

Abstract
Whether HIV from seminal cells or free HIV in semen is the origin of transmitted virus has important implications for the design of transmission prevention strategies. We found that a recent claim that HIV originates from seminal plasma and not from seminal cells was erroneous, because it was based on biological specimens that had been mislabeled, mixed-up, or contaminated. The origin of transmitted virus from semen therefore remains an open question.
In the paper by Butler et al. “The Origins of Sexually Transmitted HIV Among Men Who Have Sex with Men” (1), HIV-1 sequence data from six pairs of men were presented and the authors concluded that “cell-free seminal plasma is the origin of transmitted HIV.” However, as we describe below, the sequences that were ascribed to seminal cells and some components of blood appear to have been mixed-up, mislabeled, or contaminated, which led to inappropriate conclusions regarding this important event in HIV transmission biology.
The large genetic distances observed in the phylogenetic trees of individual subjects shown in (1) are atypical of HIV populations that evolve from single transmission events. This observation led us to evaluate their sequences, along with 93 unrelated subtype B and D sequences from the Los Alamos HIV sequence database (2), to better evaluate the genetic relationships among them. A single phylogenetic tree was constructed under the maximum likelihood model GTR + G + I (Fig. 1). Although recipient sequences from each pair of men clustered with some of their source partner’s sequences, other source sequences were outliers, scattered among unrelated circulating subtype B sequences throughout the tree. In most cases, the outlier sequences were attributed to the seminal cells [source seminal cell–associated virus (SSC)], whereas the seminal fluid [source HIV RNA in seminal plasma (SSP)] and blood RNA [source HIV RNA in blood plasma (SBP)] sequences were grouped with the recipient’s sequences.
Fig. 1.
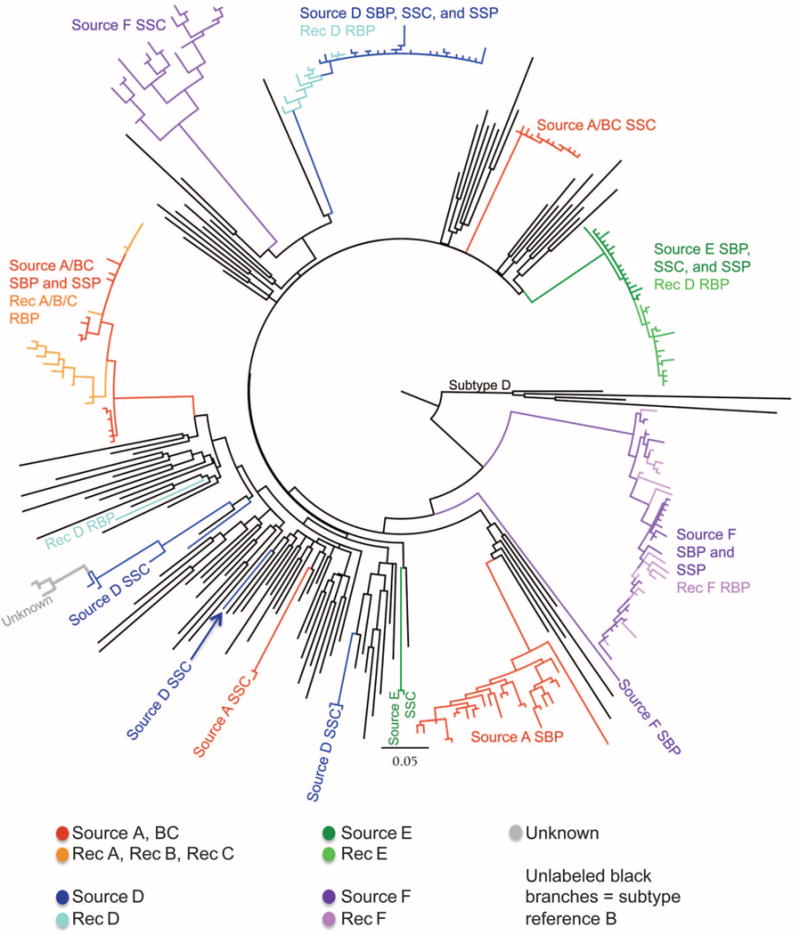
Maximum likelihood tree with the sequences from (1) available in GenBank plus 89 subtype B reference sequences and four subtype D reference sequences. Sequences from (1) are color-coded as source and recipient; clades are labeled according to content. Sequences labeled “unknown” (gray) did not have subject identifiers in GenBank, but rather were designated “unknown source” in (1). The clade corresponding to the subtype D reference sequences is labeled; all other black branches correspond to subtype B reference sequences.
As an example, we will describe the analysis of three transmission pairs. One man, source A/BC, was the source of infection to three recipients, recipients B and C and, 2 years later, A. This source was sampled at two time points, near each transmission event, with sequences reported for each of the two time points (BC and A). Sequences from the three recipients clustered with some of the source A/BC blood and seminal plasma sequences. However, source A/BC sequences were also detected in three other unlinked clades composed of 13 sequences from source A/BC seminal cells, 26 sequences from source A blood plasma, and 2 source A seminal cell sequences.
Although Butler et al. made the claim that their data “did not show evidence for superinfection within hosts,” they did not explain how this was determined. Dual or superinfection can in fact result in multiple clades separated in a tree by unrelated sequences (3). However, Butler’s results are not consistent with superinfection for the following reasons: (i) the divergent clade or clades were most often confined to one specific cell population (SSC); (ii) source A/BC sequences were found in four distinct clades, implying an extremely unlikely four different sources of infection in this subject; (iii) source D SSC sequences were also found in four separated clades and recipient D sequences were in one of these plus one sequence at a unique position, for a suspected total of five independent virus sources from this pair; and (iv) multiple clades occurred in each source from the six transmission cases, which would represent an extraordinary high prevalence of multiple infection (4, 5), whereas only one clade was found in five of the recipients [the sixth recipient (D) had an outlier sequence that was unrelated to the four clades from his source].
Figure 2A illustrates the diversity distribution (gray bars) across the reference subtype B C2V3 sequences included in Fig. 1, which included no transmission pairs or otherwise epidemiologically linked subjects. The distances between these unrelated sequences formed a normal distribution, with a mean around 0.14. Figure 2A also shows the genetic diversity from the same region of env within a single representative untreated patient (MACS2) at ~6 months (v02) and at 6 years (v12) after infection (6). At the early time point, distances were relatively small, whereas at the later time point, the distribution shifted to greater distances, reflecting viral diversification. Even after 6 years of infection, however, diversity in the individual remained much lower than across the reference subtype B sequences.
Fig. 2.
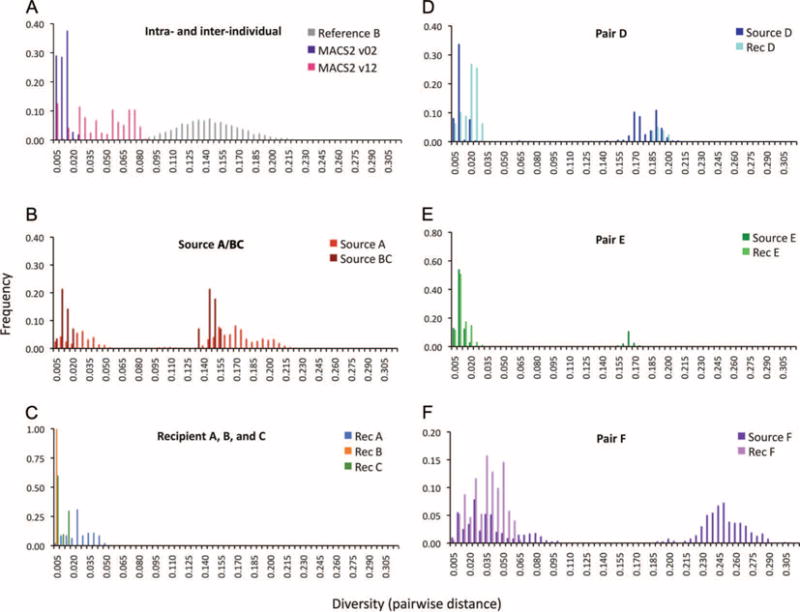
Pairwise distance histograms illustrating the diversity distribution for each source-recipient pair. For each study subject, the pairwise distance (nucleotide changes per site) between every possible pair of sequences within each individual was calculated, with the same alignment and model for calculating the tree. (A) For the reference distribution (gray bars), all pairwise distances from each subtype B reference sequence in the tree were calculated. MACS2 v02 and v12 were derived from analogous sequences derived from a subject in (6), taken ~6 months and 6 years after infection, respectively. (B to F) Sequences from each transmission pair described in (1).
A pairwise comparison of the two source A and BC time points revealed a bimodal distribution (Fig. 2B), whereas each recently infected recipient showed very low diversity (Fig. 2C). Bimodal distributions are also noted in the histograms of the other three sources (Fig. 2, D and E) and in recipient D (with one sequence outside of his main clade) (Fig. 1). The lower mode—the distribution closer to zero—is representative of the distances between sequences from one person, whereas the upper mode (further right) is representative of the distances between unrelated sequences.
The clustering of an individual’s sequences across multiple unconnected clades in the tree and the large distances between sequences attributed to single individuals provide compelling evidence that the results reported in (1) were caused by sample mix-up, mislabeling, or contamination. Because most of the divergent sequences were labeled seminal cells, the authors were led to the unwarranted conclusion that cell-free virus transmitted the infection. We did not find evidence for a common source of contamination, and the study sequences also did not match any previously published sequences in the Los Alamos database. So, the multiple origins of the unlinked sequences are unclear.
Erroneous conclusions drawn from issues of this type have been described previously, as have methods for avoiding such problems (7–9). Recently, new web-based tools have been provided to make it easier for investigators to discover and investigate anomalies before publication (10, 11).
Acknowledgments
Funding: This work was supported by NIH grants AI47734 and AI27727 (Computational Biology Core of the University of Washington Center for AIDS Research).
Footnotes
Competing interests: The authors declare no conflicts of interest.
REFERENCES AND NOTES
- 1.Butler DM, Delport W, Kosakovsky Pond SL, Lakdawala MK, Cheng PM, Little SJ, Richman DD, Smith DM. The origins of sexually transmitted HIV among men who have sex with men. Sci Transl Med. 2010;2:18re1. doi: 10.1126/scitranslmed.3000447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Los Alamos HIV Database, http://www.hiv.lanl.gov/
- 3.Jost S, Bernard MC, Kaiser L, Yerly S, Hirschel B, Samri A, Autran B, Goh LE, Perrin L. A patient with HIV-1 superinfection. N Engl J Med. 2002;347:731–736. doi: 10.1056/NEJMoa020263. [DOI] [PubMed] [Google Scholar]
- 4.Smith DM, Richman DD, Little SJ. HIV superinfection. J Infect Dis. 2005;192:438–444. doi: 10.1086/431682. [DOI] [PubMed] [Google Scholar]
- 5.Gottlieb GS, Heath L, Nickle DC, Wong KG, Leach SE, Jacobs B, Gezahegne S, van ’t Wout AB, Jacobson LP, Margolick JB, Mullins JI. HIV-1 variation before seroconversion in men who have sex with men: Analysis of acute/early HIV infection in the multicenter aids cohort study. J Infect Dis. 2008;197:1011–1015. doi: 10.1086/529206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shankarappa R, Margolick JB, Gange SJ, Rodrigo AG, Upchurch D, Farzadegan H, Gupta P, Rinaldo CR, Learn GH, He X, Huang XL, Mullins JI. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J Virol. 1999;73:10489–10502. doi: 10.1128/jvi.73.12.10489-10502.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Korber BT, Learn G, Mullins JI, Hahn BH, Wolinsky S. Protecting HIV databases. Nature. 1995;378:242–244. doi: 10.1038/378242a0. [DOI] [PubMed] [Google Scholar]
- 8.Learn GH, Jr, Korber BT, Foley B, Hahn BH, Wolinsky SM, Mullins JI. Maintaining the integrity of human immunodeficiency virus sequence databases. J Virol. 1996;70:5720–5730. doi: 10.1128/jvi.70.8.5720-5730.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frenkel LM, Mullins JI, Learn GH, Arcuino L Manns, Herring BL, Kalish ML, Steketee RW, Thea DM, Nichols JE, Liu SL, Harmache A, He X, Muthui D, Madan A, Hood L, Haase AT, Zupancic M, Staskus K, Wolinsky S, Krogstad P, Zhao J, Chen I, Koup R, Ho D, Korber B, Apple RJ, Coombs RW, Pahwa S, Roberts NJ., Jr Genetic evaluation of suspected cases of transient HIV-1 infection of infants. Science. 1998;280:1073–1077. doi: 10.1126/science.280.5366.1073. [DOI] [PubMed] [Google Scholar]
- 10.Deng W, Nickle DC, Learn GH, Maust B, Mullins JI. ViroBLAST: A stand-alone BLAST web server for flexible queries of multiple databases and user’s datasets. Bioinformatics. 2007;23:2334–2336. doi: 10.1093/bioinformatics/btm331. [DOI] [PubMed] [Google Scholar]
- 11.B. T. Korber, http://www.hiv.lanl.gov/content/sequence/QC/index.html [accessed 2010]