

Abstract
Background:
Acquired immunodeficiency syndrome caused by human immunodeficiency virus (HIV) is an immunosuppressive disease. Over the past decades, it has plagued human health due to the grave consequences in its harness.
Objective:
For this reason, anti-HIV agents are imperative, and the search for the same from natural resources would assure the safety.
Materials and Methods:
In this investigation we have performed molecular docking, molecular property prediction, drug-likeness score, and molecular dynamics (MD) simulation to develop a novel anti-HIV drug. We have screened 12 alkaloids from a medicinal plant Toddalia asiatica for its probabilistic binding with the active site of the HIV-1-reverse transcriptase (HIV-1-RT) domain (the major contributor to the onset of the disease).
Results:
The docking results were evaluated based on free energies of binding (ΔG), and the results suggested toddanol, toddanone, and toddalenone to be potent inhibitors of HIV-1-RT. In addition, the alkaloids were subjected to molecular property prediction analysis. Toddanol and toddanone with more rotatable bonds were found to have a drug-likeness score of 0.23 and 0.11, respectively. These scores were comparable with the standard anti-HIV drug zidovudine with a model score 0.28. Finally, two characteristic protein-ligand complexes were exposed to MD simulation to determine the stability of the predicted conformations.
Conclusion:
The toddanol-RT complex showed higher stability and stronger H-bonds than toddanone-RT complex. Based on these observations, we firmly believe that the alkaloid toddanol could aid in efficient HIV-1 drug discovery.
SUMMARY
In the present study, the molecular docking and MD simulations are performed to explore the possible binding mode of HIV 1 RT with 12 alkaloids of T. asiatica. Molecular docking by AutoDock4 revealed three alkaloids toddanol, toddanone, and toddalenone with highest binding affinity towards HIV 1 RT. The drug likeness model score revealed a positive score for toddanol and toddanone which is comparable to the drug likeness score of the standard anti HIV drug zidovudine. Results from simulation analysis revealed that toddanol RT complex is more stable than toddanone RT complex inferring toddanol as a potential anti HIV drug molecule.
Abbreviations used: HIV: Human immunodeficiency virus, HIV 1 RT: HIV 1 reverse transcriptase, RNase H: Ribonuclease H, MD: Molecular dynamics, PDB: Protein databank, RMSD: Root mean square deviation, RMSF: Root mean square fluctuation.
Keywords: Alkaloids, Autodock v4.0, drug-likeness, human immunodeficiency virus-1 reverse transcriptase, molecular dynamics simulation, molecular properties, toddanol, toddanone
INTRODUCTION
Acquired immunodeficiency syndrome is caused by the notorious human immunodeficiency virus (HIV)[1] that infects vital cells in the human immune system specifically CD4+ T cells and dendritic cells.[2] There are two different types of HIV, namely HIV-1 and HIV-2 that differ in their virulence. HIV-2 is less pathogenic and restricted to West Africa; the former is a cause of serious concern, and it is most commonly known as HIV. HIV-1 has various strains, which are further classified into groups and subtypes.[3] There are three key enzymes needed for the development of virion, and one among them is HIV-1-reverse transcriptase (HIV-1-RT). HIV-1-RT has a significant role in the initial stages of HIV replication.[4,5] Transformation of single-stranded viral RNA (the genetic material) into double-stranded DNA is a crucial step in the course of HIV replication, and it is catalyzed by RT. RT has two distinct enzymatic activities such as DNA-polymerase activity to copy either RNA or DNA template, and ribonuclease H (RNase H) activity to cleave the RNA strand in the DNA-RNA heteroduplex.[6,7] HIV-1-RT is a heterodimer with P66 and P51 subunits. The P66 subunit contains both the polymerase and RNase H domains, whereas the P51 subunit lacks the C-terminal RNase domain. Thus, targeting P66 domain could serve as a better therapeutic strategy against HIV. Several clinically proven anti-HIV agents are nucleosides, and their use is inadequate due to severe toxicity and the intrinsic ability of HIV to develop resistance. Hence, there is a dire need for safer alternatives (natural products), that might prove effective in targeting the HIV-1-RT, the major cause of the onset and manifestation of the pathology.
The plant-derived natural products have been in use for thousands of years and remain an essential component of the pharmaceutical industry.[8,9] The secondary metabolites from most of the plants are recognized to have antimicrobial, anticancer, and anti-inflammatory activities.[10] Among them, 20% of alkaloids from plant species have a nitrogen group containing a low-molecular weight compound that intimate the information about the structure and function of alkaloids such as resistance against herbivores, virus, and microbes.[11] In India and China, Toddalia asiatica (Rutaceae) commonly known as an orange climber, forest pepper, and lopez root has been used as a medicine, especially for its bioactive alkaloids.[12,13,14] From aerial to root part, the plant has several medicinal values, such as antiviral, antiplatelet, and analgesic activities.[15] Nitidine is an important alkaloid that is obtained from the roots of T. asiatica.[16] It acts as an antimalarial drug, and it has also been reported to inhibit human lymphoblastoid cell killing by HIV-1. Magnoflorine, an another alkaloid from the members of Rutaceae (Fagara, Phebalium, and Zanthoxylum) has less anti-HIV activity with a maximum cytoprotection of 50%.[17]
In the present investigation, attempts have been made to explore the interaction of 12 alkaloids of T. asiatica with the active site of RT particularly encompassing the P66 subunit using Autodock v4.0 software to propose its efficiency as an anti-HIV agent. Furthermore, the alkaloids were subjected to molecular properties prediction and drug-likeness score to filter and confirm for their anti-HIV activity. The dynamics study was performed to understand the stability and flexibility of the protein-ligand complex to identify pharmacological targets. This is the first report on in silico approach on figuring out the probable interactions of various alkaloids of T. asiatica with HIV-1-RT.
MATERIALS AND METHODS
Receptor and ligand preparation
The three-dimensional (3D) structure of HIV-1-RT (protein databank [PDB] ID: 1REV) was obtained from the PDB[18] Co-crystallized ligands were identified and removed from the structure of 1REV[19] and then crystallographic water molecules were excluded from the 3D coordinate file. Phytochemical alkaloids (ligands) nitidine and magnoflurine from T. asiatica[20] which were already reported as anti-HIV alkaloid, were taken for in silico docking studies and when compared with other alkaloids of the same species such as toddaline, toddaquinoline, toddasine, toddanol, toddanone, toddalenone, flindersine, toddacoumalone, toddacoumaquinone, and toddalenol. The structures of the alkaloids [Figure 1] were drawn using chemsketch.[21] A geometry optimization for each compound was performed using UCSF Chimera.[22] The UCSF Chimera program added Gasteiger atomic partial charges and was run for 10,000 steps of energy minimization.[23]
Figure 1.

Two-dimensional structure of alkaloids used in this study (a) nitidine (b) magnoflurine (c) toddaline (d) toddaquinoline (e) toddasin (f) toddanol (g) toddanone (h) toddalenone (i) flindersine (j) toddacoumalone (k) toddacoumaquinone and (l) toddalenol
Active site prediction
A small region or cleft where the ligand molecule can bind to the receptor protein and produce the preferred outcome is termed as an active site/catalytic site. Identification of this active site residue in the target protein structure has a great range of applications in molecular docking and de novo drug designing. Accurate identification of this catalytic binding site is difficult due to the constant conformational changes of the target protein.[24] The catalytic residue of protein 1REV was examined with the help of a Q-Site Finder, which uses a simple van der Waals probe and the interaction energy to locate energetically favorable binding sites.[25]
Molecular docking using Autodock
The flexible docking study was carried out using Autodock v4.0. The 3D structure of HIV-1-RT (1REV) and the alkaloids were submitted in PDB format with default parameters. Essential hydrogen atoms, Kollman united atom type charges, and solvation parameters were added with the help of Autodock tools.[26] With the help of the chimera program, the 3D structures of the ligand molecules were built, optimized, and changed into a Mol2 file. Furthermore, the nonpolar hydrogen atoms were assigned to the atom and the resulting files were saved as PDBQT files. The binding site for the ligands on HIV-1-RT was identified using Q-Site Finder online server. The grid-based approach was used to minimize the run time; the grid size was set to 60 × 60 × 60 xyz points with a grid spacing of 0.385 Å. The docking simulation was done using the Lamarckian genetic algorithm.[27] Each docking experiment was derived from 10 different conformations. The interaction analysis of protein- ligand complexes and their amino acid position with bond distances were calculated and visualized through the PyMol.[28] Hydrophobic contacts were analyzed using Ligplot.[29]
Prediction of physiochemical properties
Generally, molecules comprising functional groups have certain properties that are similar to known drugs. Therefore, calculating the molecular property is significant in producing a good oral drug, and it is shown to be an important feature in drug discovery and development. Molinspiration server[30] was used to predict the molecular property of the alkaloids that predicts both physiochemical and pharmacological properties such as LogP, hydrogen bonding characteristics, molecular size, and rotatable bonds. The combined effect of physicochemical properties, pharmacokinetics and pharmacodynamics, and the drug-likeness model score were identified using molsoft server. Thus, the alkaloids were subjected to Molinspiration and molsoft online server analysis. Furthermore, the scores were compared with the standard anti-HIV drug. Lipinski's rule of five[31] was used to evaluate the acceptability of the alkaloids.
Protein-Ligand complex simulation
The simulation of the ligand-enzyme complex was performed using the GROMACS 4.5.5 software.[32] Ligand-enzyme complexes with the lowest binding energy were selected for molecular dynamics (MD) simulation. The ligand parameters were analyzed using PRODRG online server[33] in the framework of the GROMOS force-field 43a1.[34,35] The ligand-enzyme complex was solvated using a simple point charge[36] water box under periodic boundary conditions using 1.0 nm distance from the protein to the box faces. The system was then neutralized by Cl− or Na+ counter ions for toddonal-RT and toddanone-RT complex systems, respectively. Energy minimization was done for 1,000 steps by the steepest descent method.[37] For the sake of flowing energy minimization, the systems were equilibrated under constant number of particles, volume, and temperature conditions for 100 ps at 300K, followed by 100 ps under constant number of particles, pressure, and temperature conditions. All the covalent bonds with hydrogen atoms were constrained using the Linear Constraint Solver algorithm. The electrostatic interactions were treated using the Particle Mesh Ewald method.[38] Finally, MD simulation was performed for 20 ns to check the stability of the ligand-enzyme complexes.
The potential of each trajectory produced after MD simulations were analyzed using g_rms, g_rmsf, and g_hbond of GROMACS utilities,[39] and the root mean square deviation (RMSD), root mean square fluctuation (RMSF), and the number of H-bonds formed between the ligand and the enzyme were obtained. The graphs were produced using the XM grace tool.[40]
RESULTS AND DISCUSSION
Binding site and ligand conformation
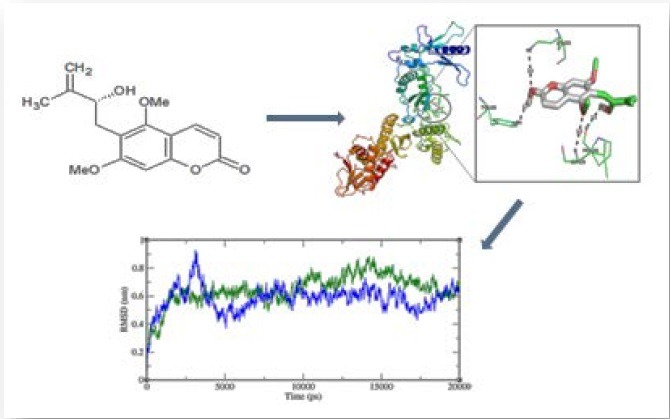
The 3D structure of HIV-1-RT is analyzed, and alkaloids are optimized to have minimal potential energy using chimera. The catalytic site amino acid residues are identified for HIV-1-RT using Q-site Finder. Among the 10 sites obtained from Q-site Finder, site 2 is selected for docking analysis. The catalytic site amino acids in domain A of HIV-1-RT are Pro-95, Pro-97, Gly-99, Leu-100, Lys-101, Lys-102, Lys-103, Val-106, Arg-172, Ile-178, Val-179, Ile-180, Tyr-181, Tyr-183, Tyr-188, Val-189, Gly-190, Pro-225, Phe-227, Tyr-229, Leu-234, His-235, Pro-236, Asp-237, and Tyr-318. These catalytic site residues are loaded as an input in Autodock4.0, which deletes other amino acid residues and select only the catalytic site amino acids of the HIV-1-RT. Evaluation of binding mode and its stability of alkaloids with HIV-1-RT are performed using Autodock. From docking analysis, we listed ligand binding conformation of alkaloids based on binding energy [Table 1]. The binding conformation for each compound into the HIV-1-RT is determined, and the one having the lowest binding energy among the different conformations is generated. The lower energy scores represent better protein-ligand binding affinity as compared to higher energy values. Among the all ligands, toddanol, toddanone, and toddalenone are found to have a lower binding energy value than the other alkaloids. Toddanol has the least binding affinity with HIV-1-RT (binding energy value = −7.76 kcal/mol), binding energy value of toddanone is −6.02 kcal/mol, and the binding energy value for toddalenone is 5.94 kcal/mol. From the docking analysis, the differences are noted in the binding mode of toddanol, toddanone, and toddalenone with HIV-1-RT.
Table 1.
Docking results of alkaloids from Toddalia asiatica with HIV-1-RT
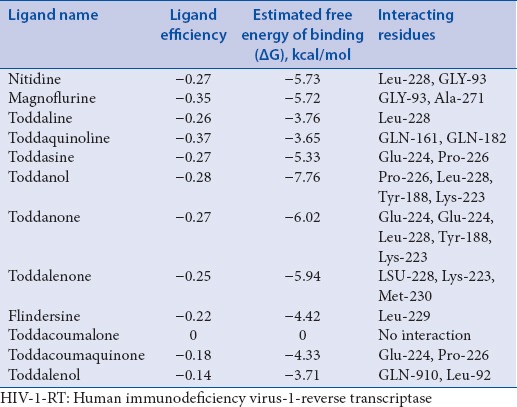
The binding affinity of toddanol, toddanone, and toddalenone with HIV-1-RT is investigated in detail. On analysis of the binding mode of toddanol into the catalytic site of HIV-1-RT, it is found that the residues Pro-226, Leu-228, Tyr-188, and Lys-223 are involved in the H-bond interaction, and they contributed four hydrogen bonds [Figure 2]. Interaction analysis of toddanone with HIV-1-RT, the residues Lys-223, Tyr-188, Leu-228, and Glu-224 are in H-bond contact with toddanone and they shared six hydrogen bonds [Figure 3]. The binding mode of toddalenone with HIV-1-RT reveals that only three amino acid residues, Leu-228, Met-230, and Lys-223 are involved in the formation of only three hydrogen bonds [Figure 4]. From the interaction analysis of toddanol, toddanone, and toddalenone with HIV-1-RT, it is found that polar and aromatic amino acids (Tyr-188, Lys-223) play an important role in ligand binding interaction.
Figure 2.

Docking results of toddanol against 1REV. (a) Binding mode of toddanol in 1REV. (b) Toddanol interaction with 1REV. Ligand atoms are colored by their type. The interacted amino acid residues and hydrogen bond networks in the binding pocket are shown
Figure 3.
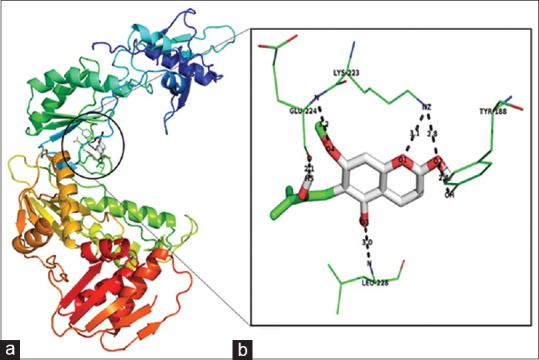
Docking results of toddanone against 1REV. (a) Binding mode of toddanone in 1REV (b) Toddanone interaction with 1REV. Ligand atoms are colored by their type. The interacted amino acid residues and hydrogen bond networks in the binding pocket are shown
Figure 4.
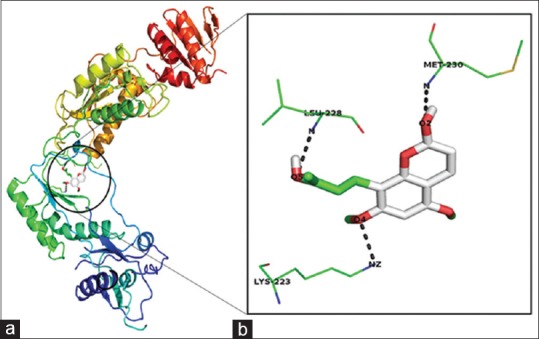
Docking results of toddanolenone against 1REV. (a) Binding mode of toddanolenone in 1REV. (b) Toddanolenone interaction with 1REV. Ligand atoms are colored by their type. The interacted amino acids residues and hydrogen bond networks in the binding pocket are shown
Hydrophobic interaction
Hydrophobic interactions play a significant role in the ligand-enzyme interaction. The residues of HIV-1-RT participating in the hydrophobic contact with toddanol, toddanone, and toddalenone are analyzed using Ligplot tool. In toddanol HIV-1-RT complex analysis, Trp-226, Tyr-181, Tyr-188, val-189, Gly-190, Val-179, Leu-100, Val-106, Lys-102, Pro-236, Tyr-318, Phe-227, Lys-101, His-235, and Leu-234 are formed hydrophobic bonds with toddanol [Figure 5a]. From the toddanone HIV-1-RT complex analysis, it is found that the residues Tyr-181, Trp-229, Phe-227, Leu-100, Leu-234, Lys-101, Tyr-318, Pro-238, His-235, Tyr-188, Val-179, Val-189, Val-106, and Gly-196 are participating in hydrophobic interactions with toddanone [Figure 5b]. On hydrophobic interaction analysis of toddalenone HIV-1-RT analysis, the residues Leu-282, Gln-278, and Arg-227 are involved in hydrophobic contact with toddalenone [Figure 5c]. On comparing the three complexes, more number of residues contributed to hydrophobic interaction with toddanol than the other two compounds toddanone and toddalenone. From the Figure 5, it is observed that the residues Tyr-181, Val-189, Val-179, Leu-100, Val-106, Phe-227, and His-235 are involved in hydrophobic interaction in toddanol-HIV-1-RT and toddanone-HIV-1-RT complexes revealing that these residues have a crucial role in ligand binding.
Figure 5.
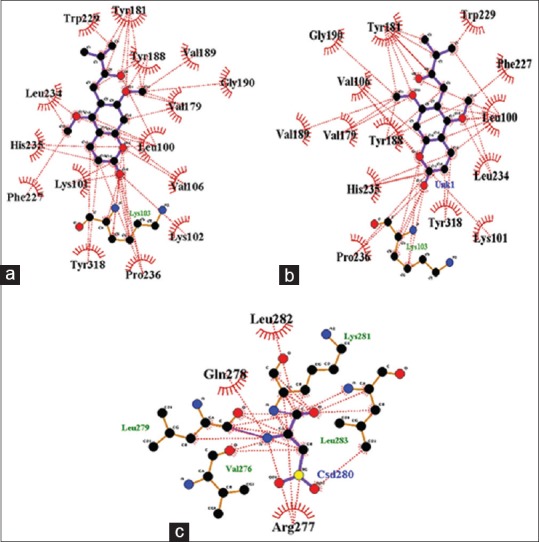
Schematic representation of the hydrophobic interaction between (a) Toddanol and human immunodeficiency virus-1-reverse transcriptase (b) Toddanone and human immunodeficiency virus-1-reverse transcriptase and (c) Toddalenone and human immunodeficiency virus-1-reverse transcriptase generated using the Ligplot program. Hydrophobic bonds are indicated by an arc with spokes radiating toward the ligand atoms they contact. The interacting atoms are those with their spokes radiating back
LogP value and “Lipinski's rule of five” properties
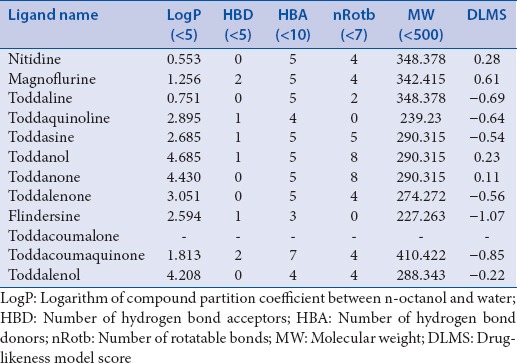
The physiochemical and pharmaceutical properties such as LogP value (octanol/water), molecular weight, number of hydrogen bond acceptors, number of hydrogen bond donors, and number of rotatable bonds for each alkaloid are analyzed. These properties are evaluated based on Lipinski's rule of five that predicts drug-likeness/identifies the biological or pharmacological activity that would aid in making a good oral active drug. Lipinski's rule of five states that most of the molecules with good membrane permeability will have LogP ≤5, molecular weight ≤500, the number of hydrogen bond acceptors ≤10, and the number of hydrogen bond donors ≤5.[41,42] Hence, the alkaloids from T. asiatica except toddacoumalone, which had no interaction with HIV-1-RT are evaluated for various parameters that would help to adjudge the particular substance to be a probable drug. According to Lipinski's rule, 12 alkaloids are found to possess possible drug-like characteristics based on Lipinski's rule of 5. A number of rotatable bonds are important for conformational changes of molecules and ultimately for the binding of receptors or channels. It has been reported that the number of rotatable bonds should be ≤10 for passing oral bioavailability criteria.[43] The alkaloids under investigation had low to high number of rotatable bonds (0–8 in general), with toddanol and toddanone having eight rotational bonds. We took toddanol and toddanone for further analysis as they can exhibit large conformational flexibility due to more number of rotable bonds [Table 2].
Table 2.
Molecular properties of alkaloids obtained from molinspiration
The computed drug-likeness model score is shown in Figure 6, where green color indicates no drug-like behavior and blue color indicates drug-like behavior. Compounds having zero or negative value should not be considered as drug-like. Computed drug-likeness scores are negative for most of the alkaloids, except toddanol and toddanone which had a score of 0.23 and 0.11. These scores are much comparable to the standard anti-HIV drug zidovudine (0.18) [Table 2].[44] This result assures that toddanol and toddanone are good bioactive molecules that can be used as inhibitors of HIV-1-RT.
Figure 6.

Drug-likeness model score of alkaloids when compared with standard anti-human immunodeficiency virus drug. (a) Toddanol (b) toddanone (c) Anti-human immunodeficiency virus drug: Zidovudine
Molecular dynamic simulations
MD simulations are carried out to determine the structural stability within a nanosecond time scale for toddonol-RT and toddanone-RT complexes. These two complexes are selected based on their least binding energy and are subjected to 20 ns MD simulations, and the results are analyzed.
Root mean square deviation
RSMD, a crucial parameter to analyze the equilibration of MD trajectories, is estimated for backbone atoms of the toddanol-RT and toddanone-RT complexes. Measurements of the backbone RMSD for the two complexes provided insights into the conformational stability. The comparisons of the RMSD value of toddanol-RT and toddanone-RT complexes are shown in Figure 7. Until 3,120 ps, there is a slight increase in the RMSD value of the toddanol-RT complex structure to 0.91 nm. Then the RMSD value of toddanol-RT dropped down to 0.37 nm between 3,150 ps and 5,054 ps. From 14,000 ps to 18,000 ps there are clear noticeable deviations in the backbone RMSD value of the toddanol-RT complex structure. After that, there are not many deviations in the RMSD value of the toddanol-RT complex structure. The toddanone-RT complex shows a slight rise in the RMSD value at 1,000 ps; after that, it attained the RMSD value of 0.7 nm. It shows the highest RSMD value of 0.8 nm at 14,196 ps. Both the trajectories converge at 20,000 ps. Based on these results, we conclude that the stability of the toddanol-RT complex is higher than the toddanone-RT complex.
Figure 7.
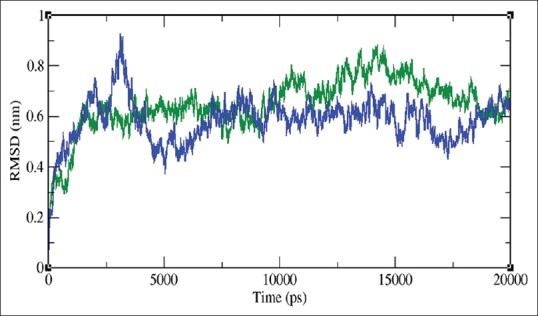
Root mean square deviation of the backbone atoms of docked complexes at 300K, Green color indicates toddanol-reverse transcriptase complex and toddanone-reverse transcriptase complex is shown in violet
Root mean square fluctuations
The RMSF of the backbone atoms of each residue in the toddanol-RT and toddanone-RT complex was analyzed to observe the flexibility of the enzyme backbone structure. The high RMSF value shows more flexibility whereas low RMSF value shows limited movements. The RMSF graph for toddanol-RT and toddanone-RT complex is shown in Figure 8. Toddanone-RT complex attained a high level of fluctuation in the residue positions 138, 251, 288, 436, and 543. The residues 288 and 555 are also seen to show great fluctuation up to 0.7 nm and 1.20 nm. The fluctuations at certain residues are seen to be very high and large. In the toddanol-RT complex the residues 87, 452, and 471 are shown to have maximum fluctuation. The RMSF analysis revealed that the flexibility of toddanone-RT is overall higher than the toddanol-RT complex system.
Figure 8.
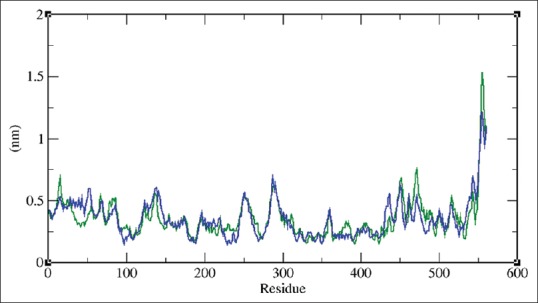
Root mean square fluctuation of the backbone atoms of docked complexes at 300K, green color indicates toddanol-reverse transcriptase complex and toddanone-reverse transcriptase complex is shown in violet
H-bond
The intermolecular H-bonding between the protein and ligand plays a vital role in stabilizing the protein-ligand complexes. The stability of hydrogen bond network formed between toddanol and RT is calculated throughout the 20 ns simulation period. The total number of H-bonds in the toddanol-RT and toddanone-RT complexes versus time at 300K is shown in Figure 9. Toddanol-RT complex exhibited four H-bonds throughout the simulation time period, which indicates that the toddanol has stable and strong H-bonds with RT.
Figure 9.

Total number of intermolecular H-bonds between the ligand and enzyme complex versus time at 300K (a) H-bond between toddanol-reverse transcriptase is shown in green color (b) H-bond between toddanone-reverse transcriptase is shown in violet color (c) inter-molecular H-bonds of both the docked complexes. For clarity's sake, each complex is also shown separately in (a) and (b)
CONCLUSIONS
In the present study, the molecular docking and MD simulations are performed to explore the possible binding mode of HIV-1-RT with 12 alkaloids of T. asiatica. The best ligand conformation is chosen based on binding free energy value, hydrogen bonding, and hydrophobic interaction. The conclusion drawn from the docking analysis is that toddanol, toddanone, and toddalenone have the highest binding affinity with HIV-1-RT. Subsequently, the molecular property screening of 12 alkaloids satisfied the Lipinski's rule of five. The drug-likeness model score revealed a positive score for toddanol and toddanone which is comparable to the drug-likeness score of the standard anti-HIV drug zidovudine. Furthermore, MD simulation is performed to analyze the binding stability of toddanol-RT and toddanone-RT complexes. RMSD, RMSF, and H-bond results reveal that toddanol-RT complex is highly stable as compared to the toddanone-RT complex. Though there are several reports on the medicinal use of alkaloids from T. asiatica, there is no in silico study predicting the anti-HIV activity of toddanol. Our study is perhaps the first to attempt inferring that toddanol is a potential anti-HIV drug molecule. Hopefully, the proposed molecule can be put forward for a constructive concept of designing HIV inhibitors. Thus, it might be a useful candidate for HIV therapy.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
ABOUT AUTHORS

R. Priya
Priya R is doing Ph.D. in VIT University. Her research interests include plant bioinformatics and molecular docking.

Rajendrarao Sumitha
Rajendra Rao Sumitha is doing Ph.D. at National Institute of Mental Health and Neuro Sciences.

C. George Priya Doss
Dr. George Priya Doss. C received his Ph.D. in Bioinformatics from the VIT University, India in 2009. His research interests include SNP analysis, Structural Bioinformatics, and Protein Dynamics.

C. Rajasekaran
Dr. Rajasekaran. C received his Ph.D. from Hemwati Nandan Bahuguna Garhwal University, India. His research interests include medicinal plants and conservation strategies.

S. Babu
Dr. Babu. S received his Ph.D. from Tamil Nadu Agricultural University, India. His research interests include genomics, proteomics, plant-microbe interaction.

R. Seenivasan
Dr. Seenivasan R received his Ph.D. from CAS Marine Biology, Annamalai University. His research interests include marine biology and medicinal plants.

R. Siva
Dr. Siva. R received his Ph.D. in Plant Biotechnology from the Bharathidhasan University, India in 2003. His research interests include plant pigments, dyes and plant molecular biology.
Acknowledgments
The authors are thankful to the VIT University for providing all the necessary facilities to carry out the research work.
REFERENCES
- 1.Douek DC, Roederer M, Koup RA. Emerging concepts in the immunopathogenesis of AIDS. Annu Rev Med. 2009;60:471–84. doi: 10.1146/annurev.med.60.041807.123549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cunningham AL, Donaghy H, Harman AN, Kim M, Turville SG. Manipulation of dendritic cell function by viruses. Curr Opin Microbiol. 2010;13:524–9. doi: 10.1016/j.mib.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 3.Yadav IK, Jaiswal D, Singh HP, Mishra A, Jain DA. Anti-HIV drugs from natural sources. Pharm Res. 2009;1:93–109. [Google Scholar]
- 4.Zhang H, Vrang L, Bäckbro K, Lind P, Sahlberg C, Unge T, et al. Inhibition of human immunodeficiency virus type 1 wild-type and mutant reverse transcriptases by the phenyl ethyl thiazolyl thiourea derivatives trovirdine ‘ MSC-127. Antiviral Res. 1995;28:331–42. doi: 10.1016/0166-3542(95)00056-9. [DOI] [PubMed] [Google Scholar]
- 5.Hehl EA, Joshi P, Kalpana GV, Prasad VR. Interaction between human immunodeficiency virus type 1 reverse transcriptase and integrase proteins. J Virol. 2004;78:5056–67. doi: 10.1128/JVI.78.10.5056-5067.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coffin JM, Hughes SH, Varmus HE. Cold Spring Harbor (NY) Cold Spring Harbor Laboratory Press; 1997. Retroviruses. [PubMed] [Google Scholar]
- 7.Oz Gleenberg I, Goldgur Y, Hizi A. Ile178 of HIV-1 reverse transcriptase is critical for inhibiting the viral integrase. Biochem Biophys Res Commun. 2007;364:48–52. doi: 10.1016/j.bbrc.2007.09.086. [DOI] [PubMed] [Google Scholar]
- 8.Siva R, Palackan MG, Maimoon L, Geetha T, Bhakta D, Balamurugan P, et al. Evaluation of antibacterial, antifungal, and antioxidant properties of some food dyes. Food Sci Biotechnol. 2011;20:7–13. [Google Scholar]
- 9.Bhakta D, Sivaramakrishna A, Siva R. Bioactive iridoid glycoside isolated from Morinda tinctoria (Roxb.) roots exhibit therapeutic efficacy. Ind Crops Prod. 2013;42:349–56. [Google Scholar]
- 10.Vázquez R, Riveiro ME, Vermeulen M, Mondillo C, Coombes PH, Crouch NR, et al. Toddaculin, a natural coumarin from Toddalia asiatica, induces differentiation and apoptosis in U-937 leukemic cells. Phytomedicine. 2012;19:737–46. doi: 10.1016/j.phymed.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 11.Facchini PJ. Alkaloid biosynthesis in plants: Biochemistry, cell biology, molecular regulation, and metabolic engineering applications. Annu Rev Plant Physiol Plant Mol Biol. 2001;52:29–66. doi: 10.1146/annurev.arplant.52.1.29. [DOI] [PubMed] [Google Scholar]
- 12.Iwasaki H, Oku H, Takara R, Miyahira H, Hanashiro K, Yoshida Y, et al. The tumor specific cytotoxicity of dihydronitidine from Toddalia asiatica Lam. Cancer Chemother Pharmacol. 2006;58:451–9. doi: 10.1007/s00280-005-0183-4. [DOI] [PubMed] [Google Scholar]
- 13.Orwa JA, Jondiko IJ, Minja RJ, Bekunda M. The use of Toddalia asiatica (L) Lam.(Rutaceae) in traditional medicine practice in East Africa. J Ethnopharmacol. 2008;115:257–62. doi: 10.1016/j.jep.2007.09.024. [DOI] [PubMed] [Google Scholar]
- 14.Siva R. Statues of natural dyes and dye-yielding plants in India. Curr Sci. 2007;92:916–25. [Google Scholar]
- 15.Thirugnanasampandan R, Jayakumar R, Prabhakaran M. Analysis of chemical composition and evaluation of antigenotoxic, cytotoxic and antioxidant activities of essential oil of Toddalia asiatica (L.) Lam. Asian Pac J Trop Biomed. 2012;2:S1276–9. [Google Scholar]
- 16.Rajumar M, Chandra RH, Veeresham C. Production of Nitidine from callus cultures of Toddalia asiatica. Int J Pharm Sci Nanotechnol. 2010;3:1028–33. [Google Scholar]
- 17.Rashid MA, Gustafson KR, Kashman Y, Cardellina JH 2nd, McMahon JB, Boyd MR. Anti-HIV alkaloids from Toddalia asiatica. Nat Prod Lett. 1995;6:153–6. [Google Scholar]
- 18.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–42. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sengupta D, Verma D, Naik PK. Docking mode of delvardine and its analogues into the p66 domain of HIV-1 reverse transcriptase: Screening using molecular mechanics-generalized born/surface area and absorption, distribution, metabolism and excretion properties. J Biosci. 2007;32:1307–16. doi: 10.1007/s12038-007-0140-y. [DOI] [PubMed] [Google Scholar]
- 20.Singh IP, Bharate SB, Bhutani KK. Anti-HIV natural products. Curr Sci. 2005;89:269–90. [Google Scholar]
- 21.Li Z, Wan H, Shi Y, Ouyang P. Personal experience with four kinds of chemical structure drawing software: Review on ChemDraw, ChemWindow, ISIS/Draw, and ChemSketch. J Chem Inf Comput Sci. 2004;44:1886–90. doi: 10.1021/ci049794h. [DOI] [PubMed] [Google Scholar]
- 22.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera – A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 23.Bieganski RM, Yarmush ML. Novel ligands that target the mitochondrial membrane protein mitoNEET. J Mol Graph Model. 2011;29:965–73. doi: 10.1016/j.jmgm.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liao JJ, Andrews RC. Targeting protein multiple conformations: A structure-based strategy for kinase drug design. Curr Top Med Chem. 2007;7:1394–407. doi: 10.2174/156802607781696783. [DOI] [PubMed] [Google Scholar]
- 25.Laurie AT, Jackson RM. Q-SiteFinder: An energy-based method for the prediction of protein-ligand binding sites. Bioinformatics. 2005;21:1908–16. doi: 10.1093/bioinformatics/bti315. [DOI] [PubMed] [Google Scholar]
- 26.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–91. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhakta D, Siva R. Morindone, an anthraquinone, intercalates DNA sans toxicity: A spectroscopic and molecular modeling perspective. Appl Biochem Biotechnol. 2012;167:885–96. doi: 10.1007/s12010-012-9744-2. [DOI] [PubMed] [Google Scholar]
- 28.Lill MA, Danielson ML. Computer-aided drug design platform using PyMOL. J Comput Aided Mol Des. 2011;25:13–9. doi: 10.1007/s10822-010-9395-8. [DOI] [PubMed] [Google Scholar]
- 29.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–34. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 30.Jarrahpour A, Fathi J, Mimouni M, Hadda TB, Sheikh J, Chohan Z, et al. Petra, Osiris and Molinspiration (POM) together as a successful support in drug design: Antibacterial activity and biopharmaceutical characterization of some azo schiff bases. Med Chem Res. 2012;21:1–7. [Google Scholar]
- 31.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 32.Hess B, Bekker H, Berendsen HJ, Fraaije JG. LINCS, A linear constraint solver for molecular simulations. J Comput Chem. 1997;18:1463–72. [Google Scholar]
- 33.Schüttelkopf AW, van Aalten DM. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 8):1355–63. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 34.van Gunsteren WF, Billeter SR, Eising AA, Hunenberger PH, Krüger PK, Mark AE, et al. Biomolecular simulation: The GROMOS96 manual and user guide. Zurich: Verlag der Fachvereine; 1996. pp. 1–1024. [Google Scholar]
- 35.Oostenbrink C, Villa A, Mark AE, van Gunsteren WF. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J Comput Chem. 2004;25:1656–76. doi: 10.1002/jcc.20090. [DOI] [PubMed] [Google Scholar]
- 36.Berendsen HJ, Postma JP, van Gunsteren WF, Hermans J. Interaction models for water in relation to protein hydration. In: Pullman B, editor. Intermolecular Forces. Dordrecht: D Reidel Publishing Company; 1981. pp. 331–42. [Google Scholar]
- 37.Attouch H, Cominetti R. A dynamical approach to convex minimization coupling approximation with the steepest descent method. J Differ Equ. 1996;128:519–40. [Google Scholar]
- 38.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577–93. [Google Scholar]
- 39.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: Fast, flexible, and free. J Comput Chem. 2005;26:1701–18. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 40.Turner PJ. Beaverton: Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology; 2005. XMGRACE. Ver. 5.1.19. [Google Scholar]
- 41.Ahsan MJ, Samy JG, Khalilullah H, Nomani MS, Saraswat P, Gaur R, et al. Molecular properties prediction and synthesis of novel 1,3,4-oxadiazole analogues as potent antimicrobial and antitubercular agents. Bioorg Med Chem Lett. 2011;21:7246–50. doi: 10.1016/j.bmcl.2011.10.057. [DOI] [PubMed] [Google Scholar]
- 42.Mazumder J, Chakraborty R, Sen S, Vadra S, De B, Ravi TK. Synthesis and biological evaluation of some novel quinoxalinyl triazole derivatives. Der Pharma Chem. 2009;1:188–98. [Google Scholar]
- 43.Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45:2615–23. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 44.Li W, Wu J, Zhan P, Chang Y, Pannecouque C, De Clercq E, et al. Synthesis, drug release and anti-HIV activity of a series of PEGylated zidovudine conjugates. Int J Biol Macromol. 2012;50:974–80. doi: 10.1016/j.ijbiomac.2012.02.019. [DOI] [PubMed] [Google Scholar]