which directional ion migration was induced by applying an electrical voltage between two electrodes, an electric current along the transverse direction resulted from electrons that were accelerated and were injected toward the ITO side of the sample in our experiment. Ion migration driven by such an electric current will be mainly along the transversal direction. Ions of different charges can move either toward (CH3NH3+) or away from (I–) the top surface of the film that is exposed to e-beam in vacuum. For instance, upon intense injection of electrons, CH3NH2 can form at the top surface and be quickly removed from the surface by the high vacuum. Therefore, thinning in film thickness can be more pronounced than broadening in grain boundaries. Iodide ions, on the other hand, can be driven toward the perovskite glass interface by the same electric current. It is worth noting that local stoichiometric variations in iodide content is recently found to influence PL and CL properties of perovskite films.25 During this process, vacancies and defects are generated in the film. Hence, directional morphology changes and PL and CL property changes can be expected from the electric current transversely applied through perovskite films by the e-beam.
In contrast, without externally applied electric current, light-induced internal charge generation and relocation result in random local charge currents that are only limited by defects, charge traps, and grain boundaries in the perovskite film. The random ion migration determines that degradation products, both CH3NH2 and HI, can form and can be removed at all exposed surfaces including the top surface and the surface of grain boundaries. Therefore, photoinduced degradation in perovskites results mainly in a decreasing PL intensity. Moreover, the residues will be mainly limited by the crystal grains in the film so that randomly scattered particle-like residues can be observed (Figure 2h). The correlation between the residue particle sizes and the grain size, however, requires further investigation. It is worth noting that the surfaces and interfaces play important roles during the material degradation, as readily shown by several studies.11,12
Hence, the degradation-induced changes in PL properties can be attributed to two factors. On the one hand, populated defects and vacancies are created in the perovskite during material degradation, leading to different charge carrier dynamics.16 This is evident from the altered PL intensities and the shortened PL lifetimes by the e-beam-driven degradation (Figure S13). On the other hand, the directional ion migration driven by the electric current transversely through the perovskite film can result in self-doping to form p–n junctions,9,26 changes of chemical composition,27 and thinning in dimensions, thus altering the band gaps. Furthermore, the observed increasing CL under e-beam scanning can be a direct consequence of degradation due to the enriched presence of PbI2, which shows brighter CL under identical conditions (Figure S14). Our results provide unambiguous evidence for the extreme sensitivity of polycrystalline perovskite films to an e-beam, even under a moderate electric current of tens of picoamperes in a conventional SEM analysis. Furthermore, the observations above imply the important role that ion migrations may play in the photoelectric properties of perovskites and in their degradation process.
To further verify the essential role of ion migration, we applied the e-beam and laser beam simultaneously to unravel the competition between the two driving forces in perovskite degradation by varying the laser light intensity (Figure S15). As one would expect, upon an excitation power density of 500 W/cm2, e-beam-driven degradation dominated the structural change, leading to an almost static morphology in SEM images (Figure S15c). However, nanocrystals started to appear, grow, and vanish kinetically with time at a much stronger excitation power density of 4.2 kW/cm2 (Figure S15d–g). Such a kinetic structural evolution on the perovskite surface is direct evidence of massive ion migration in the material (video S2). Moreover, the very different structural change observed under the strong light illumination suggests that light-driven ion migration overwhelms the one driven by the e-beam. Therefore, ion migration in perovskites plays an important role in e-beam- or light-driven degradation pathways and the corresponding changes in luminescence properties.
Moreover, we notice heterogeneities in perovskite degradation correlating with morphology in the simultaneous SEM and PL study. Figure 3 shows the in situ evolution of the morphology and PL of two perovskite cuboid clusters supported on an ITO-coated glass slide under moderate light (140 W/cm2) and e-beam (86 pA, 10 kV) illumination. The SEM image and PL image at the beginning of the measurement are given in panels a and b of Figure 3, respectively. The large cluster, outlined with red solid lines, initially showed spatially heterogeneous PL emission localized at regions of abundant grain boundaries. The small cluster, highlighted by a black circle, showed low PL. Over time, both clusters showed an immediate drop in PL intensity caused by the e-beam followed by a PL recovery to reach a maximum before the emission intensity further decreased in Figure 3c, resembling the observations on perovskite films. Their rates of PL increase and decrease during both subsequent phases, however, were very different from each other. The different rates of PL changes are strongly correlated to their degradation speed, evident from the simultaneous SEM and optical image time-series in Figure 3d–l. The larger cluster showed much faster reshaping and degradation than the small cluster (video S3). Moreover, the emission sites also evolved spatially along with structural reorganizations in the large cluster. The small cluster, in contrast, showed slower PL and structural changes. Its dimensions shrank approximately 40 nm in each direction when its emission reached the maximum, shown in Figure 3m,n.
Figure 3.

Structural and PL evolution of two cuboid clusters upon degradation. (a) SEM micrograph of the two clusters before the optical measurement. The large cluster is outlined with red solid lines. The small cluster is highlighted with a black circle. (b) PL image on the identical area at the beginning of the optical measurement. The excitation power density was 140 W/cm2. (c) PL time traces of the large cluster (in red color) and the small cluster (in black color). (d–j) Overlapped SEM and PL images on the identical area at 10, 20, 100, 140, 220, 300, and 380 s, respectively. (k) SEM micrograph on the small cluster before the optical measurement. The orange solid line outlines the shape of the cluster. (l) The SEM image on the small cluster when its PL intensity reaches its maximum at 380 s. Compared with the original shape outlined with the orange solid lines, the dimensions of the cluster shrank almost evenly at each direction for about 40 nm.
The heterogeneous photo- and electrical stability found in perovskite cuboids is most likely due to differences in their photo- and electrical properties, which are determined by crystal size, defects, and crystallinity.16,28 The superior stability observed in the small cluster may benefit from better crystalline packing in single-crystalline particles. In contrast, the large cluster seems intergrow polycrystalline, which contains more defects and instable grain boundaries. The demonstrated influence of structure and photo- and electrical stability of perovskites give tantalizing clues as to the potential to improve the material’s photo- and electrical stability by optimizing morphology and crystallinity.
During the revision of this work, Gradečak and co-workers reported similar observations on e-beam-induced correlating changes in CL, PL, and mass–thickness observed by scanning transmission electron microscopy (STEM).25 By energy-dispersive spectroscopy (EDS), local stoichiometric variations due to e-beam-induced ion segregation were revealed. The PL and CL properties are found to correlate with local iodide content.25 The heterogeneous PL, CL, and stoichiometric variations also imply heterogeneity in material stability of the same perovskite sample film under the same e-beam.25 It is worth mentioning that the nanoampere e-beam currents and acceleration voltages (120 kV)25 used in STEM are drastically different from those used in our study (10 kV and 86 pA).
Acknowledgments
We acknowledge financial support from the Research Foundation-Flanders (FWO, Grant G.0990.11, G.0855.14, G.0197.11, G.0962.13, postdoctoral fellowship to H.Y., E.D., K.J., C. S. and G. L.); KU Leuven Research Fund (GOA2011/03, OT/12/059); the Flemish government through long-term structural funding Methusalem (Meth/08/04)- the Hercules foundation (HER/08/021, HER/11/14)- the Belgian Federal Science Policy Office (IAP-VI/27)- the EC through the Marie Curie ITN project iSwitch (GA-642196); and the ERC projects FLUOROCODE (GA-291593), LIGHT (GA-307523), and PLASMHACAT (GA-280064). H.U. acknowledges the Japan Science and Technology Agency PRESTO program for financial support. P.S. acknowledges the International Center for Frontier Research in Chemistry (icFRC).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpclett.5b02828.
Additional description of synthesis, instruments, and results (PDF)
Drawing the “T” pattern using the e-beam (AVI)
Interplay of electrical and photodegradation under weak and strong light illuminations (AVI)
Degradation of two clusters of perovskite cuboids under simultaneous optical and electron microscopy (AVI)
The authors declare no competing financial interest.
Supplementary Material
References
- Lee M. M.; Teuscher J.; Miyasaka T.; Murakami T. N.; Snaith H. J. Efficient Hybrid Solar Cells Based on Meso-Superstructured Organometal Halide Perovskites. Science 2012, 338, 643–647. 10.1126/science.1228604. [DOI] [PubMed] [Google Scholar]
- Burschka J.; Pellet N.; Moon S. J.; Humphry-Baker R.; Gao P.; Nazeeruddin M. K.; Gratzel M. Sequential Deposition as a Route to High-Performance Perovskite-Sensitized Solar Cells. Nature 2013, 499, 316–319. 10.1038/nature12340. [DOI] [PubMed] [Google Scholar]
- Zhou H.; Chen Q.; Li G.; Luo S.; Song T. B.; Duan H. S.; Hong Z.; You J.; Liu Y.; Yang Y. Interface Engineering of Highly Efficient Perovskite Solar Cells. Science 2014, 345, 542–546. 10.1126/science.1254050. [DOI] [PubMed] [Google Scholar]
- Yang W. S.; Noh J. H.; Jeon N. J.; Kim Y. C.; Ryu S.; Seo J.; Seok S. I. High-Performance Photovoltaic Perovskite Layers Fabricated through Intramolecular Exchange. Science 2015, 348, 1234–1237. 10.1126/science.aaa9272. [DOI] [PubMed] [Google Scholar]
- Jeon N. J.; Noh J. H.; Yang W. S.; Kim Y. C.; Ryu S.; Seo J.; Seok S. I. Compositional Engineering of Perovskite Materials for High-Performance Solar Cells. Nature 2015, 517, 476–480. 10.1038/nature14133. [DOI] [PubMed] [Google Scholar]
- Hodes G.; Cahen D. Photovoltaics: Perovskite Cells Roll Forward. Nat. Photonics 2014, 8, 87–88. 10.1038/nphoton.2014.5. [DOI] [Google Scholar]
- Noh J. H.; Im S. H.; Heo J. H.; Mandal T. N.; Seok S. I. Chemical Management for Colorful, Efficient, and Stable Inorganic–Organic Hybrid Nanostructured Solar Cells. Nano Lett. 2013, 13, 1764–1769. 10.1021/nl400349b. [DOI] [PubMed] [Google Scholar]
- Misra R. K.; Aharon S.; Li B.; Mogilyansky D.; Visoly-Fisher I.; Etgar L.; Katz E. A. Temperature- and Component-Dependent Degradation of Perovskite Photovoltaic Materials under Concentrated Sunlight. J. Phys. Chem. Lett. 2015, 6, 326–330. 10.1021/jz502642b. [DOI] [PubMed] [Google Scholar]
- Xiao Z.; Yuan Y.; Shao Y.; Wang Q.; Dong Q.; Bi C.; Sharma P.; Gruverman A.; Huang J. Giant Switchable Photovoltaic Effect in Organometal Trihalide Perovskite Devices. Nat. Mater. 2014, 14, 193–198. 10.1038/nmat4150. [DOI] [PubMed] [Google Scholar]
- Christians J. A.; Miranda Herrera P. A.; Kamat P. V. Transformation of the Excited State and Photovoltaic Efficiency of CH3NH3PbI3 Perovskite Upon Controlled Exposure to Humidified Air. J. Am. Chem. Soc. 2015, 137, 1530–1538. 10.1021/ja511132a. [DOI] [PubMed] [Google Scholar]
- Hwang I.; Jeong I.; Lee J.; Ko M. J.; Yong K. Enhancing Stability of Perovskite Solar Cells to Moisture by the Facile Hydrophobic Passivation. ACS Appl. Mater. Interfaces 2015, 7, 17330–17336. 10.1021/acsami.5b04490. [DOI] [PubMed] [Google Scholar]
- Li X.; Ibrahim Dar M.; Yi C.; Luo J.; Tschumi M.; Zakeeruddin S. M.; Nazeeruddin M. K.; Han H.; Grätzel M. Improved Performance and Stability of Perovskite Solar Cells by Crystal Crosslinking with Alkylphosphonic Acid Ω-Ammonium Chlorides. Nat. Chem. 2015, 7, 703–711. 10.1038/nchem.2324. [DOI] [PubMed] [Google Scholar]
- Smith I. C.; Hoke E. T.; Solis-Ibarra D.; McGehee M. D.; Karunadasa H. I. A Layered Hybrid Perovskite Solar-Cell Absorber with Enhanced Moisture Stability. Angew. Chem. 2014, 126, 11414–11417. 10.1002/ange.201406466. [DOI] [PubMed] [Google Scholar]
- Xu J.; et al. Perovskite-Fullerene Hybrid Materials Suppress Hysteresis in Planar Diodes. Nat. Commun. 2015, 6, 7081. 10.1038/ncomms8081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutes Y.; Ye L.; Zhou Y.; Pang S.; Huey B. D.; Padture N. P. Direct Observation of Ferroelectric Domains in Solution-Processed CH3NH3PbI3 Perovskite Thin Films. J. Phys. Chem. Lett. 2014, 5, 3335–3339. 10.1021/jz501697b. [DOI] [PubMed] [Google Scholar]
- deQuilettes D. W.; Vorpahl S. M.; Stranks S. D.; Nagaoka H.; Eperon G. E.; Ziffer M. E.; Snaith H. J.; Ginger D. S. Impact of Microstructure on Local Carrier Lifetime in Perovskite Solar Cells. Science 2015, 348, 683–686. 10.1126/science.aaa5333. [DOI] [PubMed] [Google Scholar]
- Christians J. A.; Manser J. S.; Kamat P. V. Multifaceted Excited State of Ch3nh3pbi3. Charge Separation, Recombination, and Trapping. J. Phys. Chem. Lett. 2015, 6, 2086–2095. 10.1021/acs.jpclett.5b00594. [DOI] [PubMed] [Google Scholar]
- Tian Y.; Merdasa A.; Peter M.; Abdellah M.; Zheng K.; Ponseca C. S.; Pullerits T.; Yartsev A.; Sundström V.; Scheblykin I. G. Giant Photoluminescence Blinking of Perovskite Nanocrystals Reveals Single-Trap Control of Luminescence. Nano Lett. 2015, 15, 1603–1608. 10.1021/nl5041397. [DOI] [PubMed] [Google Scholar]
- Bischak C. G.; Sanehira E. M.; Precht J. T.; Luther J. M.; Ginsberg N. S. Heterogeneous Charge Carrier Dynamics in Organic–Inorganic Hybrid Materials: Nanoscale Lateral and Depth-Dependent Variation of Recombination Rates in Methylammonium Lead Halide Perovskite Thin Films. Nano Lett. 2015, 15, 4799–4807. 10.1021/acs.nanolett.5b01917. [DOI] [PubMed] [Google Scholar]
- Tian Y.; Peter M.; Unger E.; Abdellah M.; Zheng K.; Pullerits T.; Yartsev A.; Sundstrom V.; Scheblykin I. G. Mechanistic Insights into Perovskite Photoluminescence Enhancement: Light Curing with Oxygen Can Boost Yield Thousandfold. Phys. Chem. Chem. Phys. 2015, 17, 24978–24987. 10.1039/C5CP04410C. [DOI] [PubMed] [Google Scholar]
- Deretzis I.; Alberti A.; Pellegrino G.; Smecca E.; Giannazzo F.; Sakai N.; Miyasaka T.; La Magna A. Atomistic Origins of CH3NH3PbI3 Degradation to PbI2 in Vacuum. Appl. Phys. Lett. 2015, 106, 131904. 10.1063/1.4916821. [DOI] [Google Scholar]
- Tian Y.; et al. Enhanced Organo-Metal Halide Perovskite Photoluminescence from Nanosized Defect-Free Crystallites and Emitting Sites. J. Phys. Chem. Lett. 2015, 6, 4171–4177. 10.1021/acs.jpclett.5b02033. [DOI] [PubMed] [Google Scholar]
- Wu K.; Bera A.; Ma C.; Du Y.; Yang Y.; Li L.; Wu T. Temperature-Dependent Excitonic Photoluminescence of Hybrid Organometal Halide Perovskite Films. Phys. Chem. Chem. Phys. 2014, 16, 22476–22481. 10.1039/C4CP03573A. [DOI] [PubMed] [Google Scholar]
- Chin X. Y.; Cortecchia D.; Yin J.; Bruno A.; Soci C. Lead Iodide Perovskite Light-Emitting Field-Effect Transistor. Nat. Commun. 2015, 6, 7383. 10.1038/ncomms8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentz O.; Zhao Z.; Gradečak S.. Impacts of Ion Segregation on Local Optical Properties in Mixed Halide Perovskite Films. Nano Lett. 2016. DOI: 10.1021/acs.nanolett.5b05181. [DOI] [PubMed] [Google Scholar]
- Frolova L. A.; Dremova N. N.; Troshin P. A. The Chemical Origin of the P-Type and N-Type Doping Effects in the Hybrid Methylammonium-Lead Iodide (MAPbI(3)) Perovskite Solar Cells. Chem. Commun. 2015, 51, 14917–14920. 10.1039/C5CC05205J. [DOI] [PubMed] [Google Scholar]
- Tian Y.; Scheblykin I. G. Artifacts in Absorption Measurements of Organometal Halide Perovskite Materials: What Are the Real Spectra?. J. Phys. Chem. Lett. 2015, 6, 3466–3470. 10.1021/acs.jpclett.5b01406. [DOI] [PubMed] [Google Scholar]
- Grancini G.; Srimath Kandada A. R.; Frost J. M.; Barker A. J.; De Bastiani M.; Gandini M.; Marras S.; Lanzani G.; Walsh A.; Petrozza A. Role of Microstructure in the Electron–Hole Interaction of Hybrid Lead Halide Perovskites. Nat. Photonics 2015, 9, 695–701. 10.1038/nphoton.2015.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.