

Abstract
Genomic deletion of tumor suppressor genes (TSG) is a rite of passage for virtually all human cancers. The synthetic lethal paradigm has provided a framework for the development of molecular targeted therapeutics that are functionally linked to the loss of specific TSG functions. In the course of genomic events that delete TSGs, a large number of genes with no apparent direct role in tumor promotion also sustain deletion as a result of chromosomal proximity to the target TSG. In this perspective, we review the novel concept of “collateral lethality”, which has served to identify cancer-specific therapeutic vulnerabilities resulting from co-deletion of passenger genes neighboring TSG. The large number of collaterally deleted genes, playing diverse functions in cell homeostasis, offers a rich repertoire of pharmacologically targetable vulnerabilities presenting novel opportunities for the development of personalized anti-neoplastic therapies.
Synthetic lethality in Cancer Therapeutics
Genomic deletions are a key driver of virtually all cancers. Large scale genomic characterization efforts such as the Cancer Genome Atlas (TCGA) and International Cancer Genome Consortium (ICGC) have generated a detailed compendium of the landscape of recurrent genome alterations underlying carcinogenesis [1-4]. These efforts have identified recurrent activating events (point mutations, aberrant fusions, and amplifications of proto-oncogenes) as well as loss-of-function events (point mutations, genomic deletions, and aberrant fusions of tumor suppressor genes (TSGs)). A key focus and critical challenge in the field is the conversion of such data into effective cancer-specific therapies. Most molecular targeted cancer therapeutics have been directed at activated oncogenes or “gain of function” mutations [5-8]. Indeed, personalized molecular therapies targeting amplified, mutated or otherwise activated oncogenes have demonstrated meaningful clinical results [9-11]. By contrast, no therapy targeting loss-of-function or inactivating genetic events has yet been approved by the USA Food and Drug Administration (FDA).
Extensive attempts have been made to target loss-of-function mutations and deletions of TSG using a synthetic lethality approach. This term was coined in the early 20th century and it arose from genetic experiments with Drosophila species [12]. It is used to describe the interaction between two genes in which inactivation of either of them is compatible with life but co-inactivation in the same cell or organism leads to death. In the past decade this concept was applied to cancer research, stemming from the hypothesis that inactivating mutations would provide tumors with specific vulnerabilities that normal tissues do not have [13]. Synthetic lethal screens using small interfering/small hairpin (si/shRNA) and small molecules have been conducted for loss-of-function of major suppressor genes such as TP53 [13], RB [14], VHL [15], SMAD4 [16, 17] and others [15, 18]. These efforts have yielded intriguing preclinical findings which may inform clinical trials. One notable example of success is the finding that BRAC1/2 loss-of-function results in selective sensitization to poly ADP ribose polymerase (PARP) inhibitors (see below). To date, however, the impact of synthetic lethal approaches have been somewhat limited and this may be due to the fact that the interaction between the tumor suppressors and their partners was not “synthetic lethal” but rather “synthetic sick”, meaning that the co-occurrence of events leads to a growth impairment that is still compatible with life [19]. On the genetic level, ongoing efforts continue to search for true synthetic lethal gene partners, where the combined loss is incompatible with cellular viability, for loss of major cell-cycle regulatory TSGs such as TP53, PTEN, and CDKN2A. We speculate that the limited success of these searches may be attributed in part to the fact that these genes do not perform functions that are essential for cell survival (so called “housekeeping” functions).
The most robust and clinically advanced synthetic lethal therapy involves targeting tumors harboring BRCA1/2 loss-of-function mutations or deletions with inhibitors of PARP [20]. Loss of BRCA sensitizes cancer cells to PARP inhibitors because both of these genes play redundant roles in a specific DNA double strand break repair [21]. The strongly synthetic lethal relationship between PARP and BRCA is supported by genetic data: knockout of PARP1 in the context of mutant BRCA1 is lethal, while both knockouts are viable on their own [22]. It is possible that the comparative success of this example is due to the fact that loss of function of BRCA, unlike the above-mentioned tumor suppressor genes, compromises DNA repair, a major housekeeping function [23]. While most TSG are not critical for cellular housekeeping functions, genes playing such functions can be inactivated as passengers, if a TSG is subject to genomic deletion.
Genomic deletions and “passenger” genes
Genomic deletions are a common pathway by which TSGs are inactivated. Such deletions occur stochastically and clonally expand if they confer a biological advantage to the aspiring cancer cell. Whole arm, cytogenetically visible deletions causing heterozygous loss of hundreds of genes were first reported in the 1970s in the context of Knudson's two-hit hypothesis [24], whereby deletion of the chromosome arm containing a wild-type allele (with the other allele containing a loss-of-function mutation) of a TSG would result in complete inactivation. Such deletions cause “loss of heterozygosity” (LOH), whereby loci that are heterozygous in somatic tissues become hemizygous in cancer due to loss of one allele by genomic deletion. LOH-studies, especially in the context of familial cancers with a pre-existing mutation in a major TSG, led to the identification of key driver events such as loss of RB1 [25], WT1 [26], NF1 [27], APC [28] and PTEN [24, 29]. It is generally agreed that chromosome arm heterozygous deletions promote tumorigenesis by eliminating the remaining functional allele against the backdrop of a mutationally compromised TSG on the retained chromosome. Different tumor types exhibit highly recurrent and specific heterozygous deletions, which reflects that these genomic alterations are ‘signature’ events for the development of such tumor types [30, 31] [32]. Additionally, common heterozygous deletions often encompass multiple tumor suppressor genes, such as those found on chromosome 8p [36].
Homozygous deletions were first observed in 1990s when an area on chromosome 9 was found to be deleted in a subset of acute lymphoblastic leukemias and melanomas [33, 34]. Analysis of the minimal common deleted area led to the discovery of the cell cycle regulators (p16INK4A and p15INK4B) and the tumor suppressor (ARF) which are encoded by the CDKN2A/B locus [35]. Homozygous deletions occur less frequently and are more focal than heterozygous deletions, affecting between one and several dozen genes (Box 1). While heterozygous deletions are ubiquitous in human cancer, homozygous deletions are more restricted and their frequency varies between cancer types (Box 1). Within the same cancer type, tumors with high copy number alterations (including high load of homozygous deletions) also trend towards a worse prognosis [37].
Since the advent of genome-wide analysis, a large amount of effort has been dedicated towards identifying genes that constitute driver events that promote tumorigenesis as opposed to passenger alterations that simply neighbor the intended target or occur in “fragile sites” of the genome [38]. Contrary to drivers, passenger deletions were generally thought to be of little or no biological and therapeutic significance. However, a more in-depth analysis reveals that many passenger deleted genes play important roles in diverse metabolic and housekeeping functions, yet cell viability is maintained due to functional redundancy of related gene families. A particularly interesting case is the homozygous deletion of the 1p36 locus (Figure 1), which targets multiple tumor suppressor genes [39] but can also include ENO1 (glycolysis), NMNAT1 (NAD+ biosynthesis), and PGD (pentose phosphate shunt). Additionally, at the 10q locus, homozygous deletions targeting the well-known PTEN tumor suppressor can include PAPSS2 (sulfate metabolism), ATAD1 (mitochondrial protein folding), PANK1 (Coenzyme A biosynthesis) and several others. Metabolic pathways regulated by these deleted genes are critical for cell viability, yet such deletions are tolerated by cancer cells, likely because of the extensive genetic redundancy of the mammalian genome [40, 41].
Figure 1. Collateral Deletion of Metabolic Genes Neighboring a Major Tumor Suppressor Locus.
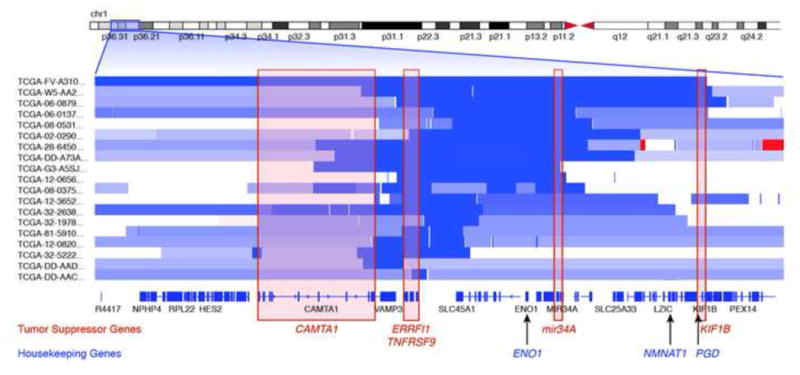
Genomic deletions inactivate tumor suppressor genes but may also collaterally delete chromosomal neighbors, if their deletion does not unduly compromise cell viability. Each row starting with the designator “TCGA-“ represents copy number data from a primary tumor from the TCGA in the 1p36 region, with segments in light blue representing regions of heterozygous deletion and dark blue, homozygous deletion. Homozygous deletions (dark blue) target tumor suppressor genes, such as mir34A [85], ERBB Receptor Feedback Inhibitor 1 (ERRFI1) [86], Tumor Necrosis Factor Receptor Superfamily, Member 9 (TNFRSF9) [46], Kinesin Family Member 1B (KIF1B) [87], Calmodulin Binding Transcription Activator 1 (CAMTA1) [88] and probably others [88] [89]. In addition, neighboring genes, such as the metabolic housekeeping enzymes Enolase 1 (ENO1), Nicotinamide Nucleotide Adenylyltransferase 1 (NMNAT1), 6-phosphogluconate dehydrogenase (PGD), can be co-deleted by virtue of chromosomal proximity to the aforementioned tumor suppressor genes.
Collateral lethality: Synthetic Lethal Targeting of Passenger Deleted Genes
The highly recurrent deletion of bystander genes encoding cell-essential functions that are members of multi-gene families prompted us to propose the concept of ‘collateral lethality’ which we define as a framework to discover new cancer-specific vulnerabilities offered by passenger deletion or inactivation of non-TSG genes. Below we summarize several different methodologies by which one may identify potential collateral lethality partners for given passenger deleted genes. These involve knowledge-based methods as well as bioinformatic data mining of big datasets.
Targeting redundant paralogues of gene families performing cell essential functions
Passenger deletion of critical metabolic enzymes appear to be tolerated by cancer cells due to the co-expression of partially redundant, closely related paralogues which serve to maintain these essential cellular metabolic reactions. In such cases, the tumor cell would be reliant on one paralogue whereas the normal tissue would still have a complement of two or more. If the remaining paralogue were specifically inhibited, the essential metabolic function would be compromised and should result in cancer cell lethality (Figure 2). The normal host cells should be less affected as inhibition of one of the two paralogues would still leave one member to carry out the essential function. Hints of this idea can be traced to a theoretical article by Alexander Kamb based on differential expression of homologues which are often synthetic lethal in yeast [42].
Figure 2. Pharmacological Vulnerability Exposed by Homozygous Deletions in Essential-Redundant Paralogues.
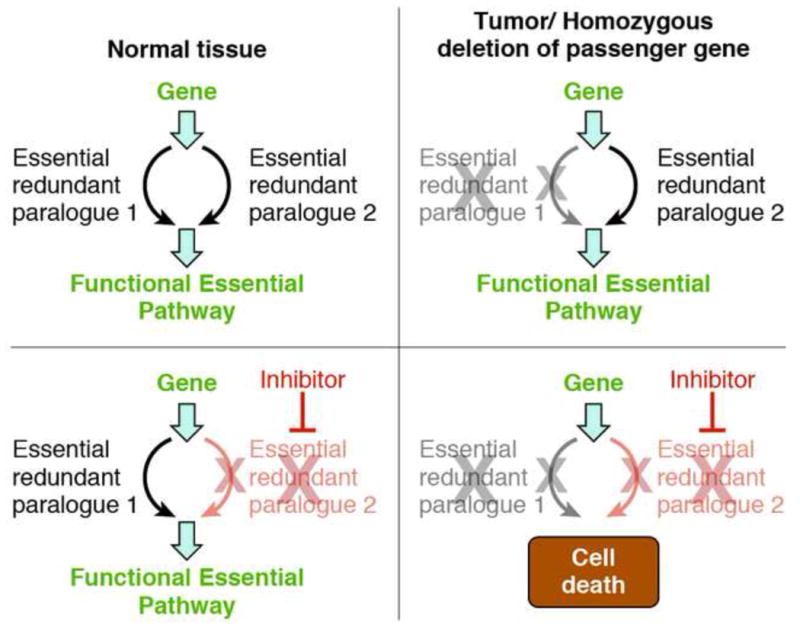
Homozygous deletions targeting tumor suppressor genes sometimes contain passenger genes that carry out a cell essential function but whose deletion is tolerated because of redundant action of a paralogue. In such cases, it may be possible to selectively target cancer cells by using small-molecule inhibitors targeting the non-deleted redundant paralogue. Proof-of-principle of this concept was demonstrated in glioma cells harboring deletion of Enolase 1 (ENO1), resulting in dramatic sensitization to ablation of Enolase 2 (ENO2) [50].
In a recent study, we surveyed the glioblastoma TCGA dataset [4] to identify examples of paralogous genes that constitute an essential cell function based on invertebrate genetic data [43-45], although we recognized that this approach is limited by differences in biology of yeast and mammalian cells. We prioritized validation of candidate deleted genes based on the strength of the invertebrate genetic data as well as the availability of genomically profiled human cell lines. As such, the glycolytic gene ENO1, which is homozygously deleted as part of the 1p36 tumor suppressor locus [46], was chosen for proof-of-concept. Enolase is a key enzyme for cellular bioenergetics, without which glycolysis cannot produce ATP. It is encoded by three redundant homologues with glioma cells only expressing ENO1 and ENO2, the latter gene is located on chromosome 12. We demonstrated that glioma cells with deletion of ENO1 are exceptionally sensitive to the ablation of ENO2, by both genetic (shRNA) and pharmacologic means (the small molecule Enolase inhibitor, phosphonoacetohydroxamate [47]). In contrast, ENO2 ablation is inconsequential to ENO1-intact glioma cells and Eno2 knockout mice have no described deleterious phenotypes [48]. Together, these data strongly suggest that ENO1-deleted tumors would be highly sensitive to an Enolase inhibitor, displaying a significant therapeutic index.
While homozygous deletion of ENO1 was initially identified in glioblastoma, TCGA data indicate that this also occurs in hepatocellular carcinoma and cholangiocarcinoma with a frequency of about 2.5% [49]. Given the central importance of glycolysis in cellular metabolism, we proposed that the same paradigm would hold in these other cancer types as well.
The ENO1/ENO2 collateral lethality example may represent the founding proof of concept of a more generalizable approach. Additional candidates identified through the same methodology include additional metabolic passenger deleted genes NMNAT1, PANK1 and ACO1 [50], which have redundant paralogues located elsewhere in the genome. For all these genes, model organism genetic data support the fact that paralogues are dispensable but total loss of activity is not compatible with cell viability.
Further data supporting this concept derives from four independent publications showing that loss-of-function mutations/deletions of the chromatin remodeling helicase SMARCA4 sensitizes lung cancer cells to ablation of its paralogue, SMARCA2 [51-54]. This relationship was identified as synthetic lethal both as a result of a broad genetic screen and using a knowledge-based approach [60,63]. Although conceptually similar to the ENO1/ENO2 collateral lethality pair, SMARCA4 is a true tumor suppressor and its inactivation is a driver rather than passenger event. That said, unlike most tumor suppressors, SMARCA2/4 exert a genuine cellular housekeeping function rather than merely restricting cellular growth.
A recent paper from Chris Sander's research team devised an algorithm to systematically identify essential gene families in which one member is homozygously deleted in a representative panel of major cancer types [55]. Analogous to our approach, the authors used a second filter consisting of genetic essentiality data from invertebrates and identified a possible 2706 targetable vulnerabilities. While we believe that this prediction may be too optimistic as it overestimates the number of homozygously deleted genes and does not distinguish between cell-essential versus organismal-essential genes, it nevertheless strongly suggests that many collateral deleted genes may lend themselves to selective homologue-based-targeting. The development of CRISPR may allow a refined definition of what constitutes a cell-essential process in mammalian systems [56-58], and as such, allow more accurate prediction of what deleted genes can be employed for selective sensitization based on homologue targeting.
Targeting passenger deleted genes based on non-homologue based genetic/biochemical redundancy
While targeting paralogues of essential-redundant genes deleted in cancer is a robust method for exploiting vulnerabilities generated by passenger deletions, these criteria are narrow and the number of genes meeting them is likely to be limited. However, many passenger deleted genes could cause biochemical alterations that render otherwise non-essential biochemical pathways essential even if they are not members of an essential/redundant gene pair (Figure 3). While there is presently no standard way of establishing such relationships, yeast genetic data, as well as biochemical pathway knowledge, can be used to infer them. Consider the deletion of 6-phosphoglucinate dehydrogenase (PGD) on the 1p36 locus as an illustrative example (Figure 1). PGD is a key enzyme of the oxidative pentose-phosphate shunt, but is not a cell-essential gene per se because NADP+ can be produced from other sources and ribose-5-phosphate, which is essential for nucleotide synthesis, can still be produced from the non-oxidative pentose phosphate shunt (Figure 4). Indeed, yeast data indicate that while PGD enzymatic activity is dispensable, the loss of PGD combined with any ablation of the non-oxidative pentose phosphate shunt (e.g. transketolase, transaldolase, ribulose epimerase or ribulose isomerase) is not tolerated [59]. As such, it is highly likely that tumors with 1p36 deletion covering PGD will be extremely sensitive to inhibitors of enzymes of the non-oxidative pentose phosphate shunt.
Figure 3. Redundant Biochemical Pathways-based collateral lethality.
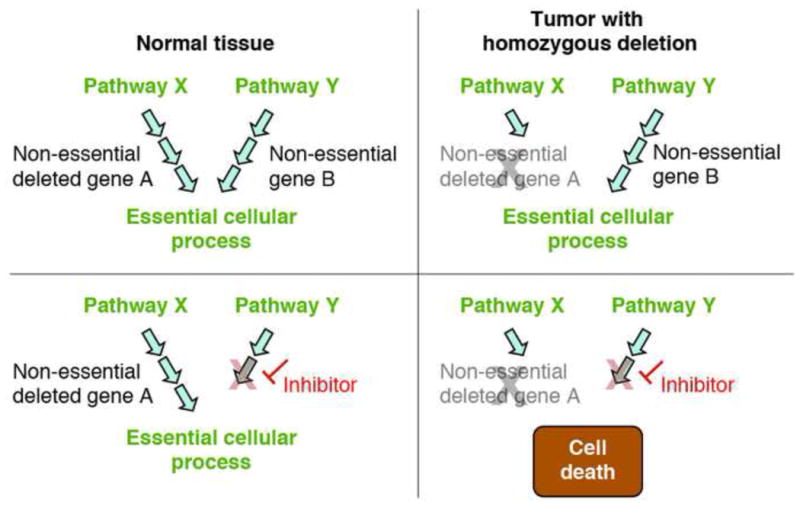
Two (or greater) biochemical pathways can lead to the same essential cellular process. Passenger homozygous deletions can affect one of those pathways while leaving the other intact, thus having no detrimental effect on cancer cell viability but causing the remaining pathway to become essential. Using selective inhibitors against key enzymes of the non-deleted pathway could lead to tumor specific cell death while leaving normal tissues unharmed. While this concept is yet to be proved experimentally in cancer, it is strongly support by model organism data [59]. An example is the production of ribose-5-phosphate as a backbone for nucleic acid synthesis, which can be obtained from either the oxidative or non-oxidative pathways of the pentose phosphate shunt (see Figure 4). 1p36 deleted tumors often harbor deletions in 6-phosphogluconate dehydrogenase (PGD), which is part of the oxidative pathway. These cancers are expected to be highly sensitive to inhibition of the non-oxidative pathway.
Figure 4. Deletion of PGD renders cells reliant on the non-oxidative Pentose Phosphate Shunt for Nucleotide Biosynthesis.
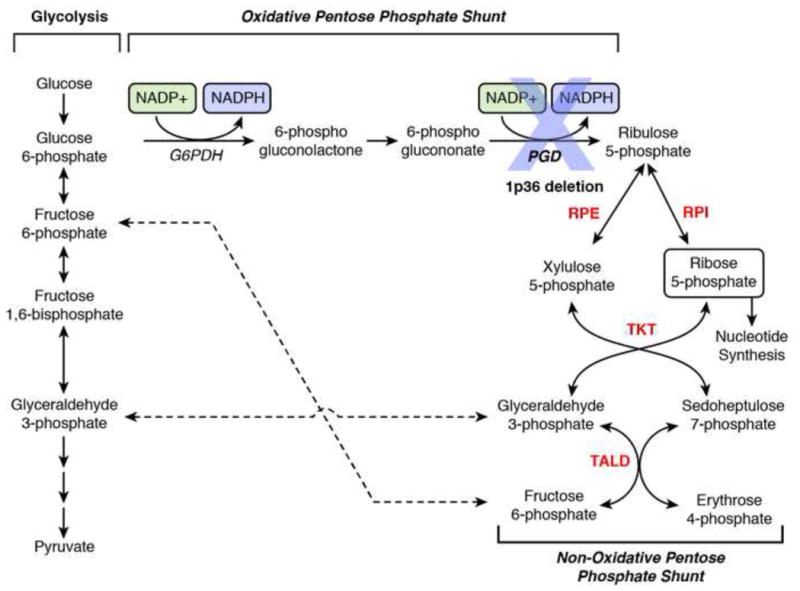
Nucleotide biosynthesis for DNA and RNA requires Ribose-5-phosphate, which can be derived from either Glucose-6-phosphate through the oxidative arm of the pentose phosphate shunt or the glycolytic intermediates, fructose-6-phosphate and glyceraldehyde-3-phosphate, through the non-oxidative arm. Genetic data in yeast indicate that both the non-oxidative and oxidative arms are on their own, dispensable for cell viability. However, yeast do not tolerate simultaneous inactivation of both arms of the pathway, such as the combined loss of 6-phosphogluconate dehydrogenase (PGD) with either transketolase (TKT), transaldolase (TALD) or Ribulose Isomerase/Epimerase (RPI/ RPE), shown in Red. Given that no alternative pathways of Ribose-5-phosphate synthesis are known, it is very likely that cancer cells with 1p36 deletion encompassing PGD (Figure 1) would be entirely dependent on the non-oxidative arm of the pentose phosphate shunt, and as such, highly sensitive to inhibition of either TKT, TALD, RPI or RPE.
Biochemical vulnerabilities can also manifest in decreased resistance to specific toxins or environmental conditions. As an example, the antioxidant gene Cu, Zn Superoxide dismutase (SOD1) is homozygously deleted in lung squamous cell carcinoma. It is well established that SOD1 null cells in yeast, bacteria and mice are up to 1000-times more sensitive to redox cycling agents that produce superoxide, such as paraquat and menadione [60]; it is therefore likely that SOD1 deleted lung tumors could be selectively vulnerable to such superoxide generating agents.
A large body of work has been dedicated to exploiting biochemical vulnerabilities exposed by deletion of methylthioadenosine phosphorylase (MTAP). MTAP is immediately adjacent to CDKN2A and is one of the most frequently homozygously deleted genes in human cancer. Whether MTAP is a genuine tumor suppressor gene remains an area of active investigation [61, 62]. While the full biochemical consequences of MTAP deletion remain to be elucidated, there is general agreement that it plays a role in salvage of methionine and adenosine. Attempts have been made to exploit these vulnerabilities, in particular using inhibitors of purine biosynthesis [63-65]. While this area has great potential, these attempts have yet to bear fruit, possibly due to the high extracellular concentrations of MTAP produced by stromal cells which may serve as a reservoir [66].
Attempts have been made to use biocomputational modeling approaches to infer relationships of biochemical/genetic redundancy between tumor suppressor genes and potential drug targets [67]. With the integration of the plentiful genetic and biochemical data from yeast [68], such approaches are becoming increasingly sophisticated. An example of a successful implementation of this approach concerns loss-of-function mutation in fumarate hydratase (FH) and haeme oxygenase [69]. Inactivating mutations of FH cause hereditary leiomyomatosis and renal-cell cancer. FH is a critical enzyme in the tricarboxylic acid (TCA) cycle and inactivating mutations cause severe metabolic disruptions, including massive accumulation of fumarate. How to exploit this therapeutically is non-obvious and a metabolic modeling approach was employed to show that that haeme oxygenase is a lynchpin for metabolic bypass of inactivated FH. As such, it was demonstrated that shRNA ablation of haeme oxygenase is selectively toxic to FH mutant cells, and that haeme oxygase inhibitors are a potential targeted therapeutic for cancers driven by such mutations. At the same time, it is notable that such prediction studies have almost exclusively focused on driver events (loss-of-function of tumor suppressor genes) rather than passenger deleted genes, some of which are likely to be much better candidates for targeting biochemical redundancy, such as PGD deletion mentioned above. Indeed, such approaches may be quite fruitful when applied to highly conserved passenger deleted genes whose exact function remains poorly understood. Examples of this type include ATAD1 near PTEN as well as MTAP itself. Yet, experimental validation will still be required. A possible source of information for further triangulation constitutes broad spectrum genomic or pharmacologic screens (Box 2)
Collateral lethality targeting of heterozygously deleted genes
Drug-induced haplolethality
Heterozygous deletions occur much more frequently in cancers than homozygous deletions. For instance, while the 1p36 homozygous deletion is found in around 2.5% of glioblastomas cases (typically cover an average of ten genes), the heterozygous deletion is found in 25% of glioblastomas and around 15% of all cancers (covering hundreds of genes). Additionally, these deletions affect very large chromosomal segments, occasionally entire chromosome arms and can occur so frequently that they are quasi-diagnostic of a specific tumor. Such examples include the 10q arm-deletion in glioblastomas [90], 1p and 19q double deletion in oligodendrogliomas [92], 5q deletion in myelodysplastic syndrome [106], 3p deletion in renal clear cell carcinoma [107] and whole chromosome 3 loss in uveal melanomas [93]. Chromosome arms can harbor more than one thousand genes, many of which serve essential housekeeping functions. As such, cancers experiencing heterozygous deletions are likely to be excellent candidates for collateral lethality-based strategies, provided a strategy can be devised to exploit heterozygous rather than homozygous deletions.
In the yeast S. cerevisiae, drug screens on heterozygous strain libraries have repeatedly shown that deletion of an essential enzyme dramatically sensitizes the strain to drugs targeting it, in a phenomenon termed “drug-induced haploinsufficiency” or “drug-induced haplolethality” [70]. A strain with a pre-existing 50% enzyme deficiency requires considerably lower doses of a drug to inhibit the target enzyme below toxic threshold (Figure 5a). Such haploinsufficiency screens have been widely used to identify novel drug-target interactions, such as pathways targeted by anti-cancer drugs [70], anti-anginal medications [71] and antimicrobials [72].
Figure 5. Heterozygous passenger deletions as targets for collateral lethality.
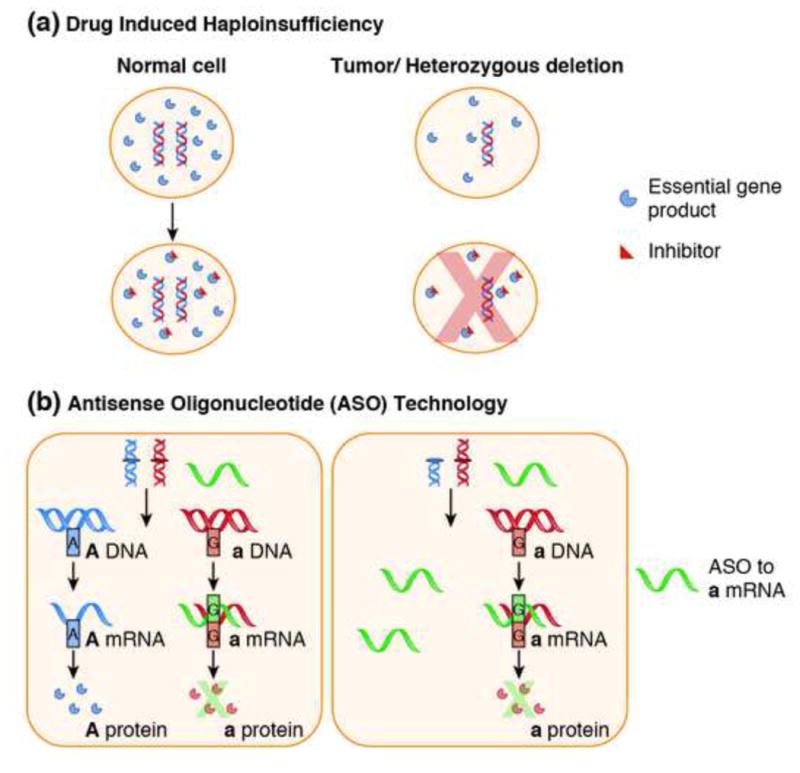
Panel A. “Drug-induced haploinsufficency” refers to sensitization that a heterozygous deletion causes to a specific inhibitor of the deleted gene, provided this gene exerts an essential function, its inhibition exhibits threshold toxicity and its expression is proportional to genomic copy number. As such a 50% deficiency means much less inhibitor is required to reach toxic threshold and consequently, cells harboring such deletions are sensitized to such an inhibitor. Drug-induced haploinsufficiency has been studied for decades in the context of model organisms but its application to cancer therapy is just beginning to be realized. Panel B: Genetic variation occurs in about 1 in 300 nucleotides in the human genome [75]. This variation is present in different alleles of the same gene, causing alterations that have neither significant phenotypic nor functional effects. Antisense oligonucleotide (ASO) technology can be used to target essential genes that are heterozygously deleted in cancers. These oligonucleotides are able to target polymorphisms that only differ by as little as one pair from each other, thereby inactivating one allele while leaving the other intact. In the figure above, an essential gene is encoded by alleles A and a. Tumor cells contain a heterozygous deletion in allele A. By introducing an antisense-oligonucleotide (ASO) directed against allele a (green), it is possible to cause selective death in cancer cells while leaving normal tissues intact.
Prior work experimentally supports this idea. ENO1-heterozygously deleted cell lines exhibit a ∼8 fold increase in sensitivity to a universal enolase inhibitor as compared to ENO1-WT lines [50]. This sensitivity could be dose-dependently reversed by overexpression of ENO1 or ENO2. Similarly, a recent study revealed a vulnerability specific to cancer cells with heterozygous deletions in 17p13 [73]. The authors noticed that the gene POLR2A, which encodes for the largest subunit of the RNA polymerase II complex, is a close chromosomal neighbor and is often co-deleted with TP53. This enzyme is inhibited by the toxin α-amanitin and POLR2A heterozygously deleted cancer cells showed approximately 8-fold increased sensitivity as compared to genomically intact counterparts. Although α-amanitin is too toxic to be used systemically, α-amanitin-based antibody-drug conjugates were at least 10-times more effective against POLR2A heterozygously deleted versus genomically intact xenografted tumors [73].
A screening tool using RNAi has also unveiled genes for which suppression causes toxicity only in cells harboring heterozygous deletions. These hits were termed CYCLOPS (Copy number alterations Yielding Cancer Liabilities Owing to Partial losS), and they are enriched in genes carrying out essential functions that are part of spliceosome, ribosome and proteasome complexes [74]. The gene PSMC2, a key member of the 19S proteasome, was chosen for validation in this case. Cancer cells with heterozygous deletions were found to be highly sensitive to shRNA-mediated inhibition of PSMC2 as opposed to wild-type controls. This held true in murine xenograft models of ovarian cancer as well [74]. Drawing on the ubiquity of drug-induced haplolethality relations in the S. cerevisiae literature and the high frequency of heterozygous deletions in human cancer, we speculate that this approach is likely to be widely applicable.
Allele-specific targeting
Genomic variation between different alleles of the same gene can be another way to exploit specific vulnerabilities provided by heterozygous deletions via an approach called “variagenic targeting” [75]. This involves selectively targeting tumors with loss of heterozygosity by allele-specific, oligonucleotide based knock-down of essential genes that are heterozygous in somatic tissues but become hemizygous by virtue of deletion in cancer (Figure 5b). We term this approach SNiPER (for Single Nucleotide Polymorphic Experimental Remedy).
There is one SNP every 300 bases in the human genome [75], and as such, heterozygosity is present for a large number of loci, including in genes that carry out essential cellular functions. Tumors that through deletions become hemizygous in such genes are left vulnerable to inhibition of the remaining allele, if it were possible to devise a way to selectively target one allele over the other. Initial proof of principle of this concept was done by targeting specific variants of the replication protein RPA70 [75] using anti-sense oligonucleotides. While selective inhibition of cell proliferation was seen in vitro, in vivo xenograft data was not presented. Despite its highly innovative concept, the paper did not generate extensive follow up. It is likely that with anti-sense technology of the time (1999), the SNiPER concept would not have been technically achievable in practice. We believe that a number of advances in both oligonucleotide chemistry and drug delivery have come together that invite a second look.
The chemistry of antisense oligonucleotides has dramatically improved since the publication of the initial paper [76]. There are now FDA-approved oligonucleotides to treat familial hypercholesterolemia [77] and promising late stage clinical results for many other conditions, including cancer [78-80]. Novel constructs are able to generate up to 100-fold discrimination based on a single nucleotides mismatch [81]. Recent developments in liposomal drug delivery systems may allow for higher plasma oligonucleotide concentration, better delivery to target tissues and less degradation by nucleases [82]. Additionally, a much more complete portrait of genomic deletions in cancer is now available and the frequency and incidence of polymorphisms in essential genes is known with great detail [83]. Finally, heterozygous deletions can be cancer initiating events, thus clonally distributed in the tumor. Because heterozygous deletions usually remove extended portions of the genome, it is likely that it will be possible to use combination therapies. All these considerations make allele-specific targeting of polymorphic, heterozygously deleted essential genes a very promising therapeutic approach.
Tumor heterogeneity and the effectiveness of collateral lethality targeting
As with all molecular targeted therapies, the issue of tumor heterogeneity is a critical complication. Thus, for collateral lethality to be 100% effective, all tumor cells must carry the genetic event being targeted, otherwise a non-deleted resistance clone will likely grow out. Therefore it is likely that multiple modalities targeting different genetic events will ultimately have to be combined to achieve true tumor elimination, in a manner reminiscent to the cocktail therapy for HIV. That said, in specific instances, targeting genomic deletions may yield a very strong response even as a monotherapy. For example, tumor cells with deletions are in general more likely to survive as they are more malignant than other cell populations. Indeed, there are many examples where specific deletions are associated with either metastasis or poor prognosis [103, 104]. Metastases may be particularly attractive to target because, having experienced a considerable genetic bottleneck, they may show more homogenous distribution of highly malignant deletions. However, under ideal conditions, the event being targeted should be as close as possible to the cancer-initiating genetic event, such that it is present in all subsequent tumor cells (the deletion is “clonal” rather than “sub-clonal”).
Examples where a specific deletion is clearly the tumor-initiating event include: deletion of SMARCB1 in rhabdoid brain tumors [105], 5q in myelodysplastic syndrome [106], and 3p in renal clear cell carcinoma [107]. It is likely that the chromosome arm LOH-events in familial cancers caused by point mutations in major tumor suppressor genes such as RB1 (13q) and BRCA1 (17q) constitute tumor initiating events, whereby tumor initiation occurs when a heterozygous germ-line point mutation is turned into a full loss-of-function by heterozygous deletion of the other intact allele [23, 108]. The 1p/19q deletion in oligodendrogliomas likely occurs very early during tumor development [109] which also looks to be the case for the 10q deletion in glioblastoma [110]. Beyond this, the phylogeny of deletions in the various cancers remains unclear. As more tumors have whole genome sequencing performed at a deeper coverage, these questions will yield more definitive answers. Mapping of break-points will allow easy integration with tumor mutation phylogeny as well as estimation of the exact percent of tumor cells carrying specific deletions.
Concluding Remarks
Molecular targeted therapy aims to exploit genetic differences between tumor and normal tissues with the goal of generating more specific and effective treatment of cancer. These therapies have largely focused on mutant-activated or amplified genes, rather than loss-of-function genetic events. Genomic deletions represent a clear genetic difference between tumor and normal tissue. Furthermore, tumors that are driven by copy number alterations (deletions) tend to have worst prognosis and remain the most difficult to treat [37, 84]. Yet to date, comparatively little effort has been undertaken to target genomic deletions for personalized therapy. Identifying robust synthetic lethal partners for deleted major tumor suppressor genes has proved challenging.
In this perspective, we have laid out several strategies through which genomic deletions can be harnessed for molecular targeted therapy in cancer, by expanding the focus on collateral or passenger deleted genes. Recent advances in cancer genomics, cancer metabolism, bioinformatics as well as genome editing tools provide an opportunity to systematically discover selective vulnerabilities brought about by passenger deletions. While we have provided illustrative examples largely focused on metabolic genes, it is important to emphasize that passenger deleted genes have a wider range in function (see Outstanding Questions). As such, they are likely to shape diverse aspects of tumor biology, allowing for selective targeting using diverse modalities under the framework of collateral lethality.
Trends box.
Genomic deletion of tumor suppressor genes is a key driver of tumorigenesis which frequently also results in the collateral or passenger deletion of chromosomal neighbors not playing an active role in tumor progression.
Such “passenger” deleted genes play diverse cellular housekeeping roles but their deletion is tolerated because of genetic redundancy.
Collateral Lethality is a novel molecular targeted therapeutic strategy, using passenger deleted genes as points of selective vulnerability.
Several recent reports have presented proof-of-principal experiments demonstrating that both homozygous passenger and heterozygous passenger deletions may serve as points of cancer-selective vulnerability.
Given the large number of passenger deleted genes in the cancer genome, Collateral Lethality may be a widely applicable therapeutic strategy.
Box 1. Regions of Recurrent Homozygous Deletions in Human Cancers.
Genomic studies have identified thousands of homozygously deleted genes in the cancer genome, yet only certain loci are targeted with high frequency. The most frequently homozygously deleted locus is at the 9p21 region with the minimal common region centered on the CDKN2A/B locus. CDKN2A/B homozygous deletions can be found in most human cancers, with some cancer types showing frequencies as high as 40% of all cases, such as GBM [90]. The frequent deletion of this locus likely derives from the fact that three critical tumor suppressor genes are located there (p16INK4A and p15INK4B, ARF, [91]). The 9p21 deletions vary in size and can include up to 25 genes as passengers. This includes MTAP, interferon clusters, ACO1 and many others. The second most recurrently deleted locus in human cancer is 10q23, with the minimal common region centered on the PTEN tumor suppressor. PTEN/10q23 deletions can be found in diverse human cancers, including GBM [90] and can include up to 30 passenger genes. Other examples of recurrent homozygous loci include 18q21 (SMAD4) and 1p36 (Several TSG).
Box 2. Deducing Collateral Lethality Relationships from Genome Screening Data.
Efforts are underway to perform whole genome loss-of-function analyses in diverse cancer cell lines using shRNA [95] and CRISPR [58, 96]. Since cancer cell lines differ in genetic makeup, loss of-function of specific genes exhibiting cell-line specific deleterious effects could be a useful source for identifying drug targets exposed by specific passenger deletions. Although difficulties with shRNA screens have been pointed out [95, 97], these issues are being addressed. shRNA dataset such as the ACHILES project are likely to be a very useful tool [98]. In addition, CRISPR whole genome loss of function screens have been initiated [96, 99] and are likely to prove exceptionally useful for identifying genetic/biochemical redundancies with passenger deleted genes, once the number of different cell lines reaches a critical threshold. The only limiting factor in such approaches is that established cancer cell lines have fairly homogenous genetic profiles, and that genetic events that are deleterious in cell culture conditions will not be represented. Many genetic events known to occur in primary tumors are either rarely or never found in established cell lines. An illustrative example would be mutation of IDH1/2 which occur in >80% of grade II-III gliomas, but have not been reported in any established glioma cell lines [100]. It is likely that large scale homozygous deletions, such as those stretching from PTEN to PANK1 and eliminating >20 genes are not tolerated in cell culture conditions. Thus, despite TCGA data showing PANK1 deletion in Ovarian, Prostate, Glioblastoma and other cancers, no PANK1 deletions are present in the ∼1000 cell lines of the CCLE [101] or ∼600 of the Sanger collection [102].
Outstanding Questions.
Can passenger deletions of non-metabolic genes (such as interferon clusters, chemotherapy resistance genes) confer specific vulnerabilities?
Is combination therapy using a collateral lethality approach more effective than targeting single genes?
How likely is development of resistance to collateral lethality-based therapies in primary patient tumors, which are more heterogeneous than xenografted murine models?
Acknowledgments
We thank all members of the DePinho, Draetta and Chin labs for critical input and discussions. This work was funded by NIH-NCI grants 7P01CA095616-10 (R.A.D), CPRIT RP140612 (R.A.D), NIH CDP SPORE P50CA127001-07 (F.L.M.). We apologize to those authors whose work could not be included because of space limitations.
Glossary box
- Homozygous deletion
A genomic deletion resulting in inactivation of both alleles of a gene or chromosomal region in a diploid genome.
- Heterozygous deletion
A genomic deletion resulting in inactivation of one allele of a gene or chromosomal region in a diploid genome.
- Hemizygous
A gene or chromosome in a diploid genome with only one copy
- Tumor Suppressor Gene
A gene which functions to limit cell proliferation and whose inactivation promotes tumor progression.
- Synthetic Lethality
The lethal phenotype of combined loss-of-function of two or more genes, where loss of each gene alone does not cause lethality. In the field of cancer research, this is currently understood as combination of a therapeutic modality with a loss-of-function mutation in a tumor suppressor gene.
- Collateral Lethality
Synthetic lethality relationship involving passenger or collateral deleted genes rather than driver TSG.
- Passenger alterations
Genomic alterations (deletions, mutations, amplifications) in genes that do not promote tumor progression. These occur as a result of genomic instability, mutator phenotypes or due to chromosomal proximity to genes that constitute important “driver” events.
- Driver alterations
Genomic alterations in genes that directly contribute to tumor progression
- Loss of heterozygosity
Occurs when a cell which is heterozygous (polymorphic) at a specific locus becomes hemizygous due to loss of one of the alleles by genomic deletion
- The Cancer Genome Atlas
A comprehensive, large-scale genetic, epigenetic and transcriptomic characterization effort aimed at providing an “atlas” of alterations in cancer. This project began in 2005 with the analysis of lung, ovarian cancer and glioblastoma. It has since then expanded to over 20 different tumor types; data is publically available for genome sequence, copy number alterations, gene expression and others [49].
- Clustered, regularly interspaced, short palindromic repeat (CRISPR) technology
Genome editing technique based on a system of acquired immunity in bacteria, whereby viral DNA integrated into the host genome can be cleaved. Its applications include rapid development of transgenic animal models and genomic loss of function screens.
- Antisense oligonucleoties
Short (∼15-25 nucleotides), single-stranded modified-DNA molecules that are complementary to unique mRNA sequences. Once introduced into a cell, they bind to the target mRNA and cause its degradation by cellular nucleases, thereby preventing translation of that mRNA into protein.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Colen RR, et al. Imaging genomic mapping of an invasive MRI phenotype predicts patient outcome and metabolic dysfunction: a TCGA glioma phenotype research group project. BMC Med Genomics. 2014;7:30. doi: 10.1186/1755-8794-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brennan CW, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462–77. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Noushmehr H, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17(5):510–22. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arellano-Rodrigo E, Alvarez-Larran A. JAK inhibition in myelofibrosis. N Engl J Med. 2010;363(25):2464. doi: 10.1056/NEJMc1011635. author reply 2464-5; discussion 2465. [DOI] [PubMed] [Google Scholar]
- 6.Gerber DE, Minna JD. ALK inhibition for non-small cell lung cancer: from discovery to therapy in record time. Cancer Cell. 2010;18(6):548–51. doi: 10.1016/j.ccr.2010.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim T, Kim J, Lee MG. Inhibition of mutated BRAF in melanoma. N Engl J Med. 2010;363(23):2261. doi: 10.1056/NEJMc1010755. author reply 2261-2. [DOI] [PubMed] [Google Scholar]
- 8.Soulieres D, et al. Multicenter phase II study of erlotinib, an oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with recurrent or metastatic squamous cell cancer of the head and neck. J Clin Oncol. 2004;22(1):77–85. doi: 10.1200/JCO.2004.06.075. [DOI] [PubMed] [Google Scholar]
- 9.Shaw AT, Solomon BJ. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. 2015;372(7):683–4. doi: 10.1056/NEJMc1415359. [DOI] [PubMed] [Google Scholar]
- 10.Krause SW, Holler E. Imatinib in chronic myeloid leukemia. N Engl J Med. 2007;356(17):1780. doi: 10.1056/NEJMc063767. author reply 1780. [DOI] [PubMed] [Google Scholar]
- 11.Piccart-Gebhart MJ, et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med. 2005;353(16):1659–72. doi: 10.1056/NEJMoa052306. [DOI] [PubMed] [Google Scholar]
- 12.Nijman SM. Synthetic lethality: general principles, utility and detection using genetic screens in human cells. FEBS Lett. 2011;585(1):1–6. doi: 10.1016/j.febslet.2010.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krastev DB, et al. A systematic RNAi synthetic interaction screen reveals a link between p53 and snoRNP assembly. Nat Cell Biol. 2011;13(7):809–18. doi: 10.1038/ncb2264. [DOI] [PubMed] [Google Scholar]
- 14.Gordon GM, Du W. Targeting Rb inactivation in cancers by synthetic lethality. Am J Cancer Res. 2011;1(6):773–86. [PMC free article] [PubMed] [Google Scholar]
- 15.Turcotte S, et al. A molecule targeting VHL-deficient renal cell carcinoma that induces autophagy. Cancer Cell. 2008;14(1):90–102. doi: 10.1016/j.ccr.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lehmann BD, Pietenpol JA. Targeting mutant p53 in human tumors. J Clin Oncol. 2012;30(29):3648–50. doi: 10.1200/JCO.2012.44.0412. [DOI] [PubMed] [Google Scholar]
- 17.McCubrey JA, et al. Targeting survival cascades induced by activation of Ras/Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways for effective leukemia therapy. Leukemia. 2008;22(4):708–22. doi: 10.1038/leu.2008.27. [DOI] [PubMed] [Google Scholar]
- 18.Shackelford DB, et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell. 2013;23(2):143–58. doi: 10.1016/j.ccr.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaelin WG., Jr The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5(9):689–98. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 20.Fong PC, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–34. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 21.Dedes KJ, et al. Synthetic lethality of PARP inhibition in cancers lacking BRCA1 and BRCA2 mutations. Cell Cycle. 2011;10(8):1192–9. doi: 10.4161/cc.10.8.15273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X, et al. Haploinsufficiency of Parp1 accelerates Brca1-associated centrosome amplification, telomere shortening, genetic instability, apoptosis, and embryonic lethality. Cell Death Differ. 2007;14(5):924–31. doi: 10.1038/sj.cdd.4402105. [DOI] [PubMed] [Google Scholar]
- 23.Welcsh PL, King MC. BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum Mol Genet. 2001;10(7):705–13. doi: 10.1093/hmg/10.7.705. [DOI] [PubMed] [Google Scholar]
- 24.Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer. 2001;1(2):157–62. doi: 10.1038/35101031. [DOI] [PubMed] [Google Scholar]
- 25.Friend SH, et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986;323(6089):643–6. doi: 10.1038/323643a0. [DOI] [PubMed] [Google Scholar]
- 26.Little M, Wells C. A clinical overview of WT1 gene mutations. Hum Mutat. 1997;9(3):209–25. doi: 10.1002/(SICI)1098-1004(1997)9:3<209::AID-HUMU2>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 27.Cawthon RM, et al. A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell. 1990;62(1):193–201. doi: 10.1016/0092-8674(90)90253-b. [DOI] [PubMed] [Google Scholar]
- 28.Su LK, et al. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science. 1992;256(5057):668–70. doi: 10.1126/science.1350108. [DOI] [PubMed] [Google Scholar]
- 29.Frohling S, Dohner H. Chromosomal abnormalities in cancer. N Engl J Med. 2008;359(7):722–34. doi: 10.1056/NEJMra0803109. [DOI] [PubMed] [Google Scholar]
- 30.Weith A, et al. Neuroblastoma consensus deletion maps to 1p36.1-2. Genes Chromosomes Cancer. 1989;1(2):159–66. doi: 10.1002/gcc.2870010209. [DOI] [PubMed] [Google Scholar]
- 31.Steck PA, et al. Functional and molecular analyses of 10q deletions in human gliomas. Genes Chromosomes Cancer. 1999;24(2):135–43. doi: 10.1002/(sici)1098-2264(199902)24:2<135::aid-gcc6>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 32.Reifenberger J, et al. Molecular genetic analysis of oligodendroglial tumors shows preferential allelic deletions on 19q and 1p. Am J Pathol. 1994;145(5):1175–90. [PMC free article] [PubMed] [Google Scholar]
- 33.Fountain JW, et al. Homozygous deletions within human chromosome band 9p21 in melanoma. Proc Natl Acad Sci U S A. 1992;89(21):10557–61. doi: 10.1073/pnas.89.21.10557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Diaz MO, et al. Deletions of interferon genes in acute lymphoblastic leukemia. N Engl J Med. 1990;322(2):77–82. doi: 10.1056/NEJM199001113220202. [DOI] [PubMed] [Google Scholar]
- 35.Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366(6456):704–7. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- 36.Xue W, et al. A cluster of cooperating tumor-suppressor gene candidates in chromosomal deletions. Proc Natl Acad Sci U S A. 2012;109(21):8212–7. doi: 10.1073/pnas.1206062109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hieronymus H, et al. Copy number alteration burden predicts prostate cancer relapse. Proc Natl Acad Sci U S A. 2014;111(30):11139–44. doi: 10.1073/pnas.1411446111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bignell GR, et al. Signatures of mutation and selection in the cancer genome. Nature. 2010;463(7283):893–8. doi: 10.1038/nature08768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Henrich KO, et al. CAMTA1, a 1p36 tumor suppressor candidate, inhibits growth and activates differentiation programs in neuroblastoma cells. Cancer Res. 2011;71(8):3142–51. doi: 10.1158/0008-5472.CAN-10-3014. [DOI] [PubMed] [Google Scholar]
- 40.Vavouri T, Semple JI, Lehner B. Widespread conservation of genetic redundancy during a billion years of eukaryotic evolution. Trends Genet. 2008;24(10):485–8. doi: 10.1016/j.tig.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 41.Nowak MA, et al. Evolution of genetic redundancy. Nature. 1997;388(6638):167–71. doi: 10.1038/40618. [DOI] [PubMed] [Google Scholar]
- 42.Kamb A. Consequences of nonadaptive alterations in cancer. Mol Biol Cell. 2003;14(6):2201–5. doi: 10.1091/mbc.E02-11-0732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chan DA, et al. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci Transl Med. 2011;3(94):94ra70. doi: 10.1126/scitranslmed.3002394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weidle UH, Maisel D, Eick D. Synthetic lethality-based targets for discovery of new cancer therapeutics. Cancer Genomics Proteomics. 2011;8(4):159–71. [PubMed] [Google Scholar]
- 45.Deutscher D, et al. Multiple knockout analysis of genetic robustness in the yeast metabolic network. Nat Genet. 2006;38(9):993–8. doi: 10.1038/ng1856. [DOI] [PubMed] [Google Scholar]
- 46.Ichimura K, et al. 1p36 is a preferential target of chromosome 1 deletions in astrocytic tumours and homozygously deleted in a subset of glioblastomas. Oncogene. 2008;27(14):2097–108. doi: 10.1038/sj.onc.1210848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anderson VE, Weiss PM, Cleland WW. Reaction intermediate analogues for enolase. Biochemistry. 1984;23(12):2779–86. doi: 10.1021/bi00307a038. [DOI] [PubMed] [Google Scholar]
- 48.Kobayakawa K, et al. Innate versus learned odour processing in the mouse olfactory bulb. Nature. 2007;450(7169):503–8. doi: 10.1038/nature06281. [DOI] [PubMed] [Google Scholar]
- 49.Cerami E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Muller FL, et al. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature. 2012;488(7411):337–42. doi: 10.1038/nature11331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Orvis T, et al. BRG1/SMARCA4 inactivation promotes non-small cell lung cancer aggressiveness by altering chromatin organization. Cancer Res. 2014;74(22):6486–98. doi: 10.1158/0008-5472.CAN-14-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hoffman GR, et al. Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRG1-deficient cancers. Proc Natl Acad Sci U S A. 2014;111(8):3128–33. doi: 10.1073/pnas.1316793111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wilson BG, et al. Residual complexes containing SMARCA2 (BRM) underlie the oncogenic drive of SMARCA4 (BRG1) mutation. Mol Cell Biol. 2014;34(6):1136–44. doi: 10.1128/MCB.01372-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oike T, et al. A synthetic lethality-based strategy to treat cancers harboring a genetic deficiency in the chromatin remodeling factor BRG1. Cancer Res. 2013;73(17):5508–18. doi: 10.1158/0008-5472.CAN-12-4593. [DOI] [PubMed] [Google Scholar]
- 55.Aksoy BA, et al. Prediction of individualized therapeutic vulnerabilities in cancer from genomic profiles. Bioinformatics. 2014;30(14):2051–9. doi: 10.1093/bioinformatics/btu164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koike-Yusa H, et al. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol. 2014;32(3):267–73. doi: 10.1038/nbt.2800. [DOI] [PubMed] [Google Scholar]
- 57.Shalem O, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343(6166):84–7. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang T, et al. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343(6166):80–4. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Folger O, et al. Predicting selective drug targets in cancer through metabolic networks. Mol Syst Biol. 2011;7:501. doi: 10.1038/msb.2011.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Han ES, et al. The in vivo gene expression signature of oxidative stress. Physiol Genomics. 2008;34(1):112–26. doi: 10.1152/physiolgenomics.00239.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Curtis C, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–52. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kadariya Y, et al. Mice heterozygous for germ-line mutations in methylthioadenosine phosphorylase (MTAP) die prematurely of T-cell lymphoma. Cancer Res. 2009;69(14):5961–9. doi: 10.1158/0008-5472.CAN-09-0145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tang B, Testa JR, Kruger WD. Increasing the therapeutic index of 5-fluorouracil and 6-thioguanine by targeting loss of MTAP in tumor cells. Cancer Biol Ther. 2012;13(11):1082–90. doi: 10.4161/cbt.21115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bertino JR, et al. Targeting tumors that lack methylthioadenosine phosphorylase (MTAP) activity: current strategies. Cancer Biol Ther. 2011;11(7):627–32. doi: 10.4161/cbt.11.7.14948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li W, et al. Status of methylthioadenosine phosphorylase and its impact on cellular response to L-alanosine and methylmercaptopurine riboside in human soft tissue sarcoma cells. Oncol Res. 2004;14(7-8):373–9. doi: 10.3727/0965040041292332. [DOI] [PubMed] [Google Scholar]
- 66.Astrid RB, et al. Methylthioadenosine (MTA) Rescues Methylthioadenosine Phosphorylase (MTAP)-Deficient Tumors from Purine Synthesis Inhibition in Vivo via Non-Autonomous Adenine Supply. Journal of Cancer Therapy, 2011 2011 [Google Scholar]
- 67.Deshpande R, et al. A comparative genomic approach for identifying synthetic lethal interactions in human cancer. Cancer Res. 2013;73(20):6128–36. doi: 10.1158/0008-5472.CAN-12-3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li XJ, et al. Syn-lethality: an integrative knowledge base of synthetic lethality towards discovery of selective anticancer therapies. Biomed Res Int. 2014;2014:196034. doi: 10.1155/2014/196034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Frezza C, et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature. 2011;477(7363):225–8. doi: 10.1038/nature10363. [DOI] [PubMed] [Google Scholar]
- 70.Baetz K, et al. Yeast genome-wide drug-induced haploinsufficiency screen to determine drug mode of action. Proc Natl Acad Sci U S A. 2004;101(13):4525–30. doi: 10.1073/pnas.0307122101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lum PY, et al. Discovering modes of action for therapeutic compounds using a genome-wide screen of yeast heterozygotes. Cell. 2004;116(1):121–37. doi: 10.1016/s0092-8674(03)01035-3. [DOI] [PubMed] [Google Scholar]
- 72.Parsons AB, et al. Exploring the mode-of-action of bioactive compounds by chemical-genetic profiling in yeast. Cell. 2006;126(3):611–25. doi: 10.1016/j.cell.2006.06.040. [DOI] [PubMed] [Google Scholar]
- 73.Liu Y, et al. TP53 loss creates therapeutic vulnerability in colorectal cancer. Nature, 2015. doi: 10.1038/s41586-021-03664-3. [DOI] [PubMed] [Google Scholar]
- 74.Nijhawan D, et al. Cancer vulnerabilities unveiled by genomic loss. Cell. 2012;150(4):842–54. doi: 10.1016/j.cell.2012.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Basilion JP, et al. Selective killing of cancer cells based on loss of heterozygosity and normal variation in the human genome: a new paradigm for anticancer drug therapy. Mol Pharmacol. 1999;56(2):359–69. doi: 10.1124/mol.56.2.359. [DOI] [PubMed] [Google Scholar]
- 76.Yamamoto Y, et al. Generation 2.5 antisense oligonucleotides targeting the androgen receptor and its splice variants suppress enzalutamide-resistant prostate cancer cell growth. Clin Cancer Res. 2015;21(7):1675–87. doi: 10.1158/1078-0432.CCR-14-1108. [DOI] [PubMed] [Google Scholar]
- 77.Kole R, Krainer AR, Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov. 2012;11(2):125–40. doi: 10.1038/nrd3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Buller HR, et al. Factor XI antisense oligonucleotide for prevention of venous thrombosis. N Engl J Med. 2015;372(3):232–40. doi: 10.1056/NEJMoa1405760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Monteleone G, et al. Mongersen, an oral SMAD7 antisense oligonucleotide, and Crohn's disease. N Engl J Med. 2015;372(12):1104–13. doi: 10.1056/NEJMoa1407250. [DOI] [PubMed] [Google Scholar]
- 80.Dean NM, Bennett CF. Antisense oligonucleotide-based therapeutics for cancer. Oncogene. 2003;22(56):9087–96. doi: 10.1038/sj.onc.1207231. [DOI] [PubMed] [Google Scholar]
- 81.Carroll JB, et al. Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene / allele-specific silencing of mutant huntingtin. Mol Ther. 2011;19(12):2178–85. doi: 10.1038/mt.2011.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ozpolat B, Sood AK, Lopez-Berestein G. Liposomal siRNA nanocarriers for cancer therapy. Adv Drug Deliv Rev. 2014;66:110–6. doi: 10.1016/j.addr.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Genomes Project, C et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cancer Genome Atlas Research, N. et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497(7447):67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Genovese G, et al. microRNA regulatory network inference identifies miR-34a as a novel regulator of TGF-beta signaling in glioblastoma. Cancer Discov. 2012;2(8):736–49. doi: 10.1158/2159-8290.CD-12-0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ying H, et al. Mig-6 controls EGFR trafficking and suppresses gliomagenesis. Proc Natl Acad Sci U S A. 2010;107(15):6912–7. doi: 10.1073/pnas.0914930107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen ZX, et al. RNA helicase A is a downstream mediator of KIF1Bbeta tumor-suppressor function in neuroblastoma. Cancer Discov. 2014;4(4):434–51. doi: 10.1158/2159-8290.CD-13-0362. [DOI] [PubMed] [Google Scholar]
- 88.Henrich KO, Schwab M, Westermann F. 1p36 tumor suppression--a matter of dosage? Cancer Res. 2012;72(23):6079–88. doi: 10.1158/0008-5472.CAN-12-2230. [DOI] [PubMed] [Google Scholar]
- 89.Waerner T, et al. Human RERE is localized to nuclear promyelocytic leukemia oncogenic domains and enhances apoptosis. Cell Growth Differ. 2001;12(4):201–10. [PubMed] [Google Scholar]
- 90.Dunn GP, et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 2012;26(8):756–84. doi: 10.1101/gad.187922.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sharpless NE. INK4a/ARF: a multifunctional tumor suppressor locus. Mutat Res. 2005;576(1-2):22–38. doi: 10.1016/j.mrfmmm.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 92.Eckel-Passow JE, et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N Engl J Med, 2015. doi: 10.1056/NEJMoa1407279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ewens KG, et al. Genomic profile of 320 uveal melanoma cases: chromosome 8p-loss and metastatic outcome. Invest Ophthalmol Vis Sci. 2013;54(8):5721–9. doi: 10.1167/iovs.13-12195. [DOI] [PubMed] [Google Scholar]
- 94.Solimini NL, et al. Recurrent hemizygous deletions in cancers may optimize proliferative potential. Science. 2012;337(6090):104–9. doi: 10.1126/science.1219580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ngo VN, et al. A loss-of-function RNA interference screen for molecular targets in cancer. Nature. 2006;441(7089):106–10. doi: 10.1038/nature04687. [DOI] [PubMed] [Google Scholar]
- 96.An In Vivo Genome-Scale CRISPR Screen Defines Regulators of Metastasis. Cancer Discov. 2015 [Google Scholar]
- 97.Bernards R, Brummelkamp TR, Beijersbergen RL. shRNA libraries and their use in cancer genetics. Nat Methods. 2006;3(9):701–6. doi: 10.1038/nmeth921. [DOI] [PubMed] [Google Scholar]
- 98.Cheung HW, et al. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc Natl Acad Sci U S A. 2011;108(30):12372–7. doi: 10.1073/pnas.1109363108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chen S, et al. Genome-wide CRISPR Screen in a Mouse Model of Tumor Growth and Metastasis. Cell. 2015;160(6):1246–60. doi: 10.1016/j.cell.2015.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Piaskowski S, et al. Glioma cells showing IDH1 mutation cannot be propagated in standard cell culture conditions. Br J Cancer. 2011;104(6):968–70. doi: 10.1038/bjc.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Barretina J, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–7. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Forbes SA, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39(Database issue):D945–50. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.El Gammal AT, et al. Chromosome 8p deletions and 8q gains are associated with tumor progression and poor prognosis in prostate cancer. Clin Cancer Res. 2010;16(1):56–64. doi: 10.1158/1078-0432.CCR-09-1423. [DOI] [PubMed] [Google Scholar]
- 104.Yoshimoto M, et al. FISH analysis of 107 prostate cancers shows that PTEN genomic deletion is associated with poor clinical outcome. Br J Cancer. 2007;97(5):678–85. doi: 10.1038/sj.bjc.6603924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hasselblatt M, et al. High-resolution genomic analysis suggests the absence of recurrent genomic alterations other than SMARCB1 aberrations in atypical teratoid/rhabdoid tumors. Genes Chromosomes Cancer. 2013;52(2):185–90. doi: 10.1002/gcc.22018. [DOI] [PubMed] [Google Scholar]
- 106.Ebert BL. Molecular dissection of the 5q deletion in myelodysplastic syndrome. Semin Oncol. 2011;38(5):621–6. doi: 10.1053/j.seminoncol.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gerlinger M, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet. 2014;46(3):225–33. doi: 10.1038/ng.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Di Fiore R, et al. RB1 in cancer: different mechanisms of RB1 inactivation and alterations of pRb pathway in tumorigenesis. J Cell Physiol. 2013;228(8):1676–87. doi: 10.1002/jcp.24329. [DOI] [PubMed] [Google Scholar]
- 109.Yip S, Iafrate AJ, Louis DN. Molecular diagnostic testing in malignant gliomas: a practical update on predictive markers. J Neuropathol Exp Neurol. 2008;67(1):1–15. doi: 10.1097/nen.0b013e31815f65fb. [DOI] [PubMed] [Google Scholar]
- 110.Ozawa T, et al. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell. 2014;26(2):288–300. doi: 10.1016/j.ccr.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]