

Summary
Cutaneous lesions described as chilblain lupus occur in the context of familial chilblain lupus or Aicardi–Goutières syndrome. To date, seven genes related to Aicardi–Goutières syndrome have been described. The most recently described encodes the cytosolic double-stranded RNA receptor IFIH1 (also known as MDA5), a key component of the antiviral type I interferon-mediated innate immune response. Enhanced type I interferon signalling secondary to gain-of-function mutations in IFIH1 can result in a range of neuroinflammatory phenotypes including classical Aicardi–Goutières syndrome. It is of note that none of the patients with a neurological phenotype so far described with mutations in this gene was reported to demonstrate cutaneous involvement. We present a family segregating a heterozygous pathogenic mutation in IFIH1 showing dermatological involvement as a prominent feature, variably associated with neurological disturbance and premature tooth loss. All three affected individuals exhibited increased expression of interferon-stimulated genes in whole blood, and the mutant protein resulted in enhanced interferon signalling in vitro, both in the basal state and following ligand stimulation. Our results further extend the phenotypic spectrum associated with mutations in IFIH1, indicating that the disease can be confined predominantly to the skin, while also highlighting phenotypic overlap with both Aicardi–Goutières syndrome and Singleton–Merten syndrome.
Aicardi–Goutières syndrome (AGS, MIM 225750), in its most characteristic form, presents as a genetic encephalopathy, associated with basal ganglia calcification and cerebral white-matter disease. In addition to the neurological phenotype, acral chilblain lesions are seen in > 40% of cases.1 Until recently, six AGS-causative genes had been described in association with AGS, respectively encoding the DNA exonuclease TREX1 (AGS1), the three nonallelic components of the RNase H2 endonuclease complex (RNASEH2B, AGS2; RNASEH2C, AGS3; and RNASEH2A, AGS4), the deoxynucleoside triphosphate triphosphohydrolase SAMHD1 (AGS5) and the double-stranded (ds)RNA-editing enzyme ADAR (AGS6).
Recently, we and others reported heterozygous gain-of-function IFIH1 mutations (AGS7)1 as causative of a spectrum of neuroinflammatory phenotypes, including AGS and apparently nonsyndromic spastic paraparesis, in which elevated levels of interferon-stimulated genes are observed.2,3 IFIH1 encodes the protein interferon-induced helicase C domain-containing protein 1, also known as melanoma differentiation-associated protein 5 (MDA5). Interestingly, heterozygous mutations in TREX1 and SAMHD1 have also been identified in patients with familial chilblain lupus (FCL, MIM 610 448).4,5 FCL is a monogenic form of cutaneous lupus, which presents in childhood with acral ulcerating lesions that are exacerbated by cold. To date, mutations in IFIH1 have not been reported in association with FCL.
A seemingly clinically distinct condition, Singleton–Merten syndrome (SMS, MIM 182250), has been recently described as being due to a specific heterozygous point mutation in IFIH1.6 The hallmarks of SMS include aortic calcification, delayed dental eruption and early loss of permanent teeth, osteopenia and acro-osteolysis. Additionally, a number of patients have been reported with psoriasis and glaucoma, the latter being a common feature in AGS also. As for AGS and the related neuroimmunological phenotypes already described due to mutations in IFIH1, patients with SMS demonstrated a marked induction of type I interferon signalling.
Here we report a family comprising three affected individuals who harbour a heterozygous IFIH1 mutation, variably expressing a skin and a neurological phenotype. The initial presentation in each case was dermatological. Subsequently, features overlapping both AGS and SMS became evident within this single family.
Case report
The proband, a white French boy, was born at 34 weeks of gestation, with weight 2.980 kg (+ 0.34 SDs from the mean), height 49 cm (+ 0.57 SDs) and cranial perimeter 33 cm (−0.23 SDs). He was hospitalized in the neonatal period because of transitory respiratory distress. He presented to dermatologists at 1 year of age with ulcerating lesions of the ear helices, which were exacerbated by cold and healed with scarring. On clinical examination, superficial crusted and erythematous lesions of the helix were present (Fig. 1a). He was also noted to have erythematous cheeks (Fig. 1b) and multiple lentigines on the upper and lower limbs without any significant ultraviolet exposure (Fig. 1c). His nails were somewhat fragile with longitudinal striations. Histological examination of a cutaneous biopsy of the helix was consistent with lichenoid lupus, with slight interface dermatitis with a moderate infiltrate in the superficial and deep dermis, along the basement membrane and along the vessels and the sebaceous glands, associated with slight acanthosis and a few apoptotic cells in the basal layer (Fig. 1d).
Fig 1.
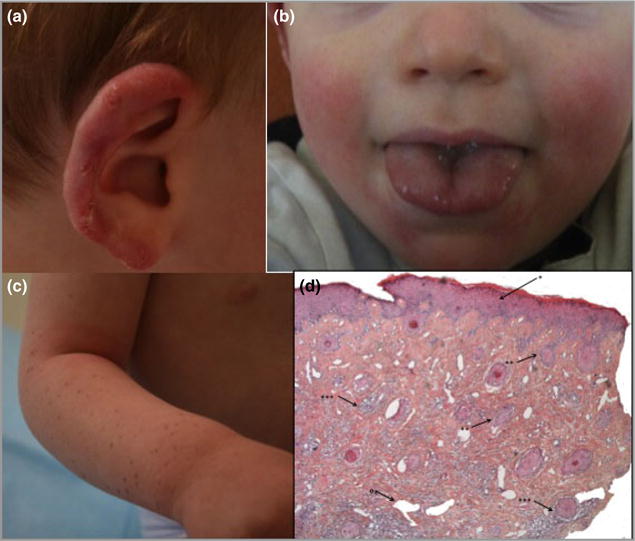
Clinical images of the proband. (a) Erythema and ulceration of the outer helix of the right ear. (b) Erythema of the cheeks bilaterally. No lesions of the tongue were noticed. (c) Lentigines of the right forearm. (d) Histology of the ear helix demonstrating slight acanthosis of the epidermis (*), interface dermatitis located mostly in the follicles (**) and superficial and deep infiltrate along the adnexae and around the vessels (***). In addition most capillaries are dilated (°) (magnification level, d:HES × 10).
Neurological examination revealed apparent stiffness of the lower limbs. He subsequently walked at age 20 months, although his parents reported leg stiffness after prolonged sitting beyond this time, and the patient complained of leg pain so that he could not climb stairs unaided at the age of 3 years. A cranial magnetic resonance imaging (MRI) scan taken at 5 years revealed a small area of hypersignal of the periventricular white matter on axial fluid-attenuated inversion recovery imaging, but was otherwise unremarkable. He did not undergo computed tomography (CT) imaging. Of note, by the age of 4 years his motor and intellectual development was considered to be within normal limits. Cardiac ultrasound was normal.
The father of the proband reported no previous family history of relevance. He described ulcerations of the ears and nose since the age of 7 months, which were worse in the winter and healed with scarring and subsequent tissue loss. He had experienced similar lesions on the legs, also healing with scarring, and similar leg stiffness after prolonged sitting as for his son. During childhood, multiple lentigines were noted on the limbs, and a degree of photosensitivity was reported. He also complained of papular lesions on the legs, which resolved leaving atrophic scars. In adulthood, ‘blister-like lesions’ were described to occur, most notably on the legs following trauma. Subsequently, hyperkeratotic lesions formed at the elbows and knees, and deformities of the metacarpophalangeal and interphalangeal joints of the hands with tendon retraction and foot deformity, including marked bilateral hallux valgus, developed without any previous pain or rheumatism. After adolescence, his permanent teeth, apart from the molars and premolars, were lost, with evident root hypoplasia (Fig. 2a). There was no history of developmental delay or neurological dysfunction, but lower-limb pain was reported with a tightening of the Achilles tendons.
Fig 2.
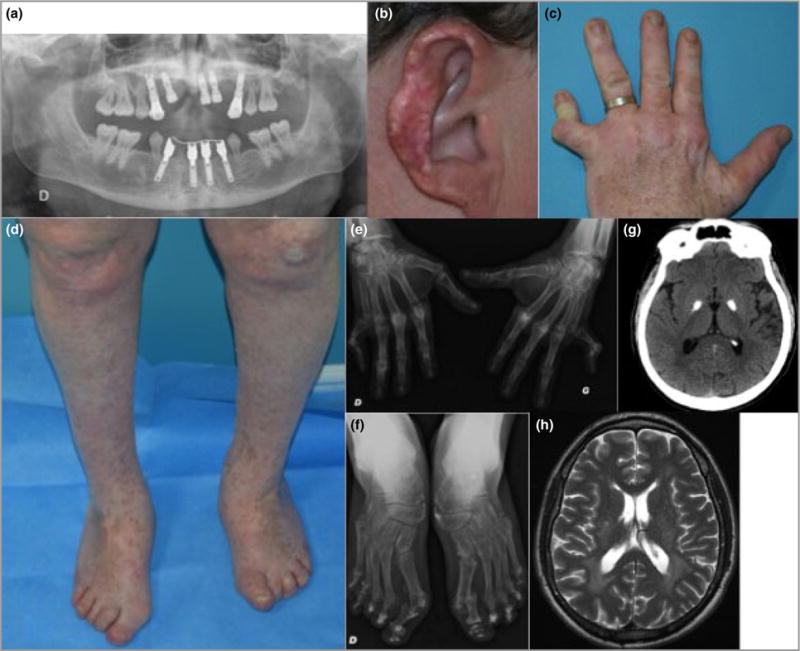
Clinical images of the 41-year-old father. (a) Pantomogram showing loss of permanent teeth. The remaining dentition, molars and premolars demonstrate root hypoplasia. (b) Scarring and tissue loss of the outer ear helix following chronic ulceration. (c) Deformities of the metacarpophalangeal and interphalangeal joints of the left hand, and multiple lentigines. (d) Hallux valgus, psoriatic lesions over the knees and multiple lentigines. (e) Radiography showing joint deformities on the hands. (f) Radiography showing joint deformities on the feet. (g) Brain imaging at age 41 years showing bilateral calcification of the globus pallidus on computed tomography. (h) Brain imaging at age 41 years illustrating high signal in the white matter around the posterior poles of the lateral ventricles on axial T2-weighted magnetic resonance imaging.
On examination at 41 years of age this man’s ears were bilaterally scarred secondary to crusted lesions (Fig. 2b). Multiple lentigines were evident on the limbs and torso without telangiectasia. His skin was fragile and covered with atrophic scars (Fig. 2c, d). Crusted, hyperkeratotic lesions were noticed on the knees (Fig. 2d). There was marked hallux valgus, and bilateral fifth-finger metacarpophalangeal contractures (Fig. 2c and d), confirmed on X-ray (Fig. 2e and f). He had a dental prosthesis. Neurological examination was normal. Eye examination revealed macular pigmentation, without atrophy or telangiectasia. There was no evidence of glaucoma. Cardiac echography showed minimal calcification of the aortic valve. At this same age, bilateral dense calcification of the globus pallidus was seen on cranial CT imaging (Fig. 2g), while MRI revealed periventricular high signal of the white matter around the posterior poles of the lateral ventricles (Fig. 2h).
The youngest brother to the proband was born at 33 weeks of gestation, with weight 2.240 kg (−0.8 SDs from the mean), height 46.5 cm (+ 0.26 SDs) and cranial perimeter 31.5 cm (−0.97 SDs). He required initial respiratory support due to respiratory distress. He presented with cold-exacerbated lesions of the ears beginning at age 15 months. On examination, crusting and erythema of the helices was noted bilaterally (Fig. 3a). He also demonstrated multiple lentigines on the forearms and legs (Fig. 3b), erythematous cheeks (Fig. 3c) and nail fragility with longitudinal striations. From 3 years of age, during the winter months, papular lesions were noticed on the legs, which resolved leaving atrophic scars (Fig. 3d). His motor development was delayed. He began to walk with help at 18 months of age. He then experienced a period of apparent motor regression, and at age 2.5 years was unable to walk or stand. He had only a few spoken words at this age. He demonstrated some useful hand function and was able to scribble, but not to draw.
Fig 3.
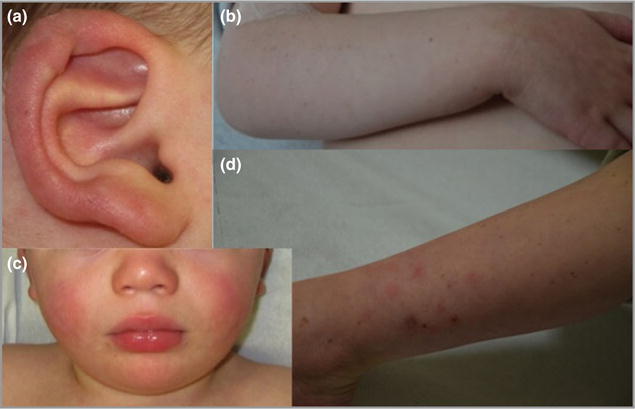
Clinical images of the younger sibling of the proband. (a) Erythema and ulceration of the outer helix of the right ear. (b) Multiple lentigines of the right forearm. (c) Bilateral erythema of the cheeks. (d) Lower-limb atrophic scars, which occurred following resolution of papular lesions.
On neurological examination, spastic tetraparesis was noticed. A cranial MRI at age 3 years demonstrated minimal increased signal changes in the white matter at the posterior poles of the ventricles. Cranial CT imaging and cardiac ultrasound were both considered normal. Due to pain and motor delay, a course of oral corticosteroid treatment was commenced at 1 mg kg−1 per day and then tapered slowly to 0.3 mg kg−1 per day after 1 month. This reduced the pain and enhanced his mobility, such that walking with the aid of a frame was possible after 1 month. In addition an improvement in speech was reported, with short sentences spoken after 1 month of treatment and speech therapy. His social behaviour was also considered to have improved. However, after only 3 months of systemic corticosteroids, he developed bilateral glaucoma, requiring cessation of therapy and a combination of medical and surgical ophthalmic management.
Despite the discrepancy in neurological involvement across the three family members, the cutaneous features suggested the possibility of a dominant interferonopathy. RNA expression of six interferon-stimulated genes in whole blood demonstrated significant upregulation in all three patients compared with controls, similarly to previously reported cases with mutations in IFIH1 (Fig. 4).2 Assessment of additional proinflammatory cytokines by quantitative polymerase chain reaction (IL6, TNFA, NFKB1, IL1B and IL10) showed equivalent expression to that observed in controls (data not shown).
Fig 4.
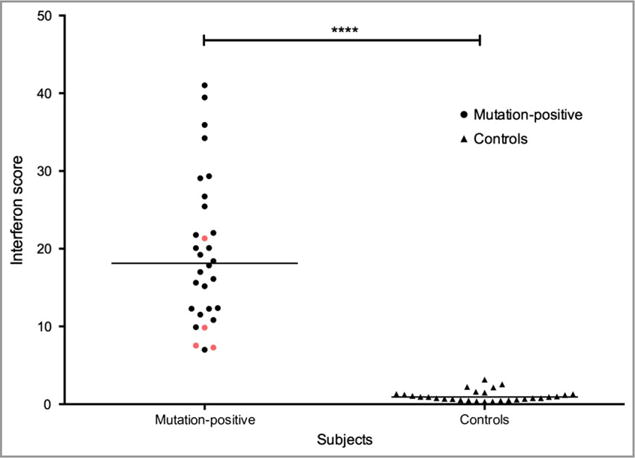
Individuals with the p.Ala489Thr interferon-induced helicase C domain-containing protein 1 (IFIH1) mutant demonstrate elevated levels of interferon-stimulated genes compared with controls. An interferon score was calculated from the median fold change in RQ value for a panel of six interferon signature genes measured using quantitative polymerase chain reaction – IFI27 (Hs01086370_m1), IFI44L (Hs00199115_m1), IFIT1 (Hs00356631_g1), ISG15 (Hs00192713_m1), RSAD2 (Hs01057264_m1) and SIGLEC1 (Hs00988063_m1) – normalized to the expression levels of HPRT1 (Hs03929096_g1) and 18S RNA (Hs999999001_s1). All three affected members of the family (red) display a score that is comparable with previously reported cases (black)7 with IFIH1-related disease, and is significantly higher than in control samples (****P < 0.0001). Levels were measured on two separate occasions in one child in the reported family.
Mutations in the two genes previously implicated in chilblain lupus inherited as an autosomal dominant trait, TREX1 and SAMHD1, were not observed. However, whole-exome sequencing identified a heterozygous missense variant in IFIH1 (c.1465G>A, p.Ala489Thr) in all three affected individuals. This variant was predicted as deleterious using in silico analysis tools, and was absent from the Exome Aggregation Consortium browser (> 120 000 control alleles) and > 600 in-house exomes. The presence of the variant was confirmed by Sanger sequencing. The variant was not seen in the blood of either of the proband’s asymptomatic paternal grandparents, thus indicating that the variant had arisen de novo, or due to gonadal mosaicism.
Mapping of the altered residue Ala489 onto the crystal structure of the 2CARD deletion construct (Δ2CARD) demonstrated that the residue lies within the Hel1 domain, one of two highly conserved core helicase domains (Fig. 5a). These domains are responsible for binding RNA and RNA-dependent ATP hydrolysis. The Ala489 residue in IFIH1 is in close proximity to the ATP binding site, and substitution of Ala489 by threonine may diminish the ATP binding and hydrolysis activities of IFIH1. Analysis of purified wild-type and mutant IFIH1 (Fig. 5b) displayed impaired ATP hydrolysis activity in the mutant (Fig. 5c) and increased stability of the mutant protein–RNA complex compared with the wild-type in the presence of ATP (Fig. 5d). Note that ATP hydrolysis by wild-type IFIH1 triggers dissociation from dsRNA, which results in dsRNA length-sensitive stability and interferon signalling activity.
Fig 5.
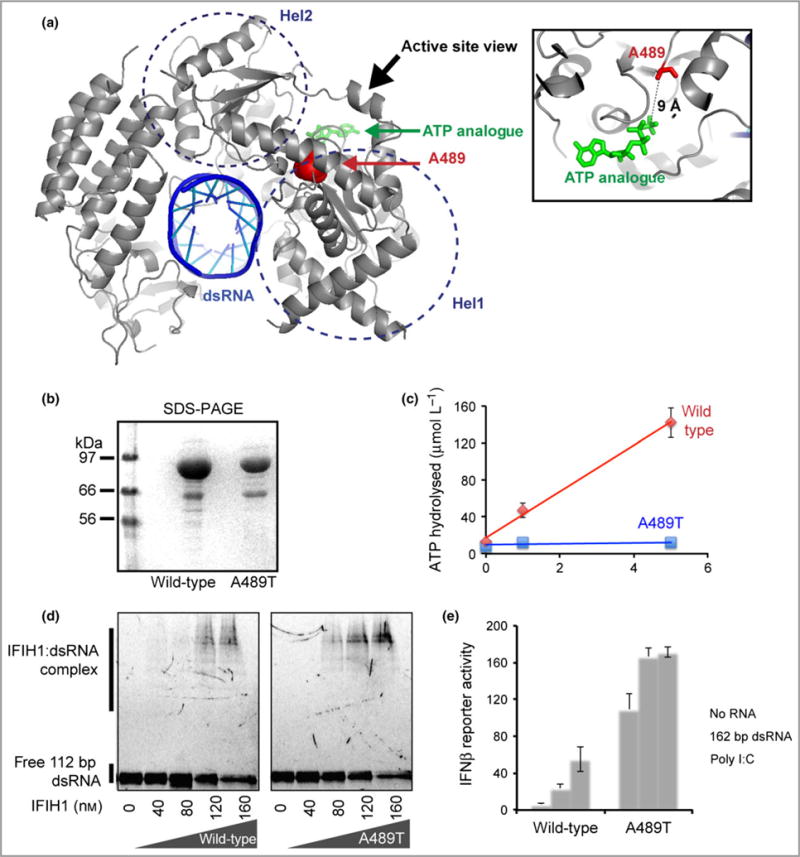
The p.Ala489Thr interferon-induced helicase C domain-containing protein 1 (IFIH1) mutant confers a gain of interferon signalling. (a) Mapping of the altered residue A489 (red sphere) onto the structure of IFIH1 Δ2CARD (grey) bound by double-stranded (ds)RNA (blue) and ATP analogue (green) (Protein Data Bank 4GL2). (b) Sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis (Coomassie stain) of the purified wild-type and mutant IFIH1. (c) ATP hydrolysis activity (mean ± SD, n = 3 biological replicates) of wild-type and mutant IFIH1. (d) Electrophoretic mobility shift assay of purified wild-type and mutant IFIH1 with 112-bp dsRNA. The gel image is representative of three independent experiments in the presence of ATP. (e) Interferon (IFN)-β reporter activity (mean ± SD, n = 3 biological replicates) of Flag-tagged wild-type and mutant IFIH1 with and without stimulation with 162-bp dsRNA or polyinosinic–polycytidylic acid (poly I:C) in HEK293T cells. The results are representative of three independent experiments.
Interferon-β reporter stimulatory activity of wild-type and mutant IFIH1 in HEK293T cells was also assessed (Fig. 5e). As described previously,7 basal levels of interferon signalling were markedly increased in the absence of exogenous RNA. Furthermore, wild-type IFIH1 was induced only upon stimulation with polyinosinic–polycytidylic acid (poly I:C), a long (> 1-kb) dsRNA, and not with short (162-bp) dsRNA, and activity was negligible in the absence of exogenous RNA. In contrast, mutant IFIH1 displayed marked interferon induction following stimulation with either long dsRNA (poly I:C) or short dsRNA.
Discussion
Here we report a family with cutaneous features resembling those of chilblain lupus,4,7 also demonstrating variable neurological involvement reminiscent of AGS1 and features of SMS6 in the affected father (Table 1).
Table 1.
Comparison of features seen in familial chilblain lupus,4,7 Aicardi–Goutières syndrome1 and Singleton–Merten syndrome6
| Familial chilblain lupus | Aicardi–Goutières syndrome | Singleton–Merten syndrome | |
|---|---|---|---|
| Clinical features | Skin predominantly affected: acral chilblains (hands, feet, ears, nose). Arthralgia (articular deformities) | Major neurological involvement: progressive encephalopathy. Skin: acral chilblains (feet) (articular deformities). Glaucoma | Aortic calcification. Hypoplastic teeth. Osteopenia. Psoriatic-type lesions. Articular deformities. Glaucoma |
| Features on cerebral imaging | Not normally present | Intracranial calcification, white-matter disease, cerebral atrophy | Not reported previously |
| Inheritance mode | Autosomal dominant | Autosomal recessive (autosomal dominant) | Autosomal dominant |
| Genetic basis | TREX1, SAMHD1 | TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, IFIH1 | IFIH1 |
The predominant clinical manifestation secondary to enhanced interferon production in this family was ulcerating lesions of the ears. These cutaneous manifestations are in keeping with those described in both AGS and FCL, but have not previously been observed in the context of mutations in IFIH1.2,3 Of note, none of the patients in this report exhibited the typical acral lesions of AGS/chilblain lupus affecting the toes and fingers. The neurological features seen in the sibling pair were not typical of the neonatal encephalopathy observed in classical AGS, although they remain within the recognized clinical and neuroimaging diagnostic spectrum of that disease.2 The neurological examination of the father was completely normal, despite the fact that he demonstrated obvious intracranial calcification and white-matter abnormalities.
A finding of particular interest in this family was the presence of acral lentigines in all three affected individuals, reminiscent of the pigmentary changes observed in dyschromatosis symmetrica hereditaria (DSH), secondary to ADAR mutations.8 DSH is allelic to AGS at the ADAR (AGS6) locus, and rare cases of ADAR-related AGS also manifest lentigines.9 The aetiology of these lesions is currently unclear, but they may provide a diagnostic clue in certain cases.
The loss of secondary dentition, marked hallux valgus, and bilateral fifth-finger metacarpophalangeal contractures seen in the father of our proband are not typical of AGS, but are in keeping with SMS. Indeed, the fifth-finger contractures (Fig. 2c) are very reminiscent of those previously published in SMS, and in some patients with FCL.4 The development of glaucoma in childhood after only 3 months of corticosteroid therapy is of note, considering that glaucoma is a recognized feature of both AGS and SMS. Consequently, we suggest that active ophthalmic monitoring is required in such families. Although echocardiography was normal in all three individuals, we note the phenotypic variability reported in IFIH1-related SMS,6 and in RIG-I (retinoic acid-inducible gene I)-associated ‘atypical SMS’.10 These observations lead us to suggest that an SMS phenotype may occur in cases beyond those with the recently described p.Arg822Gln mutation in IFIH1. The dental findings reported in SMS are interesting given the fact that single-nucleotide polymorphisms in IFIH1 have been associated with chronic periodontitis.11
The cutaneous and neurological phenotypes observed in this family demonstrate overlap with a number of other inflammatory monogenic disorders, illustrating the profound effect of inappropriate stimulation, or defective negative regulation, of the type I interferon response on the skin and nervous system. Mutations in TMEM173 confer gain of function on the cytosolic DNA sensor STING,12 which causes STING-associated vasculopathy with onset in infancy (SAVI). This interferon-driven inflammatory disease can present with a severe infantile cutaneous vasculopathy leading to extensive tissue loss, in addition to inflammatory interstitial lung disease in some cases. The erythematous cheeks observed in both children in the family presented here (Figs 1b and 3c) bear resemblance to those seen in SAVI, even though they are much less severe. A second autosomal recessive monogenic disorder, caused by TADA2A mutations, may also present with lesions of the ear helix, reminiscent of those reported here. Furthermore, TADA2A mutations can manifest with a neurological phenotype, due to small-vessel ischaemic strokes.13
Histologically, vacuolation of the basal layer, interface dermatitis, and superficial and deep dermal infiltrate can be seen either in chilblain lupus, AGS or cutaneous lupus erythematosus.14,15 The granular deposition of immunoglobulins and complement along the basement membrane is described in FCL, AGS and cutaneous lupus erythematosus, but seems to be inconstant in these first two entities.5,14–16 Mucin deposits have been reported in chilblain lupus.5,14 All of these data are consistent with the observation that both chilblain lupus and AGS are allelic, and that these monogenic pathologies share a common pathophysiology with lupus erythematosus.
The inflammatory phenotype in the family reported here was caused by a gain-of-function mutation in the cytosolic RNA sensor IFIH1, inherited as an autosomal dominant trait. The interferon signature seen in all three affected individuals was comparable with that previously observed in IFIH1-related disease (Fig. 4),2 and is indicative of a type I interferonopathy.17
IFIH1 encodes a cytoplasmic dsRNA sensor called IFIH1, or MDA5. In humans, IFIH1 has been associated with a number of inflammatory states. Thus, IFIH1 expression is markedly increased in the skin of patients with dermatomyositis, chronic discoid lupus and lichen planus,18 and IFIH1 polymorphisms have been linked with an increased risk of developing systemic lupus erythematosus, psoriasis and chronic periodontitis.11 Furthermore, apparent loss-of-function IFIH1 variants confer protection against type 1 diabetes.19
Engagement of the 2CARD oligomers of IFIH1 with the adaptor protein mitochondrial antiviral signalling protein (MAVS) induces type I interferon production, which acts as an integral component of the antiviral innate immune response.20 Pathological activation of an antiviral cytokine response mediated by type I interferons can be associated with inflammatory disease.17 In keeping with this, a gain-of-function IFIH1 mutation in mice caused a lupus-like phenotype, which was partially rescued following back-crossing with type I interferon receptor-deficient mice.21
To signal to MAVS, the IFIH1 helicase and long dsRNA form a core filament, which promotes stochastic assembly of the 2CARD oligomers of IFIH1. Disassembly of the core filament is dependent on ATP hydrolysis, and is dsRNA length dependent. The regulation of filament stability is a proposed mechanism by which aberrant signalling to MAVS is normally suppressed in response to short (< ~0.5-kb) cellular dsRNA.20 The close proximity of the Ala489 residue to the ATP substrate is therefore of interest, particularly given the observed absence of ATP hydrolysis and increased stability of the filament compared with the wild-type following the addition of ATP (Fig. 5c and d).
We have previously reported an IFIH1 mutation at residue Arg337, an amino acid in direct contact with the adenine base of ATP that stabilizes the IFIH1–ATP interaction.2 Substitution of the arginine at residue 337 by glycine produced similar effects to those seen with the p.Ala489Thr mutant described here, resulting in an absence of ATP hydrolysis and increased stability of the filament. Thus, both mutations likely result in enhanced signalling to MAVS due to abnormal stability of the IFIH1–RNA interaction. In keeping with this, interferon signalling induced by long dsRNA was increased in the mutant compared with the control, and interferon induction also occurred following short dsRNA delivery, which was not the case in the wild-type context (Fig. 5e). Furthermore, baseline levels of interferon induction were elevated with the mutant construct, suggesting the possibility that in IFIH1-related disease an undefined endogenous ligand binds and activates the mutant protein.
In summary, this report further expands the spectrum of phenotypes associated with mutations in IFIH1, and indicates that features of SMS may not be limited to a single IFIH1 point mutation. The importance of assessing interferon expression and investigating the underlying genetic aetiology in such cases will be of particular relevance as anti-interferon therapies become routinely available.
What’s already known about this topic?
Dermatological involvement is a major feature of the type I interferonopathies, which include the allelic disorders familial chilblain lupus (FCL) and Aicardi-Goutières syndrome (AGS).
Cutaneous lesions are seen in 40% of cases of AGS, but have never been described in the context of mutations in IFIH1.
What does this study add?
IFIH1 mutations can be associated with predominant cutaneous features.
This study highlights the overlap between AGS, FCL and Singleton–Merten syndrome, and emphasizes the utility of searching for an interferon signature as a diagnostic tool in this context.
Acknowledgments
The authors would like to thank the Exome Aggregation Consortium (ExAC), Cambridge, MA, U.S.A. (http://exac.broadinstitute.org).
Funding sources: T.A.B. acknowledges funding from the National Institute for Health Research and a L’Oréal–UNESCO U.K. & Ireland Fellowship for Women in Science. Y.d.T.D. was supported by fellowships from the Novartis Foundation and Swiss National Science Foundation. S.H. was funded by a National Institutes of Health R01 grant (AI106912 and AI111784). Y.J.C. acknowledges the European Union’s Seventh Framework Programme (FP7/2007-2013) under grant agreement 241779, the European Research Council (GA 309449) and a state subsidy managed by the National Research Agency (France) under the ‘Investments for the Future’ programme bearing the reference ANR-10-IAHU-01.
Footnotes
Conflicts of interest
None declared.
References
- 1.Crow YJ, Chase DS, Lowenstein Schmidt J, et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A. 2015;167A:296–312. doi: 10.1002/ajmg.a.36887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rice GI, del Toro Duany Y, Jenkinson EM, et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet. 2014;46:503–9. doi: 10.1038/ng.2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oda H, Nakagawa K, Abe J, et al. Aicardi-Goutières syndrome is caused by IFIH1 mutations. Am J Hum Genet. 2014;95:121–5. doi: 10.1016/j.ajhg.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rice G, Newman WG, Dean J, et al. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutières syndrome. Am J Hum Genet. 2007;80:811–15. doi: 10.1086/513443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Günther C, Meurer M, Stein A, et al. Familial chilblain lupus – a monogenic form of cutaneous lupus erythematosus due to a heterozygous mutation in TREX1. Dermatology. 2009;219:162–6. doi: 10.1159/000222430. [DOI] [PubMed] [Google Scholar]
- 6.Rutsch F, MacDougall M, Lu C, et al. A specific IFIH1 gain-of-function mutation causes Singleton-Merten syndrome. Am J Hum Genet. 2015;96:275–82. doi: 10.1016/j.ajhg.2014.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ravenscroft JC, Suri M, Rice GI, et al. Autosomal dominant inheritance of a heterozygous mutation in SAMHD1 causing familial chilblain lupus. Am J Med Genet A. 2011;155A:235–7. doi: 10.1002/ajmg.a.33778. [DOI] [PubMed] [Google Scholar]
- 8.Oyama M, Shimizu H, Ohata Y, et al. Dyschromatosis symmetrica hereditaria (reticulate acropigmentation of Dohi): report of a Japanese family with the condition and a literature review of 185 cases. Br J Dermatol. 1999;140:491–6. doi: 10.1046/j.1365-2133.1999.02716.x. [DOI] [PubMed] [Google Scholar]
- 9.Rice GI, Kasher PR, Forte GM, et al. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat Genet. 2012;44:1243–8. doi: 10.1038/ng.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jang MA, Kim EK, Now H, et al. Mutations in DDX58, which encodes RIG-I, cause atypical Singleton-Merten syndrome. Am J Hum Genet. 2015;96:266–74. doi: 10.1016/j.ajhg.2014.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen G, Zhou D, Zhang Z, et al. Genetic variants in IFIH1 play opposite roles in the pathogenesis of psoriasis and chronic periodontitis. Int J Immunogenet. 2012;39:137–43. doi: 10.1111/j.1744-313X.2011.01068.x. [DOI] [PubMed] [Google Scholar]
- 12.Liu Y, Jesus AA, Marrero B, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. 2014;371:507–18. doi: 10.1056/NEJMoa1312625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou Q, Yang D, Ombrello AK, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med. 2014;370:911–20. doi: 10.1056/NEJMoa1307361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kolivras A, Aeby A, Crow YJ, et al. Cutaneous histopathological findings of Aicardi-Goutières syndrome, overlap with chilblain lupus. J Cutan Pathol. 2008;35:774–8. doi: 10.1111/j.1600-0560.2007.00900.x. [DOI] [PubMed] [Google Scholar]
- 15.Sigura K, Takeichi T, Kono M, et al. Severe chilblain lupus is associated with heterozygous missense mutations of catalytic amino acids or their adjacent mutations in the exonuclease domains of 3′-repair exonuclease 1. J Invest Dermatol. 2012;132:2855–7. doi: 10.1038/jid.2012.210. [DOI] [PubMed] [Google Scholar]
- 16.Arribas MP, Albares M, Soro P, Belinchon I. Red and purple papules on the dorsum of the fingers and toes in a woman. Int J Dermatol. 2013;52:1295–6. doi: 10.1111/ijd.12102. [DOI] [PubMed] [Google Scholar]
- 17.Crow YJ. Type I interferonopathies: Mendelian type I interferon up-regulation. Curr Opin Immunol. 2015;32C:7–12. doi: 10.1016/j.coi.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 18.Zahn S, Barchet W, Rehkamper C, et al. Enhanced skin expression of melanoma differentiation-associated gene 5 (MDA5) in dermatomyositis and related autoimmune diseases. J Am Acad Dermatol. 2011;64:988–9. doi: 10.1016/j.jaad.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 19.Nejentsev S, Walker N, Riches D, et al. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–9. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goubau D, Deddouche S, Reis E, Sousa C. Cytosolic sensing of viruses. Immunity. 2013;38:855–69. doi: 10.1016/j.immuni.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Funabiki M, Kato H, Miyachi Y, et al. Autoimmune disorders associated with gain of function of the intracellular sensor MDA5. Immunity. 2014;40:199–212. doi: 10.1016/j.immuni.2013.12.014. [DOI] [PubMed] [Google Scholar]