

Abstract
Hydrogen sulfide is emerging as a critically important molecule in medicine, yet there are few methods for the long-term delivery of molecules that degrade to release H2S. In this paper the first long-term release of a thiobenzamide that degrades to release H2S is described. A series of polymers were synthesized by the copolymerization of L-lactide and a lactide functionalized with 4-hydroxythiobenzamide. A new method to attach functional groups to a derivative of L-lactide is described based on the addition of a thiol to an α,β-unsaturated lactide using catalytic I2. This reaction proceeded under mild conditions and did not ring-open the lactone. The copolymers had molecular weights from 8 to 88 kg mol−1 with PDIs below 1.50. Two sets of microparticles were fabricated from a copolymer; the average diameters of the microparticles were 0.53 and 12 μm. The degradation of the smaller microparticles was investigated in buffered water to demonstrate the slow release of thiobenzamide over 4 weeks. Based on the ability to synthesize polymers with different loadings of thiobenzamide and that thiobenzamide is a known precursor to H2S, these particles provide a polymer-based method to deliver H2S over days to weeks.
Introduction
For decades it was believed that H2S had only negative health effects because it is a highly poisonous gas that can kill at lower concentrations than carbon monoxide (CO).1 Yet, recent work has shown that H2S also has many potentially important, beneficial roles in vivo and it is widely recognized as the third gasotransmitter known in medicine – the other two gasotransmitters being CO and NO.2-9 Gasotransmitters are important gases because they have the ability to move across biological membranes, control biological pathways and functions, have short half-lives in the body, and are synthesized by enzymes.10,11 The beneficial biological effects attributed to H2S are proposed to be due to at least three different mechanisms. First, H2S is a reducing agent and neutralizes reactive oxygen species (ROS) that cause cellular damage.12-16 Second, H2S can mediate the activation of adenosine triphosphate (ATP)-sensitive potassium channels (KATP).17-19 These channels play a pivotal role in regulating biological functions including heart activity, smooth muscle tone, insulin secretion, and neurotransmitter disease. 20,21 Third, H2S can regulate the concentration of NO in cardiac tissue and affect the signaling pathways of both gasotransmitters.22-24 H2S is emerging as a key molecule in medicine that has potential to affect and treat a wide variety of diseases.
One of the challenges in the field of medicinal applications of H2S is that its concentration must be accurately controlled at nM to μM concentrations.25-27 H2S is produced in vivo by at least four different enzymes, and its concentration in vivo is generally regarded as being less than 1 μM.4,26,28,29 Its concentration is low because H2S causes pulmonary edema and olfactory paralysis at even modest concentrations of tens of μM; at concentrations >100 μM death may occur.4,30 Yet, the body produces H2S by several enzymes and maintains its concentration at less than 1 μM31 and therapeutic doses of H2S at μM concentrations has been shown to have a positive effect in cells and tissues.8 One of the challenges in the field of H2S in medicine is the delivery of low, constant doses of H2S to targeted tissue.8 In prior work, NaSH was used to deliver H2S in vivo, but its use led to large, dangerous spikes in concentration of H2S.32 To provide a slow release of H2S in vivo, numerous small molecules were investigated that release H2S by degradation (Figure 1).32-40 These molecules have been used to affect the concentration of H2S over minutes to hours, but they are rapidly cleared from the body which necessitates repeated administration of these molecules every few hours. An important advance in this field would be the development of a safe, mild method to deliver a steady, low dose of H2S over hours to weeks at desired locations in the body.8 In this paper, we report the first synthesis of a polymer with a H2S-releasing prodrug that can slowly release the prodrug over days to weeks.
Fig. 1.
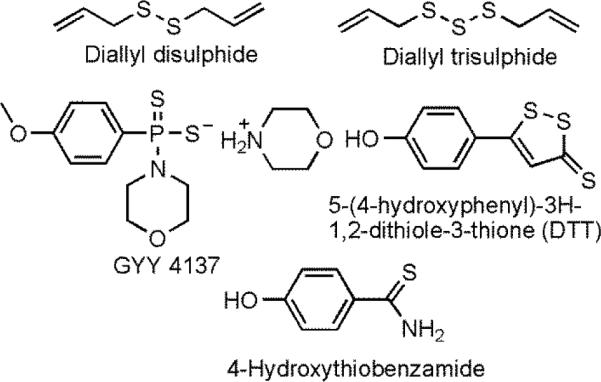
The five most common molecules used to release H2S in vivo.
Biodegradable polymers have been used in medicine to deliver steady, low doses of drugs over days to weeks.41,42 The drugs are typically encapsulated in nano- to micro-sized particles composed of a polymer that slowly degrades to release the drug.41 Typically, polyesters or polyanhydrides are used due to their ability to degrade by hydrolysis without the need for enzymes, glutathione, or other molecules.42 Poly(lactic acid) is one of the most studied polymers in this field because of its ease of synthesis, ease of forming copolymers with glycolic acid to tune its degradation profile, and its approval by the FDA for use in medical devices.41-43 The degradation of poly(lactic acid) produces lactic acid which is considered a safe molecule in vivo. Poly(lactic acid) and poly(lactic-co-glycolic acid) are used in numerous applications in medicine and are an attractive choice for the development of future applications because of their proven safety.41,43
In this paper we report the design and synthesis of a polymer based on poly(lactic acid) that has 4-hydroxythiobenzamide bonded to the backbone of the polymer. The polymer is produced by the copolymerization of L-lactide and a lactide that was modified to be covalently bonded to 4-hydroxythiobenzamide. The reaction to attach 4-hydroxythiobenzamide to lactide is new and involve a mild reaction of propanedithiol with catalytic I2. This reaction can be of use to others who need to functionalize lactide. 4-Hydroxythiobenzamide was chosen because it has been shown to release H2S in vivo and to have synergy with other drugs such as naproxen which is a non-steroidal anti-inflammatory drug (NSAID; Figure 2).34,44 The synergy of therapeutic doses of H2S with drugs such as aspirin, diclofenac, and sildenafil have shown that the simultaneous release of H2S and a drug can result in a better therapeutic effect than the delivery of the drug or H2S individually.33,35,45 4-Hydroxythiobenzamide has a phenol group that was used to attach to drugs through an ester bond that was rapidly cleaved in vivo to release a drug such as naproxen and 4-hydroxythiobenzamide. An important advantage of the polymers reported in this article is the ability to noncovalently encapsulate drugs within microparticles for the simultaneous release of a drug and 4-hydroxythiobenzamide.
Fig. 2.
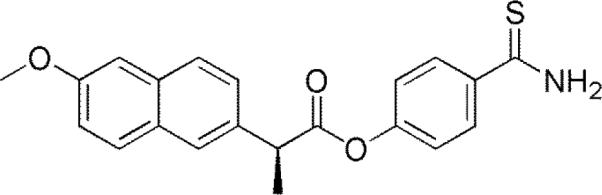
A prodrug of naproxen and 4-hydroxythiobenzamide. The ester bond is cleaved in vivo to release naproxen and the thiobenzamide that degrades to release H2S.
Results and Discussion
Synthesis of a functionalized lactide monomer
The convergent synthesis of a monomer containing 4-hydroxythiobenzamide took seven steps (Figure 3). Commercially available lactide was reacted with NBS followed by triethylamine to yield 2 in 51% yield. This sequence was first reported by Hillmyer et al. in 2008, and it proceeded as described in the literature.46-48
Fig. 3.

The convergent synthesis of molecule 7.
In prior work, 2 was reacted with cyclopentadiene to yield a monomer that could be polymerized by both the Grubbs catalyst and by tin-catalyzed ring-opening of the lactide. Other reactions to functionalize 2 have not been reported, so the reactions of 2 with different nucleophiles were investigated. Nucleophilic addition reactions at the α,β-unsaturated ester of 2 were expected to be challenging due to the facile ring-opening of the lactide with nucleophiles. Several different nucleophiles were investigated to learn which reagent would react with the α,β-unsaturated ester rather than ring-open the lactide. Initial attempts with a secondary amine (methylbenzylamine) were unsuccessful and resulted in ring-opening of 2. Similar results were obtained when 2 was reacted with propanethiol with catalytic triethylamine as a base. No reaction was observed when propanethiol and 2 were reacted with sodium bicarbonate, sodium carbonate, or at 80 °C in the absence of base.
The reaction of propanethiol with 2 proceeded to 32% yield under click reaction conditions with AiBN at 75 °C.49 Attempts to optimize this reaction failed and lead to the formation of several products that were challenging to separate. The addition of propanethiol to 2 was successful when catalytic amounts of I2 were added.50 In a control experiment to investigate if the catalytic species was I−, KI was added to a mixture of 2 and propanethiol, but no reaction was observed.
The reaction of molecule 2 with a 5 molar excess of 1,3-propanedithiol and a 5 mol % loading of I2 lead to the formation of molecule 3 in 70% yield. This reaction yielded two diastereomers as shown in Figure 4 in a 3:1 ratio. A NOESY experiment was conducted to investigate which diastereomer was formed in higher yield. A correlation between the two tertiary hydrogens on the ring was observed for the major diastereomer but not for the minor diastereomer. The major diastereomer was therefore assigned as the cis-product as shown in Figure 4.
Fig. 4.
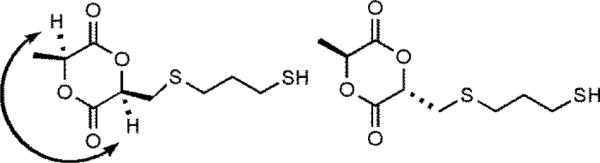
The two indicated H atoms in the major diastereomer that generated a NOESY correlation.
To complete the synthesis of the monomer, an aromatic nitrile group was converted to a thioamide in the same step an alkyl bromide was converted to a thiol (molecule 6 in Figure 3).51 This reaction yielded a thiol that was reacted with 3 under a variety of conditions including catalytic I2 and click conditions with AIBN. These reactions were unsuccessful, so the thiol on 3 was activated with N-chlorosuccinimide and then reacted with 6 to yield a lactide monomer bonded to a thiobenzamide. The overall yield over seven steps was 18%.
Copolymerization of L-lactide and molecule 7
Molecule 7 was copolymerized with lactide to yield polymers that had different amounts of thiobenzamide along the backbone (Figure 5). Many catalysts have been discovered and developed that will rapidly polymerize lactide, but this report focused on basic catalysts due to the presence of the mildly basic thioamide functional group that coordinates to Lewis acid.52,53 4-Dimethylaminopyridine (DMAP) was chosen based on its ready availability and prior work that demonstrated that it would rapidly polymerize lactide under mild reaction conditions.54 Octadecanol was chosen as the initiator for these polymerizations because the large number of methylenes in this molecule provided a method to estimate the molecular weight of the polymers using end group analysis from 1H NMR spectra.
Fig. 5.

The copolymerization of two monomers led to polymers functionalized with a thiobenzamide group.
The copolymerization of lactide and molecule 7 was first attempted in THF and CH2Cl2 for 48 h to investigate which solvent was best for the polymerization (entries 1 and 2 in Table 1). The polymerization in THF went to 50% conversion, but the polymerization in CH2Cl2 went to 98% conversion. The polydispersity (PDI) for entry 2 was high, so the concentration of monomers was cut in half (entry 3 in Table 1). This polymerization took longer (96 h) to reach a high conversion, and the molecular weight measured by end group analysis using 1H NMR spectroscopy disagreed with the molecular weight measured by SEC using multi-angle laser light scattering and refractive index detectors that allow absolute molecular weight determination. A likely explanation for this difference in observed molecular weight values is that at long reaction times of 96 h cross-linking of the polymer became significant. One possible reaction that would yield cross-links is a disulfide metathesis reaction as shown in Figure 6.55
Table 1.
Data for the copolymerizations of lactide and molecule 7.
| Entry | Initial ratioa | Time (h) | solvent | Concb (M) | M/Ic | Calc Mwd (g mol−1) | Polymer ratioe | Convf | Mng (NMR) | Mn (SEC) | Yield | PDI |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 10/1 | 48 | THF | 2.75 | 55 | 9930 | Na | 50% | Na | Na | Na | Na |
| 2 | 10/1 | 48 | CH2Cl2 | 2.75 | 55 | 9930 | 10/1 | 98% | 8670 | 13300 | 85% | 1.55 |
| 3 | 10/1 | 96 | CH2Cl2 | 1.38 | 55 | 9930 | 10/1 | 96% | 7050 | 22700 | 73% | 1.19 |
| 4 | 10/1 | 24 | CH2Cl2 | 2.75 | 55 | 9930 | 10/1 | 85% | 8530 | 7850 | 54% | 1.44 |
| 5 | 10/1 | 48 | CH2Cl2 | 2.50 | 55 | 9930 | 9/1 | 95% | 9200 | 11900 | 81% | 1.25 |
| 6 | 20/1 | 48 | CH2Cl2 | 2.75 | 63 | 10400 | 19/1 | 92% | 9340 | 11000 | 75% | 1.15 |
| 7h | 10/1 | 115 | CH2Cl2 | 2.75 | 110 | 19600 | 11/1 | 95% | 18700 | 88400 | 68% | 1.24 |
| 8h | 20/1 | 115 | CH2Cl2 | 3.15 | 126 | 20500 | 16/1 | 91% | 16200 | 69100 | 82% | 1.21 |
| 9 | 10/1 | 72 | CH2Cl2 | 2.75 | 110 | 19600 | 9/1 | 91% | 14600 | 22700 | 80% | 1.30 |
Lactide to molecule 7 ratio in the polymerizations.
Concentration of the monomers.
Ratio of the sum of the monomers to initiator.
Calculated molecular weight based on the ratio of monomers to initiator.
Observed ratio of lactide to molecule 7 in the copolymer as shown by 1H NMR spectroscopy.
Conversion of the polymerization.
The Mn was calculated from by end group analysis using 1H NMR spectroscopy.
The temperature of polymerization was 40 °C for these experiments, it was 35 °C for all other polymerizations.
Fig. 6.

The disulfide metathesis reaction that was shown to occur by the reaction of a polymer at 35 °C for 1 week yielded molecule 8.
The product of the disulfide metathesis reaction was observed after 500 mg from entry 9 in Table 1 was dissolved in CH2Cl2, exposed to light, and heated to 35 °C for 1 week. Molecule 8 was observed by 1H NMR spectroscopy and its presence was confirmed by high-resolution mass spectrometry.
To minimize the cross-linking reaction, the concentration of monomer was increased to 2.75 M or 2.50 M to lower the time necessary for completion (entries 4 and 5 in Table 1). The polymerizations were mostly complete within 24 h, and good agreement was observed between the molecular weight values measured by end group analysis and SEC. Furthermore, the ratio of lactide to molecule 7 was varied from 10/1 to 20/1 to produce polymers with varied loadings of thiobenzamide (entry 6 in Table 1).
Polymerizations to yield polymer with predicted molecular weights above 10,000 g mol−1 were limited by disagreement between the molecular weight measured by end group analysis and SEC. Polymerizations were completed to yield a polymer with a molecular weight close to 20,000 g mol−1, but reaction times of 115 h yielded polymers that had been cross-linked but still had low PDIs (entries 7 and 8 in Table 1). When the polymerization time was decreased to 72 h, the conversion was still high, and the agreement between molecular weights measured by end group analysis and SEC were much closer (entry 9 in Table 1).
Release of thiobenzamide from microparticles
Entry 5 from Table 1 was used to make two sets of microparticles with diameters of 12 ± 3.9 and 0.53 ± 0.13 μm (Figure 7). These particles were readily characterized by scanning electron microscopy (SEM) to show that they possessed smooth surfaces and spherical shapes. The zeta potential for the small particles as measured to be −20.45 mV, which is consistent with prior work with particles formed from poly(lactic acid) under these conditions.56
Fig. 7.

SEM micrographs show the a) 0.53 μm and b) 12 μm microparticles.
The degradation of the small particles was studied at 37 °C in buffered water at pH = 3.0 and 7.4 to investigate the release of thiobenzamide. After one day at pH 3.0, the microparticles released 4.1% of the thiobenzamide, but that value increased to 14.8% after one week. The particles degraded slowly, only losing 7% of their weight after one week at pH 3.0.
The degradation of the microparticles and release of thiobenzamide were controlled over 4 weeks at pH = 7.4. After one week, the microparticles released 7.5% of the available thiobenzamide increasing to 18.9% over 4 weeks as seen in Figure 8. The particles lost only 9.7% of their weight over 4 weeks – which was expected based on degradation profiles of similar microparticles based on poly(lactic acid).57,58 It is important to note that the microparticles slowly degraded and released thiobenzamide, which is well described in the literature as a molecule that releases H2S in vivo.8,34,37
Fig. 8.
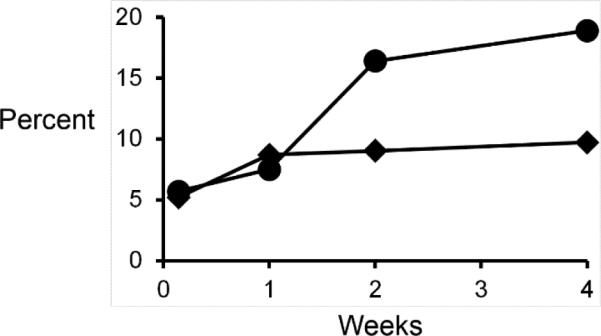
Graph of percent weight loss (diamonds) and percent of thiobenzamide released (circles) by the microparticles (0.53 μm) at pH 7.4 over a period of 4 weeks at 37 °C.
Attempts to quantify the release of H2S from the microparticles were unsuccessful although its odor was detected. This result was not surprising due to the low loading of thiobenzamide in the microparticles, the slow release of the thiobenzamide, and the rapid clearance of H2S from aqueous solutions by either evaporation (the boiling point of H2S is negative 60 °C) or oxidation. Prior work by Calderone et al. has shown the challenges of measuring the release of H2S from benzamides when they studied how a series of benzamides released H2S in aqueous buffer.44 Although 4-hydroxythiobenzamide was present at a concentration of 1 mM, the steady state concentration of H2S was less than 5 μM and rapidly declined. Importantly, this work and others demonstrated that thiobenzamides release H2S under physiological conditions.
Conclusions
This article describes a method to achieve the long-term release of a thiobenzamide that is known to degrade and release H2S in vivo. This is a critically important advance in the field of medicinal H2S because most prior methods only allow the release of small molecules that degrade to release H2S over minutes to hours, and the concentration of H2S is ill-controlled. Encapsulation of thiobenzamde in a biodegradable polymer can be used to deliver specific loadings of it to selected sites in the body. The degradation of the polymer and release of the thiobenzamide can be controlled by varying the composition of the polymer by the introduction of glycolic acid in the backbone of the polymer. In future work, we will investigate how these parameters affect the release of thiobenzamide and, subsequently, the release of H2S. Furthermore, the polymers reported in this paper have different loadings of the thiobenzamide and possessed molecular weights from 8,000 to 88,000 g mol−1 with low values for the PDI. A wide range of polymers can be envisioned that will allow some control over the degradation profile and release of thiobenzamide from particles.
A new method to functionalize a lactide monomer was also demonstrated by the reaction of 2 with thiols and catalytic I2. Lactides are important monomers in medicine; yet, it is challenging to functionalize to attach to other functional groups. This paper reports a new way to attach molecules to lactide monomers that builds on prior work by Hillmyer et al. This reaction can be used to attach different thiols to a lactide monomer to synthesize a wide range of new monomers.
Experimental
Materials
All chemicals and solvents were purchased from Aldrich or Acros at their highest purity. L-Lactide was purified by three crystallizations from ethyl acetate. The CH2Cl2 used for the polymerizations was dried under N2 using 3 Å molecular sieves. All other chemicals and solvents were used as received.
Characterization
1H NMR and 13C NMR spectra were acquired at 300 MHz and 75 MHz, respectively, using a Bruker Fourier 300 MHz spectrometer and referenced either to TMS for CDCl3 (δ=0.0) or to DMSO-d6 (δ=2.5). A high-resolution mass spectrometer used was a waters Q-Tof Premier. SEC was run using THF (1.00 mL min−1) as the mobile phase. A Waters 515 pump and two Waters Styragel HR3 THF columns were used in series. A Wyatt DAWN Heleos-II 18 angle laser light scattering detector was used to measure light scattering and a Wyatt Optilab T-rEX was used to measure change in refractive index.
(3S,6S)-3-Methyl-[6-propylthiol sulfide]-1,4-dioxane-2,5-dione (3)
(3S)-3-Methyl-6-methylene-1,4-dioxane-2,5-dione (2) (3.02 g, 21.3 mmol) was placed in 9 mL of CH2Cl2 and cooled to 0 °C. 1,3-Propanedithiol (11.5 g, 106 mmol) was added, followed by I2 (0.271 g, 1.07 mmol) and reacted for 3h. The reaction was quenched by addition of 25 mL of saturated sodium thiosulfate. The product was extracted with CH2Cl2 (3 × 50 mL) then the organic layer was washed with brine (3 × 150 mL) and evaporated. The product was purified by column chromatography using 7:3 (v:v) hexane:EtOAc. Two diastereomers were obtained with a combined yield of 3.74 g (70% yield). Two diastereomers were separated for characterization. 1H NMR (300 MHz, CDCl3) (major diastereomer): δ 5.15 (dd, 1 H, J = 6.0 Hz, 4.1 Hz), 5.07 (q, 1 H, J = 6.7 Hz), 3.20 (dd, 1 H, J = 14.9 Hz, 4.1 Hz), 3.09 (dd, 1 H, J = 14.9 Hz, 6.0 Hz), 2.82 (t, 2 H, J = 7.1 Hz), 2.65 (dt, 2 H, J = 8.0 Hz, 7.0 Hz), 1.91 (quin, 2 H, J = 7.0 Hz), 1.70 (d, 3 H, J = 6.7 Hz), 1.39 (t, 1 H, J = 8.0 Hz). 13C NMR (75 MHz, CDCl3) (major diastereomer): δ 166.44, 165.61, 76.87, 72.58, 33.15, 32.10, 32.00, 23.19, 16.12. HRMS-EI (m/z) (major diastereomer): [M]+ calc for C9H14O4S2 250.0334 ; found, 250.0320. 1H NMR (300 MHz, CDCl3) (minor diastereomer): δ 5.37 (q, 1 H, J = 7.2 Hz), 5.29 (t, 1 H, J = 4.4 Hz), 3.25 (dd, 1 H, J = 15 Hz, 4.4 Hz), 3.14 (dd, 1 H, J = 15 Hz, 4.3 Hz), 2.79 (dd, 1 H, J = 6.6 Hz, 1.4 Hz), 2.79 (dd, 1 H, J = 7.3 Hz, 0.9 Hz), 2.62 (dt, 2 H, J = 8.0 Hz, 7.0 Hz), 1.89 (quin, 2 H, J = 7.0 Hz), 1.73 (d, 3 H, J = 7.1 Hz), 1.38 (t, 1 H, J = 8.1 Hz). 13C NMR (75 MHz, CDCl3) (minor diastereomer): δ 165.53, 164.76, 77.16, 73.26, 33.09, 34.90, 32.05, 23.10, 18.32. HRMS-EI (m/z) (minor diastereomer): [M]+ calc for C9H14O4S2 250.0334 ; found, 250.0318.
(3S,6S)-3-Methyl-[(6-proplythio)-2,5-pyrrolidinedione sulfide]-1,4-dioxane-2,5-dione (4)
N-Chlorosuccinimide (1.92 g, 14.4 mmol) and pyridine (1.34 g, 17.0 mmol) were dissolved in 30 mL of CH2Cl2 and cooled 0 °C. (3S,6S)-3-Methyl-[6-proplythiol sulfide]-1,4-dioxane-2,5-dione was dissolved in 12 mL of CH2Cl2 and added to the reaction dropwise over 30 min, and allowed to react at 0 °C for 2.5 h. The organics were then washed with brine (3 × 50 mL) and dried with MgSO4. The solvent was removed under vacuum to give a colorless oil that was used without purification (4.08 g, 90% crude yield). The 1H NMR spectrum displayed a shift in succinimide to δ 2.86 indicating the formation of product.
4-(4-Bromobutoxy)-benzonitrile (5)
4-Nitrophenol (4.76 g, 40 mmol) in 15 mL of DMF was added dropwise to 1,4-dibromobutane (17.27 g, 80 mmol) and K2CO3 (8.39 g, 60 mmol) dissolved in 40 mL DMF. After heating at 70 °C for 2.5 h, the reaction was cooled to room temperature and 200 mL of CH2Cl2 was added. The organic layer was washed with brine (4 × 200 mL) and dried over MgSO4. The solvent was removed under vacuum, and the product was purified with silica gel column chromatography using 17:3 (v:v) hexane:EtOAc to yield a white solid (5.96 g, yield 59%). 1H NMR (300 MHz, CDCl3) δ 7.57 (d, 2H, J = 9.0 Hz), 6.93 (d, 2H, J = 9.0 Hz), 4.04 (t, 2H, J = 5.7 Hz), 3.48 (t, 2H, J = 6.3 Hz), 2.11-1.92 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 161.68, 133.87, 119.06, 115.00, 103.87, 67.12, 32.99, 29.10, 27.48. HRMS-EI (m/z): [M]+ calc for C11H12NOBr, 253.0102; found, 253.0106.
4-(4-Butylthiol)-benzothiolamide (6)
A solution of 4-(4-bromobutoxy)-benzonitrile (5.72 g, 22.5 mmol) in 12 mL of DMF was added to a slurry of sodium hydrosulfide hydrate (12.42 g, 135 mmol) and magnesium chloride hexahydrate (5.49 g, 27 mmol) in 30 mL DMF. The reaction was stirred at room temperature for 5 h. The product was precipitated into 80 mL of water and then filtered. The solid was taken up with 160 mL of 1 M HCl and stirred for 20 min then filtered. The solid was dried under vacuum to yield a yellow solid (5.21 g, yield 96%). The product contained 15% disulfide that was removed after the next step. 1H NMR (300 MHz, DMSO-d6): δ 9.64 (s, 1H), 9.32 (s, 1H), 7.94 (d, 2H, J = 8.9 Hz), 6.94 (d, 2H, J = 8.9 Hz), 4.03 (t, 2H, J = 6.2 Hz), 2.55 (t, 2H, J = 7.2 Hz), 2.31 (t, 1H, J = 7.7 Hz), 1.85-1.63 (m, 4H), disulfide peak (CH2SSCH2) 2.78 (t, J = 6.3). 13C NMR (75 MHz, DMSO-d6) δ 198.68, 161.44, 131.16, 129.54, 113.42, 67.41, 29.96, 27.38, 23.61, disulfide δ 37.34, 25.18. HRMS-ES+ (m/z): [M+Na]+ calc for C11H16S2NO, 242.0673; found, 242.0673.
Molecule 7
(3S,6S)-3-Methyl-[(6-proplythio)-2,5-pyrrolidinedione sulfide]-1,4-dioxane-2,5-dione (4.01 g 11.5 mmol) and 4-(4-butylthiol)-benzothiolamide (2.79 g, 11.5 mmol) were dissolved in 20 mL of dry THF and stirred at room temperature for 5 h. The solvent was removed under vacuum to yield an orange oil. The oil was dissolved in 50 mL of CH2Cl2 and cooled to precipitate the disulfide impurity from the synthesis of compound 6. The CH2Cl2 solution was then washed with brine (3 × 100 mL) to remove the succinimide. The solvent was removed under vacuum, and the product was then purified with silica gel column chromatography using 9:11 (v:v) hexane:EtOAc to yield a yellow solid (2.44 g, yield 43%) with a diastereomeric ratio of 4:1. 1H NMR (300 MHz, DMSO-d6) (major diastereomer): δ 9.31 (s, 1H), 9.63 (s, 1H), 7.94 (dt, 2H, J = 8.9 Hz, 1.8 Hz), 6.94 (dt, 2H, J = 8.9 Hz, 2.1 Hz), 5.56 (dd, 1H, J = 4.1 Hz, 6.1 Hz), 5.45 (q, 1H, J = 6.7 Hz), 4.04 (t, 2H, J = 5.7 Hz), 3.12 (dd, 1H, J = 4.1 Hz, 15 Hz), 3.01 (dd, 1H, J = 6.1 Hz, 15 Hz), 2.78 (t, 4H, J = 7.2 Hz), 2.70 (t, 2H, J = 7.2 Hz), 1.90 (quin, 2H, J = 7.6 Hz), 1.81-1.79 (m, 4H), 1.47 (d, 3H, J = 6.7 Hz). 13C NMR (75 MHz, DMSO-d6) δ 198.63, 168.00, 166.89, 161.32, 131.26, 129.53, 113.54, 75.90, 72.30, 67.35, 37.44, 36.41, 31.04, 28.45, 27.34, 25.24, 15.31. HRMS-ES+ (m/z): [M+Na]+ calc for C20H27NO5S4Na, 512.0670; found, 512.0683.
Copolymerization (entry 5 of Table 1)
Molecule 7 (1.60 g, 3.27 mmol), lactide (4.71 g, 32.7 mmol), DMAP (0.319 g, 2.61 mmol), and octadecanol (0.177 g, 0.65 mmol) were added to a dry Schlenk flask. Dry CH2Cl2 (14.4 mL) was added and the solution was stirred at 35°C for 48 h. CH2Cl2 (100 mL) was added, and the solution was then washed with 0.5 M acetic acid solution (2 × 100 mL) and brine (2 × 100 mL). The organic layer was dried with MgSO4 and the solvent was removed under vacuum to yield an orange oil. This oil was dissolved in CH2Cl2 (25 mL), precipitated with 100 mL of hexanes, and filtered. The solid was dissolved with CH2Cl2 (25 mL) and precipitated into hexanes for a second time. A yellow solid was isolated (5.28 g, 81% yield). 1H NMR (300 MHz, CDCl3) δ 7.90 (d, 10H, J = 9.6 Hz), 7.56 (s, 5H), 7.30 (s, 5H), 6.87 (d, 10H, J = 9.6), 5.16 (q, 100H, J = 7.6), 4.08-4.00 (m, 10H), 3.17-3.11 (m, 4H), 3.08-3.00 (m, 6H), 2.81-2.64 (m, 30), 2.23-1.84 (m, 30), 1.58 (d, 304H, J = 7.6), 1.30-1.23 (m, 32), 0.88 (t, 3H, J = 5.7). 13C NMR (75 MHz, CDCl3) δ 201.45, 175.44, 169.89, 167.69, 162.30, 134.29, 131.36, 129.48, 114.27, 72.68, 69.29, 67.91, 66.83, 38.70, 37.34, 32.22, 29.61, 28.07, 25.94, 22.93, 20.75, 16.92, 16.04, 14.37.
Formation of microparticles (12 μm in diameter)
Entry 5 from Table 1 (200 mg) was dissolved in 1.5 mL of CH2Cl2. This solution was added to 30 mL of 1% poly(vinyl alcohol) in water and homogenized using an Ultra Turrax T-25 basic homogenizer (IKA-WERKE, Inc., Wilmington, NC) at speed 2, 9500 min−1 for 30 s. The emulsion was stirred for 1.5-2.0 h to evaporate CH2Cl2. The particles were collected by centrifugation at 1000 rpm (164 × g) for 5 min, washed twice with nanopure water, dispersed in 5 mL of water, and lyophilized using FreeZone 4.5 freeze dry system (Labconco, Kansas City, MO). The microparticles (150 mg) were white.
Formation of microparticles (0.53 μm in diameter)
Entry 5 from Table 1 (200 mg) was dissolved in 2.0 mL of CH2Cl2. This solution was added to 8 mL of 2.5% poly(vinyl alcohol) in water and sonicated with a sonic dismembrator ultrasonic processor (Fisher Scientific, Pittsburgh, PA) at 40% amplitude for 30 s. The emulsion was poured into 22 mL of 2.5% poly(vinyl alcohol) in water. The emulsion was stirred for 1.5-2.0 h to evaporate the CH2Cl2. The particles suspension was centrifuged at 2000 rpm (657 × g) for 5 min to separate the big particles that precipitated out. The particles that remained in the supernatant, from the first centrifugation, were then collected by centrifugation at 8500 rpm (8873 × g) for 5 min, washed twice with nanopure water, dispersed in 5 mL of water, and lyophilized. The microparticles (150 mg) were white in color.
Microparticles characterization
The morphology of microparticles was examined using SEM. The lyophilized particles were re-dispersed in nanopure water and dropped on a silicon wafer which was mounted on a SEM stub. The particles were then sputter-coated with gold-palladium for 2 minutes at 10 mA and then imaged using a Hitachi S-4800 SEM (Hitachi High-Technologies, Ontario, Canada). The size of the 12 μm microparticles was obtained from measuring the particles’ diameters (200 particles) in SEM images using ImageJ 1.44p software (Wayne Rasband, National Institutes of Health, USA). The size and zeta potential of the 0.53 μm microparticles were measured in nanopure water at 25 °C using a Zetasizer nano ZS particle analyzer (Malvern Instrument Ltd., Southborough, MA).
Degradation of microparticles
For degradation at pH 3.0, four samples each containing 150 mg of either the 0.53 or 12 μm particles were weighted out and dispersed in 30 mL of 10 mM acetate buffer at pH 3.0. This suspension was then incubated at 37 °C with 300 rpm shaking speed. One sample was stopped at each time point of 24 h, 1 week, 2 weeks, and 4 weeks. For degradation at pH 7.4, the same method described previously was employed but instead of dispersing the particles in 30 mL of 10 mM acetate buffer at pH 3.0, phosphate buffered saline at pH 7.4 was used. At each specified time point, the particle suspensions were centrifuged (10,000 × g for 0.53 μm and 4,500 × g for 12 μm particles) and the supernatant which did not contain any thiobenzamide was removed. The particles were washed three times with 30 mL of nanopure water to remove any salt, lyophilized, and weighed. A 50 mg sample of the particles was washed with MeOH (3 × 10 mL) to extract any thiobenzamide. The MeOH was filtered to remove any particles and dried under vacuum to yield solids that were investigated by 1H NMR spectroscopy. The 1H NMR spectrum was obtained using a known amount of 1,4-dinitrobenzene as an internal standard to calculate the amount of thiobenzamide that was released from the particles and extracted with MeOH.
Supplementary Material
Acknowledgements
T.R.L. and N.B.B. gratefully acknowledge support from the NSF (CHE-1213325). A.K.S gratefully acknowledges support from the American Cancer Society (RSG-09-015-01-CDD), the National Cancer Institute at the National Institutes of Health (1R21CA13345-01/ 1R21CA128414-01A2/UI Mayo Clinic Lymphoma SPORE) and the Lyle and Sharon Bighley Professorship.
Notes and references
- 1.Vandiver MS, Snyder SH. J. Mol. Med. 2012;90:255. doi: 10.1007/s00109-012-0873-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martelli A, Testai L, Marino A, Breschi C, M., Da Settimo F, Calderone V. Curr. Med. Chem. 2012;19:3325. doi: 10.2174/092986712801215928. [DOI] [PubMed] [Google Scholar]
- 3.Martelli A, Testai L, Breschi MC, Blandizzi C, Virdis A, Taddei S, Calderone V. Med. Res. Rev. 2012;32:1093. doi: 10.1002/med.20234. [DOI] [PubMed] [Google Scholar]
- 4.Olson KR. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011;301:R297. doi: 10.1152/ajpregu.00045.2011. [DOI] [PubMed] [Google Scholar]
- 5.Kashfi K, Olson KR. Biochem. Pharmacol. 2013;85:689. doi: 10.1016/j.bcp.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen C, Xin H, Zhu Y. Acta Pharmacol. Sin. 2007;28:1709. doi: 10.1111/j.1745-7254.2007.00629.x. [DOI] [PubMed] [Google Scholar]
- 7.Liu YH, Lu M, Hu LF, Wong PTH, Webb GD, Bian JS. Antioxid. Redox Signaling. 2012;17:141. doi: 10.1089/ars.2011.4005. [DOI] [PubMed] [Google Scholar]
- 8.a Caliendo G, Cirino G, Santagada V, Wallace JL. J. Med. Chem. 2010;53:6275. doi: 10.1021/jm901638j. [DOI] [PubMed] [Google Scholar]; b Allan PK, Wheatley PS, Aldous D, Mohideen MI, Tang C, Hriljac JA, Megson IL, Chapman KW, Weireld GD, Vaesen S, Morris RE. Dalton Trans. 2012;41:4060. doi: 10.1039/c2dt12069k. [DOI] [PubMed] [Google Scholar]; c Foster JC, Matson JB. Macromolecules. 2014;47:5089. [Google Scholar]; d Carter JM, Qian Y, Foster JC, Matson JB. Chem. Commun. 2015;51:13131. doi: 10.1039/c5cc04883d. [DOI] [PubMed] [Google Scholar]
- 9.Wang R. Physiol. Rev. 2012;92:791. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- 10.Wang R. FASEB J. 2002;16:1792. doi: 10.1096/fj.02-0211hyp. [DOI] [PubMed] [Google Scholar]
- 11.Kasparek MS, Linden DR, Kreis ME, Sarr MG. Surgery. 2008;143:455. doi: 10.1016/j.surg.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whiteman M, Cheung NS, Zhu Y-Z, Chu SH, Siau JL, Wong BS, Armstrong JS, Moore PK. Biochem. Biophys. Res. Commun. 2005;326:794. doi: 10.1016/j.bbrc.2004.11.110. [DOI] [PubMed] [Google Scholar]
- 13.Mitsuhashi H, Yamashita S, Ikeuchi H, Kuroiwa T, Kaneko Y, Hiromura K, Ueki K, Nojima Y. Shock. 2005;24:529. doi: 10.1097/01.shk.0000183393.83272.de. [DOI] [PubMed] [Google Scholar]
- 14.Whiteman M, Armstrong JS, Chu SH, Siau J-L, Wong B-S, Cheung NS, Halliwell B, Moore PK. J. Neurochem. 2004;90:765. doi: 10.1111/j.1471-4159.2004.02617.x. [DOI] [PubMed] [Google Scholar]
- 15.Geng B, Chang L, Pan C, Qi Y, Zhao J, Pang Y, Du J, Tang C. Biochem. Biophys. Res. Commun. 2004;318:756. doi: 10.1016/j.bbrc.2004.04.094. [DOI] [PubMed] [Google Scholar]
- 16.Mishra PK, Tyagi N, Sen U, Givvimani S, Tyagi SC. Am. J. Physiol. 2010;298:H451. doi: 10.1152/ajpheart.00682.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao W, Zhang J, Lu Y, Wang R. EMBO J. 2001;20:6008. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kiss L, Deitch EA, Szabo C. Life Sci. 2008;83:589. doi: 10.1016/j.lfs.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang B, Tang G, Cao K, Wu L, Wang R. Antioxid. Redox Signal. 2010;12:1167. doi: 10.1089/ars.2009.2894. [DOI] [PubMed] [Google Scholar]
- 20.Nichols CG. Nature. 2006;440:470. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- 21.Ashcroft SJ, Ashcroft FM. Cell. Signal. 1990;2:197. doi: 10.1016/0898-6568(90)90048-f. [DOI] [PubMed] [Google Scholar]
- 22.Ali MY, Ping CY, Mok YY, Ling L, Whiteman M, Bhatia M, Moore PK. Br. J. Pharmacol. 2006;149:625. doi: 10.1038/sj.bjp.0706906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kubo S, Doe I, Kurokawa Y, Nishikawa H, Kawabata A. Toxicology. 2007;232:138. doi: 10.1016/j.tox.2006.12.023. [DOI] [PubMed] [Google Scholar]
- 24.Hosoki R, Matsuki N, Kimura H. Biochem. Biophys. Res. Commun. 1997;237:527. doi: 10.1006/bbrc.1997.6878. [DOI] [PubMed] [Google Scholar]
- 25.Goubern M, Andriamihaja M, Nubel T, Blachier F, Bouillaud F. FASEB J. 2007;21:1699. doi: 10.1096/fj.06-7407com. [DOI] [PubMed] [Google Scholar]
- 26.Whitfield NL, Kreimier EL, Verdial FC, Skovgaard N, Olson KR. Am. J. Physiol. 2008;294:R1930. doi: 10.1152/ajpregu.00025.2008. [DOI] [PubMed] [Google Scholar]
- 27.Wintner EA, Deckwerth TL, Langston W, Bengtsson A, Leviten D, Hill P, Insko MA, Dumpit R, Vanden Ekart E, Toombs CF, Szabo C. Br. J. Pharmacol. 2010;160:941. doi: 10.1111/j.1476-5381.2010.00704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Toombs CF, Insko MA, Wintner EA, Deckwerth TL, Usansky H, Jamil K, Goldstein B, Cooreman M, Szabo C. Br. J. Clin. Pharmacol. 2010;69:626. doi: 10.1111/j.1365-2125.2010.03636.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li L, Rose P, Moore PK. Annual Rev. Pharmacol. Toxicol. 2011;51:169. doi: 10.1146/annurev-pharmtox-010510-100505. [DOI] [PubMed] [Google Scholar]
- 30.Guidotti TL. Int. J. Toxicol. 2010;29:569. doi: 10.1177/1091581810384882. [DOI] [PubMed] [Google Scholar]
- 31.Kamoun P. Amino Acids. 2004;26:243. doi: 10.1007/s00726-004-0072-x. [DOI] [PubMed] [Google Scholar]
- 32.Li L, Whiteman M, Guan YY, Neo KL, Cheng Y, Lee SW, Zhao Y, Baskar R, Tan CH, Moore PK. Circulation. 2008;117:2351. doi: 10.1161/CIRCULATIONAHA.107.753467. [DOI] [PubMed] [Google Scholar]
- 33.Sparatore A, Perrino E, Tazzari V, Giustarini D, Rossi R, Rossoni G, Erdman K, Schröder H, Soldato PD. Free Radical Biol. Med. 2009;46:586. doi: 10.1016/j.freeradbiomed.2008.11.013. [DOI] [PubMed] [Google Scholar]
- 34.Wallace JL, Caliendo G, Santagada V, Cirino G. Br. J. Pharmacol. 2010;159:1236. doi: 10.1111/j.1476-5381.2009.00611.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wallace JL, Caliendo G, Santagada V, Cirino G, Fiorucci S. Gastroenterology. 2007;132:261. doi: 10.1053/j.gastro.2006.11.042. [DOI] [PubMed] [Google Scholar]
- 36.Benavides GA, Squadrito GL, Mills RW, Patel HD, Isbell TS, Patel RP, Darley-Usmar VM, Doeller JE, Kraus DW. Proc. Natl. Acad. Sci. U. S. A. 2007;104:17977. doi: 10.1073/pnas.0705710104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martelli A, Testai L, Citi V, Marino A, Pugliesi I, Barresi E, Nesi G, Rapposelli S, Taliani S, Da Settimo F, Breschi MC, Calderone V. ACS Med. Chem. Lett. 2013;4:904. doi: 10.1021/ml400239a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen W, Rosser EW, Zhang D, Shi W, Li Y, Dong W-J, Ma H, Hu D, Xian M. Org. Lett. 2015;17:2776. doi: 10.1021/acs.orglett.5b01194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ono K, Akaike T, Sawa T, Kumagai Y, Wink DA, Tantillo DJ, Hobbs AJ, Nagy P, Xian M, Lin J, Fukuto JM. Free Radical Biol. Med. 2014;77:82. doi: 10.1016/j.freeradbiomed.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang C.-t., Zhao Y, Xian M, Li J.-h., Dong Q, Bai H.-b., Xu J.-d., Zhang M.-f. Cell. Physiol. Biochem. 2014;34:1304. doi: 10.1159/000366339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mundargi RC, Babu VR, Rangaswamy V, Patel P, Aminabhavi TM. J. Controlled Release. 2008;125:193. doi: 10.1016/j.jconrel.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 42.Uhrich KE, Cannizzaro SM, Langer RS, Shakesheff KM. Chem. Rev. 1999;99:3181. doi: 10.1021/cr940351u. [DOI] [PubMed] [Google Scholar]
- 43.Nair LS, Laurencin CT. Prog. Polym. Sci. 2007;32:762. [Google Scholar]
- 44.Martelli A, Testai L, Citi V, Marino A, Pugliesi I, Barresi E, Nesi G, Rapposelli S, Taliani S, Da Settimo F, Breschi MC, Calderone V. ACS Med. Chem. Lett. 2013;4:904. doi: 10.1021/ml400239a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shukla N, Rossoni G, Hotston M, Sparatore A, Del Soldato P, Tazzari V, Persad R, Angelini GD, Jeremy JY. BJU Int. 2009;103:1522. doi: 10.1111/j.1464-410X.2009.08415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jing F, Hillmyer MA. J. Am. Chem. Soc. 2008;130:13826. doi: 10.1021/ja804357u. [DOI] [PubMed] [Google Scholar]
- 47.Fiore GL, Jing F, Young VG, Jr., Cramer CJ, Hillmyer MA. Polym. Chem. 2010;1:870. [Google Scholar]
- 48.Castillo JA, Borchmann DE, Cheng AY, Wang Y, Hu C, Garcia AJ, Weck M. Macromolecules. 2012;45:62. doi: 10.1021/ma2016387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Uygun M, Tasdelen MA, Yagci Y. Macromol. Chem. Phys. 2010;211:103. [Google Scholar]
- 50.Chu C-M, Gao S, Sastry MNV, Yao C-F. Tetrahedron Lett. 2005;46:4971. [Google Scholar]
- 51.Manaka A, Sato M. Synth. Commun. 2005;35:761. [Google Scholar]
- 52.Gupta AP, Kumar V. Eur. Polym. J. 2007;43:4053. [Google Scholar]
- 53.Kiesewetter MK, Shin EJ, Hedrick JL, Waymouth RM. Macromolecules. 2010;43:2093. [Google Scholar]
- 54.Nederberg F, Connor EF, Moeller M, Glauser T, Hedrick JL. Angew. Chem. Int. Ed. 2001;40:2712. [PubMed] [Google Scholar]
- 55.Otsuka H, Nagano S, Kobashi Y, Maeda T, Takahara A. Chem. Commun. 2010;46:1150. doi: 10.1039/b916128g. [DOI] [PubMed] [Google Scholar]
- 56.Peltonen L, Koistinen P, Karjalainen M, Hakkinen A, Hirvonen J. AAPS PharmSciTech. 2002;3:E32. doi: 10.1208/pt030432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gonzalez MF, Ruseckaite RA, Cuadrado TR. J. Appl. Polym. Sci. 1999;71:1223. [Google Scholar]
- 58.Shive MS, Anderson JM. Adv. Drug Delivery Rev. 1997;28:5. doi: 10.1016/s0169-409x(97)00048-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.