

Abstract
We briefly review the protective role of maternal antibodies during fetal development and at early postnatal stages. We describe antibody delivery to fetuses, particularly in the context of the developing blood–brain barrier (BBB), and present the essential concepts regarding the adult BBB, together with existing information on the prenatal developing BBB. We focus on maternal antibody transfer to the developing brain and the consequences of the presence of pathogenic antibodies at early stages of brain development on subsequent brain dysfunction.
Keywords: Autoimmunity, Neuroimmunology, Maternal antibodies, Developing blood–brain barrier
Protective role of maternal antibodies
Maternal antibodies provide passive immunity to developing fetuses and perinatal offspring. The sterile environment of the uterine cavity offers partial fetal protection, but maternal–fetal infections do occur and are most dangerous at the beginning of pregnancy, before there is any significant transfer of the protective maternal antibodies to the fetus. The transfer of these antibodies occurs through the placenta via the neonatal Fc receptor (FcRn) and depends on gestational stage, on the integrity of the placenta, and on the antibody level in maternal circulation, suggesting a regulated and saturable transfer system [1, 2]. In humans, maternal antibody transfer starts as early as week 13 of gestation. Antibodies bound to FcRn are protected from degradation, while free antibodies are degraded much faster. Interestingly, the level of immunoglobulin (IgG) in fetal circulation is relatively low (5–10 % of the maternal level) at weeks 17–22, reaching 50 % by week 32 and usually exceeding the maternal level at delivery [3]. Babies that are born prematurely have much lower levels of total IgG [4]. The nature of regulation of IgG in fetal circulation is currently unknown. Placental transfer of maternal antibodies depends on the class of antibody (IgG being the only class significantly transferred to the fetus) and on the subclass, or isotype. In humans, IgG1 is preferentially transported, followed by IgG4 and to a lesser extent by IgG3 and IgG2 [2, 5]. There is a clear difference, however, in maternal antibody delivery across the placenta among species; in primates and rodents, there is a substantial transfer of maternal antibody across the placenta; in contrast, in sheep, horses, cattle, and pigs, neonates receive high amounts of maternal antibody for the first time during lactation [6]. The protective role of maternal antibody extends to the newborn whose immune system is not fully developed. In addition to antibodies present at birth, antibodies are delivered postnatally in maternal milk.
Maternal antibodies and developmental abnormalities: human studies and animal models
Paradoxically, maternal antibodies can be a potential source of harm to the developing fetus. Most of the experimental evidence in studies of the pathogenic role of maternal antibodies comes from animal models and to a lesser extent from human biopsies (that can be affected by postmortem changes). Clinical studies of autoimmune diseases with high levels of self-reacting antibodies in maternal circulation provide convincing evidence of maternal antibody-transferred disease. Neonatal lupus (NLE) with cutaneous manifestation (erythema or skin rush) is the most common symptom of maternal anti-Ro/SSA (antinuclear) antibodies, followed by congenital heart block (CHB). Most symptoms of NLE are reversible and disappear with the lowering of the antibody level in the newborn, with the exception of CHB that is irreversible and detrimental to the offspring [7–10]. There is high mortality (about 20 %) and morbidity associated with cardiac NLE with more than 60 % of the children surviving CHB needing pacemaker implantation. Of note, the risk of recurrence of CHB increases tenfold for the subsequent pregnancies of the mother with CHB child [11]. NLE is associated with maternal antinuclear Ro/SSA or La/SSB antibodies, or both, in all cases reported thus far. Interestingly, the vast majority of mothers with positive serology for these antibodies deliver healthy babies. Obviously, the onset of a neonatal disease is multifactorial in pregnancies of the affected offspring and likely moderated by some mitigating factors in the healthy outcomes. One of the mitigating factors in NLE might be anti-idiotypic antibody, which reacts with anti-Ro/La (SSA/SSB) antibodies. Increased levels of anti-idiotypic antibodies have been reported in anti-La/SSB-positive mothers delivering healthy children [12]. Another factor might be that cross-reactivity leading to pathogenicity is a phenomenon that occurs only in a subset of patients, making it challenging to assess. An example of this cross-reactivity between maternal anti-Ro antibodies and serotonin receptor (5-HT4R) expressed on fetal heart cells leading to CHB was reported in a mouse model of NLE. Mice immunized with a peptide from 5-HT4R, showing homology with a sequence of SSA/Ro52 protein, but not with Ro52 peptide, manifested CHB. In addition, fetuses of mice immunized with 5-HT4R peptide displayed severe brain defects, delayed neuronal folding, absence of hindbrain or open neural tube, leading to behavioral deficits of the offspring [13, 14]. These experiments suggest that diverse outcomes may be mediated by a single antibody. While an initial human study did not find association of anti-5HT4R antibodies with CHB [15], a subsequent study declared the presence of these antibodies in maternal serum as additional risk factor for a small subset of Ro-positive SLE patients [16]. Furthermore, studies of children exposed to maternal anti-Ro antibodies revealed increased neuropsychiatric problems in the offspring: 40 % (42 of 104 total) versus 27 % (6 of 22) healthy controls [17]. Clearly, additional evidence of maternal antibodies causing disease is needed, and more clinical studies with new screening assays are necessary.
Transient neonatal forms of pemphigus, myasthenia gravis, and anti-phospholipid syndrome have been reported [18–21]. Neonatal pemphigus is thought to be caused by the transfer of maternal IgG autoantibodies to desmoglein 3 and is characterized by transient severe blisters and erosions in the skin [20]. Clinical manifestations of neonatal myasthenia gravis include muscle weakness, poor suckling, respiratory disease, and facial diplegia. They result from a neuromuscular transmission defect, analogous to that observed in adult myasthenia gravis, which is caused by anti-acetylcholine receptor (AChR) antibodies. The disease also remits as maternal antibodies decrease, but in rare and severe cases, the infants require treatment with cholinesterase inhibitors [22].
Anti-phospholipid (aPL) antibodies can cause fetal abnormalities by thrombosis of placental vessels or by binding the trophoblasts directly [21]. The most frequent complications are severe preeclampsia of pregnant women and early miscarriages or even late fetal loss. Recent data suggest learning disabilities in the viable offspring [23].
The fact that disease attenuates with a decrease in maternal antibody in the offspring and the protection provided by a higher level of anti-idiotypic antibody in maternal serum supports antibody involvement in these neonatal autoimmune diseases.
In utero or early postnatal exposure to pathogenic maternal antibodies may manifest much later in life, when the maternal antibodies are no longer present in the circulation of the offspring. This is a subject of increased interest in epidemiological studies, and in studies utilizing animal models, given that clinical data suggest a sharp increase in prevalence of neuropsychiatric disorders, such as schizophrenia and autism spectrum disorders (ASD), in children and adolescents [24]. Animal models provide evidence of maternal antibodies as a cause of ASD. Passive transfer of maternal serum, or purified IgG derived from mothers of autistic children to pregnant mice or monkeys leads to behavioral abnormalities similar to those observed in autistic patients, while sera or purified antibodies obtained from mothers of normally developed children do not lead to such alterations [25–30]. However, even though such sera have been shown to bind Purkinje cells and other neuronal structures, the actual target antigens have not been defined yet [31].
Our laboratory has developed an animal model of learning disabilities in lupus, as children of lupus mothers have a higher incidence of learning disorders [10, 32–35]. We have postulated, and later demonstrated in a mouse model, that prenatal exposure of mouse fetuses to maternal antibodies reactive with N-methyl-D-aspartate receptor (NMDAR) and also cross-reactive with double-stranded (ds) DNA, leads to abnormal fetal brain development, with cortical thinning and increased mitotic activity [36]. Cortical disorganization persists in these animals through adulthood. Young pups exposed in utero to these antibodies display a delay in negative geotaxis, and, as adults, exhibit cognitive impairment in novel object recognition and fear extinction tasks, both of which depend on the proper function of the neocortex [36]. Interestingly, these experiments demonstrate some plasticity of the developing brain as the impairment in neonatal geotaxis disappears at postnatal day 20 (P20), while other cognitive impairments persist through life [36]. The antibody effects are dose dependent, as demonstrated in experiments analyzing the offspring of mice exposed to high or low titer of antibody; only the offspring of high-titer dams showed significant difference in behavioral assessment compared with the offspring of low-titer dams (tenfold lower that the high titer) or control MAP-core-immunized mice. Similar histological results are obtained in experiments of passive transfer of anti-NMDAR/anti-dsDNA antibodies. Brains of fetuses of pregnant mice that were injected with a mouse or human monoclonal anti-NMDAR/anti-dsDNA antibody (R4A and G11, respectively) at day E12.5 of gestation also exhibit cortical abnormalities while control antibody-injected dams (MPC11 and B1, respectively) show no effect. An important aspect of this study from the point of view of prevention or therapy is that pathogenic effect of maternal anti-NMDAR antibodies could be blocked with an NMDAR specific peptide. D/EWDYS/G (DWEYS) sequence is present in both the GluN2A and GluN2B subunits of the NMDAR and is part of the antigenic epitope. The D-form of the peptide injected at the time of antibody injection to pregnant mice and repeated twice 24 h apart prevents alterations in the offspring’s brain [36]. Peptide-mediated protection has been evaluated in passive transfer studies of anti-NMDAR/dsDNA antibody pathogenicity providing the formal proof of antibody involvement. These studies suggest that maternal antibody might contribute to the increased incidence of learning disabilities observed in the children of mothers with SLE. In addition, they provide a defined antigen for the pathogenic antibodies and a putative mechanism leading to learning disabilities. Of note, anti-NMDAR/anti-dsDNA antibodies are reported in up to 40 % of lupus patients and the presence of these antibodies in the cerebrospinal fluid (CSF) is correlated with neuropsychiatric lupus (NPSLE) [37, 38].
Epidemiological and basic research studies have associated maternal autoimmune disease with offspring’s ASD [39–41]. We have performed additional studies of the potential pathogenic effect of maternal antibodies in ASD and aquaporin-4 syndrome (manuscript in preparation).
The adult blood–brain barrier
There are numerous review articles on the subject of the BBB [42–45], and we will only describe briefly the essential concepts of the BBB relevant to our studies. The BBB is a tightly controlled physiological interface between brain parenchyma and the blood. The major cellular components of the adult BBB are cerebral endothelial capillary cells that in connection with astrocytes and pericytes control the central nervous system (CNS) environment and maintain its homeostasis. In contrast to the periphery, paracellular transport to the brain is restricted by tight junctions (TJ), a series of proteins that join brain endothelial cells (EC) capillaries. Transcellular transport, or transcytosis, exists but in the homeostatic state of the CNS is rather low. In the adult brain, delivery of nutrients (to the brain) and disposal of metabolites (from the brain) are governed by an intricate and highly specific transport system. The receptors of this transport system are expressed on the luminal (blood side) or abluminal (brain side) side of the EC. The entry of the immune cells from the periphery is limited, but the interaction of the immune system and brain cells is an important part of the function of the BBB in health and disease as well as during the development of the BBB. Cytokines play critical role in that interaction. Cytokines are involved in the transient opening of the BBB during disease states including brain injury, cancer or infection. The adult BBB does not have any known active transport of antibody to the brain. In the periphery, antibody is protected from catabolic forces and permitted entry into the tissue by binding to FcRn. FcRn in the periphery is on the luminal side of the blood vessel. In the brain, however, FcRns are present on the abluminal side of the EC and are believed to facilitate the efflux of the antibody from the brain [46, 47]. There are areas of the brain near the ventricles called circumventricular organs (CVOs) that are deficient in BBB due to the presence of permeable fenestrated capillaries. CVOs include organum vasculosum of the lamina terminalis, subfornical organ, area postrema, pineal gland medial eminence, and subcommissural organ. CVOs facilitate communication between brain, CSF, and blood/peripheral organs and transduce that information to other centers of the brain. There is differential size-dependent permeability among CVOs [48]. Of note, the surface area of the BBB supersedes CVO area by several orders of magnitude.
The developing blood–brain barrier
There are limited studies that address the developing BBB, and they are inconsistent. Earlier studies stated that the developing BBB is permeable to all molecules through gestation as the major expansion of astrocytes believed to seal the developing BBB occurs postnatally. Thus, the fetal BBB was postulated to be leaky. Others proposed an immature but fully functional BBB at each stage of fetal development (see excellent review on the subject [49]). The latter view was supported in a recent report by Danemen and colleagues describing the involvement of pericytes in controlling permeability of the developing BBB [50]. It is noteworthy that studies of astrocytes is complicated by the lack of the distinct molecular markers of astrocyte precursors and of different subsets of astrocytes (see review [51]).
As mentioned above, the adult BBB does not permit antibody extravasation to the brain in appreciable quantities. How the developing brain controls this process is still not clear. Most studies regarding the BBB, whether adult or developing, were performed using dyes, Evans Blue or Trypan Blue, that bind albumin in the circulation and are in fact addressing the penetration of albumin (~68 kDa) to brain tissue. Albumin, the major protein in the blood, is a good indicator of permeability of the BBB, and the ratio of the albumin to IgG concentration in CSF is widely used to identify BBB leaks. Normally, the concentration of albumin in the brain is very low. Some recent studies used labelled dextrans of varied molecular weight, from a few kDa to 2 MDa, but dextrans are cleared more rapidly from the circulation, and lower molecular weight dextrans are washed out in certain brain preparations giving false negative results [52]. Danemen and colleagues utilized biotin in transcardiac perfusion testing the BBB permeability of pericyte-deficient mice at E18.5 and detected it with labelled streptavidin in the brain tissue of deficient mice but not in the control mice [50]. Biotin is a water-soluble molecule of the relatively low molecular weight of 244 Daltons (D). In addition, they used 3- and 70-kDa dextrans in a similar assay demonstrating size-dependent BBB permeability in pericyte-deficient mice at E18.5 [50].
It is generally accepted that the permeability of the BBB is size dependent and that many biologically vital molecules have specific transport systems on ECs. It seems logical then, that conclusions on the permeability of the BBB while using any specific marker should be made with caution. It is also quite remarkable that studies of immunoglobulin, a major protein delivered to the fetus from mother, are scarce.
When we initiated our studies, little information regarding maternal antibody transfer to the developing brain was available at any stage of gestation. The time of maternal antibody transfer in rodents was believed to occur very late in gestation, just before birth, or postnatally [53]. As exemplified in Fig. 1, we tested maternal antibody delivery to the fetuses at each day of gestation ranging from E13.5 to E 17.5 [54]. Utilizing infrared (IR)-labeled antibody that is intravenously injected to pregnant dams, we evaluated the penetration of the antibody to the fetus 1 h after antibody injection, with the use of an infrared imager (Odyssey, LI-COR), a method that is simple, quick, and effective. We imaged whole embryos without further processing, after cauterizing placental veins to prevent antibody leakage. We observed that antibody is present in fetuses at all stages of gestation, starting at E13.5, 1 h after antibody injection to pregnant dams [54] (see Fig. 1a). There was no signal in fetuses from the saline-injected control mice (Fig. 1b). These results substantiate that maternal antibodies are delivered to mouse embryos at earlier stages of gestation than previously reported [53]. We also tested the antibody transfer to the brain of fetuses at every day of gestation starting from E12.5 to E17.5, 24 h after injection to the pregnant dams. In order to eliminate possible contamination from IgG in the fetal circulation, we used fetal transcardiac perfusion to evacuate blood from the brain vessels. In our hands, this has been the only effective method of blood removal from fetal brain (Fig. 2); perfusion through the placenta is ineffective, as the placenta seems to protect the fragile fetal tissue from changes of maternal blood pressure. We have taken into consideration the fragility of developing blood vessels and adjusted the perfusion volume and pressure according to the stage of gestation. We have used a Harvard high-precision infusion pump and glass microcapillaries of defined diameter. Cardiac perfusion was performed with a surgical microscope. The volume of perfusate was 120 μl for E13.5 fetuses, which is 3 × the volume needed to obtain complete blood clearance from brain vessels (clearly visible in the embryonic brain under microscope). We gradually increased the volume to 500 μl for E17.5 fetuses. The flow rate was 0.05 ml per min for E13.5–E14.5 fetuses and 0.1 ml per min for E15.5–E17.5 fetuses. IR-labelled antibody is very stable in vivo and provides a very sensitive method for detection in blood or in brain lysates, in the range of picograms per ml. Brain lysates demonstrated the presence of maternal antibody in fetal brains analyzed at E13.5, E14.5, and E15.5 and injected at E12.5, E13.5 and E14.5, respectively [54]. However, we saw a sharp decline of antibody penetration to fetal brain analyzed at E16.5–17.5. At E17.5 the antibody is barely detectable [54]. In agreement with our observations, Danemen and colleagues also injected antibody to pregnant mice at E15.5 and found no antibody transfer to the brain tissue in detectable amount at E16.5 (personal information).
Fig. 1.

Maternal antibody is transferred to the mouse fetus starting at early stages of gestation. a Embryos at indicated ages are harvested 1 h after IR-labeled antibody was intravenously injected to pregnant dams. The embryos are imaged using a LICOR’s IR Imager as described [54]. b Embryos obtained from mice receiving saline are used as controls. The figure depicts E15.5 embryos from IR antibody-injected (left) and saline-injected (right) mice
Fig. 2.
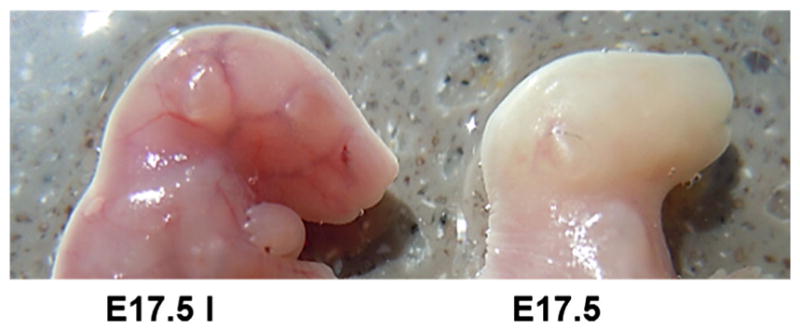
Transcardiac perfusion is effective in evacuating blood from the fetal brain of developing embryos at every stage of gestation. A non-perfused (left) and perfused (right) E17.5 embryo from the same litter. Transcardiac perfusion was performed as described [54]
We performed histological staining for Glut-1 (Fig. 3a), a glucose transporter known to be present early on the brain blood vessels, and whose expression is regulated by WNT/β-catenin signalling, an important component of the developing BBB [55]. Glut-1 is expressed abundantly at all gestational stages tested, starting at E13.5 (Fig. 3a). Similar staining was observed for the TJ proteins claudin-5, occludin, and ZO-1 on E13.5, E14.5, E15.5 E16.5, and E17.5. Although histological staining does not provide sufficient information on the substructure of TJs, which might be critical in permeability of the BBB, systematic expression of the TJ proteins was noted from E13.5 through E17.5 (Fig. 3b, c and data not shown). Analysis of the expression of the pan-EC antigen, plasmalemma vesicle-associated protein (PLVAP, PV-1) using the antibody MECA-32 showed strong expression of the molecule on brain capillaries at early stages of gestation that gradually disappears with age; it is barely present at E16.5, and, as originally described [56], it is completely absent at E 17.5 (data not shown). PLVAP is considered a marker of a leaky BBB in development and disease [57]; it has been detected as late as E18.5 in a mouse model lacking pericytes and displaying BBB impairment [50]. It is noteworthy that in our experiments, the disappearance of PLVAP coincides with the tightening of the BBB for immunoglobulin penetration.
Fig. 3.

Brain ECs express BBB markers starting early in fetal development. Histological analysis of brain sections performed at indicated stages of development. Brain sections (20 μm) are fixed with 95 % ethanol and acetone, blocked with 1 % BSA/PBS, and incubated with the proper antibody overnight at 4 °C followed by AlexaFluor conjugated secondary antibody for 1 h at RT. a Glucose transporter, Glut-1, one of the earliest BBB markers, is expressed richly through all the gestational stages tested. b–c Claudin-5 (b) and Z0-1 (c) are expressed as early as E13.5. Scale bar represents 10 μm
Summary
In our studies, we were interested in the following questions: Do maternal antibodies penetrate the fetal brain? If so, do they penetrate throughout gestation? If not, at what gestational stage are developing fetal brain capillaries no longer permeable to maternal antibodies? Can we identify endothelial cell markers associated with maternal antibody access to the developing brain that could be used in human studies? What regulates maternal antibody delivery to the fetal brain?
The factors that regulate the transfer of the antibodies to the developing brain and the role of antibodies present in the developing brain are still open questions. It is also unknown whether there is an active transport system of the antibody operating early in the developing BBB. We employed multiple precautions to avoid alteration of fetal BBB during experimentation. Assuming our techniques do not alter fetal BBB, we observed a window of opportunity for maternal antibody penetration into fetal brain at early stages of gestation. The maturing BBB progressively restricts maternal antibody penetration to the fetal brain. In mice, gestational stage E15.5–16.5 seems to be the turning point for restricting maternal antibody penetration to the fetal brain, though we cannot exclude partial restriction at some areas of the brain at early stages, or low-level leakiness at later stages.
The intriguing question is why maternal antibody is permitted at these early stages of gestation. Does the antibody play some instructive role for future brain-immune interactions? Does it bring maternal immunological experience to the developing brain? Is this a collaborative effort with the maternal gut microbiome to secure the proper BBB maturation [54]? Could the low levels of maternal antibody in human fetus at early stages of gestation (5–10 % of maternal level at week 17–22) and subsequent gradual increases at later stages of gestation (reaching 50 % by week 32) reflect a process for limiting the potential damage from maternal antibody?
It seems that PLVAP expression in developing brain is associated with a leaky BBB and that PLVAP is expressed on brain ECs early, but gradually disappears at later stages following diminished permeability of the BBB. Can anti-PLVAP antibody be used to characterize maturation of the human BBB for the benefit of human studies? The regulation or restriction of maternal antibody delivery to the fetal brain is an important and fascinating issue and should be a subject for future studies.
Preventive measures
Our studies reveal a possible new pharmacological intervention aiming at blocking the interaction of the antibody with the target brain antigen [58]. This approach using competing decoy antigens preserving the function of the target molecule warrants further exploration. It requires, however, that we understand the temporal window during which maternal antibody has access to the fetal brain and that we identify correctly the pathogenic antibodies.
Acknowledgments
This work is supported by National Institute of Health Grant 1PO1AI073693.
References
- 1.Kim J, Mohanty S, Ganesan LP, Hua K, Jarjoura D, Hayton WL, et al. FcRn in the yolk sac endoderm of mouse is required for IgG transport to fetus. J Immunol. 2009;182(5):2583–9. doi: 10.4049/jimmunol.0803247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palmeira P, Quinello C, Silveira-Lessa AL, Zago CA, Carneiro-Sampaio M. IgG placental transfer in healthy and pathological pregnancies. Clin Dev Immunol. 2012;2012:985646. doi: 10.1155/2012/985646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malek A, Sager R, Kuhn P, Nicolaides KH, Schneider H. Evolution of maternofetal transport of immunoglobulins during human pregnancy. Am J Reprod Immunol. 1996;36(5):248–55. doi: 10.1111/j.1600-0897.1996.tb00172.x. [DOI] [PubMed] [Google Scholar]
- 4.van den Berg JP, Westerbeek EA, van der Klis FR, Berbers GA, van Elburg RM. Transplacental transport of IgG antibodies to preterm infants: a review of the literature. Early Hum Dev. 2011;87(2):67–72. doi: 10.1016/j.earlhumdev.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 5.Garty BZ, Ludomirsky A, Danon YL, Peter JB, Douglas SD. Placental transfer of immunoglobulin G subclasses. Clin Diagn Lab Immunol. 1994;1(6):667–9. doi: 10.1128/cdli.1.6.667-669.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hurley WL, Theil PK. Perspectives on immunoglobulins in colostrum and milk. Nutrients. 2011;3(4):442–74. doi: 10.3390/nu3040442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kobayashi R, Mii S, Nakano T, Harada H, Eto H. Neonatal lupus erythematosus in Japan: a review of the literature. Autoimmun Rev. 2009;8(6):462–6. doi: 10.1016/j.autrev.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 8.Lee LA. Neonatal lupus erythematosus: clinical findings and pathogenesis. J Investig Dermatol Symp Proc. 2004;9(1):52–6. doi: 10.1111/j.1087-0024.2004.00827.x. [DOI] [PubMed] [Google Scholar]
- 9.Motta M, Chirico G, Rebaioli CB, Faden D, Lojacono A, Allegri F, et al. Anticardiolipin and anti-beta2 glycoprotein I antibodies in infants born to mothers with antiphospholipid antibody-positive autoimmune disease: a follow-up study. Am J Perinatol. 2006;23(4):247–51. doi: 10.1055/s-2006-939533. [DOI] [PubMed] [Google Scholar]
- 10.Tincani A, Nuzzo M, Motta M, Zatti S, Lojacono A, Faden D. Autoimmunity and pregnancy: autoantibodies and pregnancy in rheumatic diseases. Ann N Y Acad Sci. 2006;1069:346–52. doi: 10.1196/annals.1351.032. [DOI] [PubMed] [Google Scholar]
- 11.Brucato A. Prevention of congenital heart block in children of SSA-positive mothers. Rheumatology (Oxford) 2008;47(Suppl 3):iii35–7. doi: 10.1093/rheumatology/ken153. [DOI] [PubMed] [Google Scholar]
- 12.Stea EA, Routsias JG, Clancy RM, Buyon JP, Moutsopoulos HM, Tzioufas AG. Anti-La/SSB antiidiotypic antibodies in maternal serum: a marker of low risk for neonatal lupus in an offspring. Arthritis Rheum. 2006;54(7):2228–34. doi: 10.1002/art.21954. [DOI] [PubMed] [Google Scholar]
- 13.Eftekhari P, Roegel JC, Lezoualc’h F, Fischmeister R, Imbs JL, Hoebeke J. Induction of neonatal lupus in pups of mice immunized with synthetic peptides derived from amino acid sequences of the serotoninergic 5-HT4 receptor. Eur J Immunol. 2001;31(2):573–9. doi: 10.1002/1521-4141(200102)31:2<573:AID-IMMU573>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 14.Kamel R, Garcia S, Lezoualc’h F, Fischmeister R, Muller S, Hoebek J, et al. Immunomodulation by maternal autoantibodies of the fetal serotoninergic 5-HT4 receptor and its consequences in early BALB/c mouse embryonic development. BMC Dev Biol. 2007;7:34. doi: 10.1186/1471-213X-7-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buyon JP, Clancy R, Di Donato F, Miranda-Carus ME, Askanase AD, Garcia J, et al. Cardiac 5-HT(4) serotoninergic receptors, 52kD SSA/Ro and autoimmune-associated congenital heart block. J Autoimmun. 2002;19(1–2):79–86. doi: 10.1006/jaut.2002.0594. [DOI] [PubMed] [Google Scholar]
- 16.Kamel R, Eftekhari P, Clancy R, Buyon JP, Hoebeke J. Autoantibodies against the serotoninergic 5-HT4 receptor and congenital heart block: a reassessment. J Autoimmun. 2005;25(1):72–6. doi: 10.1016/j.jaut.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 17.Askanase AD, Izmirly PM, Katholi M, Mumtaz J, Buyon JP. Frequency of neuro-psychiatric dysfunction in anti-SSA/SSB exposed children with and without neonatal lupus. Lupus. 2010;19(3):300–6. doi: 10.1177/0961203309354542. [DOI] [PubMed] [Google Scholar]
- 18.Motta M. Neonates from mother with autoimmune disease. Haematol Rep. 2006;2(10):55–9. [Google Scholar]
- 19.Papazian O. Transient neonatal myasthenia gravis. J Child Neurol. 1992;7(2):135–41. doi: 10.1177/088307389200700202. [DOI] [PubMed] [Google Scholar]
- 20.Parlowsky T, Welzel J, Amagai M, Zillikens D, Wygold T. Neonatal pemphigus vulgaris: IgG4 autoantibodies to desmoglein 3 induce skin blisters in newborns. J Am Acad Dermatol. 2003;48(4):623–5. doi: 10.1067/mjd.2003.170. [DOI] [PubMed] [Google Scholar]
- 21.Tincani A, Rebaioli CB, Andreoli L, Lojacono A, Motta M. Neonatal effects of maternal antiphospholipid syndrome. Curr Rheumatol Rep. 2009;11(1):70–6. doi: 10.1007/s11926-009-0010-8. [DOI] [PubMed] [Google Scholar]
- 22.Licht C, Model P, Kribs A, Herkenrath P, Michalk DV, Haupt WF, et al. Transient neonatal myasthenia gravis. Nervenarzt. 2002;73(8):774–8. doi: 10.1007/s00115-002-1344-x. [DOI] [PubMed] [Google Scholar]
- 23.Tincani A, Danieli E, Nuzzo M, Scarsil M, Motta M, Cimaz R, et al. Impact of in utero environment on the offspring of lupus patients. Lupus. 2006;15(11):801–7. doi: 10.1177/0961203306071005. [DOI] [PubMed] [Google Scholar]
- 24.Boyle CA, Boulet S, Schieve LA, Cohen RA, Blumberg SJ, Yeargin-Allsopp M, et al. Trends in the prevalence of developmental disabilities in US children, 1997–2008. Pediatrics. 2011;127(6):1034–42. doi: 10.1542/peds.2010-2989. [DOI] [PubMed] [Google Scholar]
- 25.Bauman MD, Iosif AM, Ashwood P, Braunschweig D, Lee A, Schumann CM, et al. Maternal antibodies from mothers of children with autism alter brain growth and social behavior development in the rhesus monkey. Transl Psychiatry. 2013;3:e278. doi: 10.1038/tp.2013.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Braunschweig D, Golub, Koenig CM, Qi L, Pessah IN, Van de Water J, et al. Maternal autism-associated IgG antibodies delay development and produce anxiety in a mouse gestational transfer model. J Neuroimmunol. 2012;252(1–2):56–65. doi: 10.1016/j.jneuroim.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Camacho J, Jones K, Miller E, Ariza J, Noctor S, Van de Water J, et al. Embryonic intraventricular exposure to autism-specific maternal autoantibodies produces alterations in autistic-like stereotypical behaviors in offspring mice. Behav Brain Res. 2014;266:46–51. doi: 10.1016/j.bbr.2014.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dalton P, Deacon R, Blamire A, Pike M, McKinlay I, Stein J, et al. Maternal neuronal antibodies associated with autism and a language disorder. Ann Neurol. 2003;53(4):533–7. doi: 10.1002/ana.10557. [DOI] [PubMed] [Google Scholar]
- 29.Martin LA, Ashwood P, Braunschweig D, Cabanlit M, Van de Water J, Amaral DG. Stereotypies and hyperactivity in rhesus monkeys exposed to IgG from mothers of children with autism. Brain Behav Immun. 2008;22(6):806–16. doi: 10.1016/j.bbi.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singer HS, Morris C, Gause C, Pollard M, Zimmerman AW, Pletnikov M. Prenatal exposure to antibodies from mothers of children with autism produces neurobehavioral alterations: a pregnant dam mouse model. J Neuroimmunol. 2009;211(1–2):39–48. doi: 10.1016/j.jneuroim.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 31.Rout UK, Dhossche DM. A pathogenetic model of autism involving Purkinje cell loss through anti-GAD antibodies. Med Hypotheses. 2008;71(2):218–21. doi: 10.1016/j.mehy.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 32.Lahita RG. Systemic lupus erythematosus: learning disability in the male offspring of female patients and relationship to laterality. Psychoneuroendocrinology. 1988;13(5):385–96. doi: 10.1016/0306-4530(88)90045-5. [DOI] [PubMed] [Google Scholar]
- 33.McAllister DL, Kaplan BJ, Edworthy SM, Martin L, Crawford SG, Ramsey-Goldman R, et al. The influence of systemic lupus erythematosus on fetal development: cognitive, behavioral, and health trends. J Int Neuropsychol Soc. 1997;3(4):370–6. [PubMed] [Google Scholar]
- 34.Ross G, Sammaritano L, Nass R, Lockshin M. Effects of mothers’ autoimmune disease during pregnancy on learning disabilities and hand preference in their children. Arch Pediatr Adolesc Med. 2003;157(4):397–402. doi: 10.1001/archpedi.157.4.397. [DOI] [PubMed] [Google Scholar]
- 35.Urowitz MB, Gladman DD, MacKinnon A, Ibanez D, Bruto V, Rovet J, et al. Neurocognitive abnormalities in offspring of mothers with systemic lupus erythematosus. Lupus. 2008;17(6):555–60. doi: 10.1177/0961203308089326. [DOI] [PubMed] [Google Scholar]
- 36.Lee JY, Huerta PT, Zhang J, Kowal C, Bertini E, Volpe BT, et al. Neurotoxic autoantibodies mediate congenital cortical impairment of offspring in maternal lupus. Nat Med. 2009;15(1):91–6. doi: 10.1038/nm.1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arinuma Y, Yanagida T, Hirohata S. Association of cerebrospinal fluid anti-NR2 glutamate receptor antibodies with diffuse neuropsychiatric systemic lupus erythematosus. Arthritis Rheum. 2008;58(4):1130–5. doi: 10.1002/art.23399. [DOI] [PubMed] [Google Scholar]
- 38.Yoshio T, Onda K, Nara H, Minota S. Association of IgG anti-NR2 glutamate receptor antibodies in cerebrospinal fluid with neuropsychiatric systemic lupus erythematosus. Arthritis Rheum. 2006;54(2):675–8. doi: 10.1002/art.21547. [DOI] [PubMed] [Google Scholar]
- 39.Atladottir HO, Pedersen MG, Thorsen P, Mortensen PB, Deleuran B, Eaton WW, et al. Association of family history of autoimmune diseases and autism spectrum disorders. Pediatrics. 2009;124(2):687–94. doi: 10.1542/peds.2008-2445. [DOI] [PubMed] [Google Scholar]
- 40.Brimberg L, Sadiq A, Gregersen PK, Diamond B. Brain-reactive IgG correlates with autoimmunity in mothers of a child with an autism spectrum disorder. Mol Psychiatry. 2013;18(11):1171–7. doi: 10.1038/mp.2013.101. [DOI] [PubMed] [Google Scholar]
- 41.Croen LA, Grether JK, Yoshida CK, Odouli R, Van de Water J. Maternal autoimmune diseases, asthma and allergies, and childhood autism spectrum disorders: a case–control study. Arch Pediatr Adolesc Med. 2005;159(2):151–7. doi: 10.1001/archpedi.159.2.151. [DOI] [PubMed] [Google Scholar]
- 42.Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood–brain barrier. Neurobiol Dis. 2010;37(1):13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 43.Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood–brain barrier. Nat Rev Neurosci. 2006;7(1):41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 44.Bauer HC, Bauer H. Neural induction of the blood–brain barrier: still an enigma. Cell Mol Neurobiol. 2000;20(1):13–28. doi: 10.1023/a:1006939825857. [DOI] [PubMed] [Google Scholar]
- 45.Hawkins BT, Davis TP. The blood–brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57(2):173–85. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 46.Schlachetzki F, Zhu C, Pardridge WM. Expression of the neonatal Fc receptor (FcRn) at the blood–brain barrier. J Neurochem. 2002;81(1):203–6. doi: 10.1046/j.1471-4159.2002.00840.x. [DOI] [PubMed] [Google Scholar]
- 47.Zhang Y, Pardridge WM. Mediated efflux of IgG molecules from brain to blood across the blood–brain barrier. J Neuroimmunol. 2001;114(1–2):168–72. doi: 10.1016/s0165-5728(01)00242-9. [DOI] [PubMed] [Google Scholar]
- 48.Morita S, Miyata S. Different vascular permeability between the sensory and secretory circumventricular organs of adult mouse brain. Cell Tissue Res. 2012;349(2):589–603. doi: 10.1007/s00441-012-1421-9. [DOI] [PubMed] [Google Scholar]
- 49.Saunders NR, Liddelow SA, Dziegielewska KM. Barrier mechanisms in the developing brain. Front Pharmacol. 2012;3:46. doi: 10.3389/fphar.2012.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature. 2010;468(7323):562–6. doi: 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Davila D, Thibault K, Fiacco TA, Agulhon C. Recent molecular approaches to understanding astrocyte function in vivo. Front Cell Neurosci. 2013;7:272. doi: 10.3389/fncel.2013.00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hoffmann A, Bredno J, Wendland M, Derugin N, Ohara P, Wintermark M. High and low molecular weight fluorescein isothiocyanate (FITC)-dextrans to assess blood–brain barrier disruption: technical considerations. Transl Stroke Res. 2011;2(1):106–11. doi: 10.1007/s12975-010-0049-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pentsuk N, van der Laan JW. An interspecies comparison of placental antibody transfer: new insights into developmental toxicity testing of monoclonal antibodies. Birth Defects Res B Dev Reprod Toxicol. 2009;86(4):328–44. doi: 10.1002/bdrb.20201. [DOI] [PubMed] [Google Scholar]
- 54.Braniste V, Al-Asmakh M, Kowal C, Anuar F, Abbaspour A, Toth M, et al. The gut microbiota influences blood–brain barrier permeability in mice. Sci Transl Med. 2014;6(263):263ra158. doi: 10.1126/scitranslmed.3009759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Daneman R, Agalliu D, Zhou L, Kuhnert F, Kuo CJ, Barres BA. Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc Natl Acad Sci USA. 2009;106(2):641–6. doi: 10.1073/pnas.0805165106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hallmann R, Mayer DN, Berg EL, Broermann R, Butcher EC. Novel mouse endothelial cell surface marker is suppressed during differentiation of the blood brain barrier. Dev Dyn. 1995;202(4):325–32. doi: 10.1002/aja.1002020402. [DOI] [PubMed] [Google Scholar]
- 57.Shue EH, Carson-Walter EB, Liu Y, Winans BN, Ali ZS, Chen J, et al. Plasmalemmal vesicle associated protein-1 (PV-1) is a marker of blood–brain barrier disruption in rodent models. BMC Neurosci. 2008;9:29. doi: 10.1186/1471-2202-9-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bloom O, Cheng KF, He M, Papatheodorou A, Volpe BT, Diamond B, et al. Generation of a unique small molecule peptidomimetic that neutralizes lupus autoantibody activity. Proc Natl Acad Sci USA. 2011;108(25):10255–9. doi: 10.1073/pnas.1103555108. [DOI] [PMC free article] [PubMed] [Google Scholar]