

Effective new agents for patients with colorectal cancer (CRC) with disease progression during standard therapy regimens are needed. Single-agent olaparib delivered after failure of standard systemic therapy did not demonstrate activity, regardless of microsatellite status. Future trials testing poly ADP ribose polymerase (PARP) inhibitors against CRC should focus on DNA-damaging chemotherapy and/or radiation therapy, combined with PARP inhibitors.
Keywords: Colon cancer, PARP inhibitor
Abstract
Background.
Effective new agents for patients with colorectal cancer (CRC) with disease progression during standard therapy regimens are needed. We hypothesized that poly ADP ribose polymerase (PARP) inhibitor therapy in patients with CRC and inefficient tumor DNA repair mechanisms, such as those with high-level microsatellite instability (MSI-H), would result in synthetic lethality.
Methods.
This was an open-label phase II trial testing olaparib 400 mg p.o. b.i.d. for patients with disseminated, measurable CRC failing standard therapies with centrally confirmed tumor MSI status. The primary endpoint was the tumor response, assessed by RECIST, version 1.0. The secondary endpoints were safety/toxicity, progression-free survival (PFS), and overall survival (OS).
Results.
Thirty-three patients (20 microsatellite stable [MSS], 13 MSI-H) were enrolled. The median age for all patients was 57 years and for MSS and MSI-H patients was 51 and 61 years, respectively. All patients received at least one 28-day cycle of olaparib. No patient had a complete or partial response. Nausea (48%), fatigue (36%), and vomiting (33%) were the most commonly reported treatment-related adverse events. The median PFS for all patients was 1.84 months. No statistically significant differences were found in the median PFS or OS for the MSS group compared with the MSI-H group.
Conclusion.
Single-agent olaparib delivered after failure of standard systemic therapy did not demonstrate activity for CRC patients, regardless of microsatellite status. Future trials, testing PARP inhibitors in patients with CRC should focus on the use of DNA-damaging chemotherapy and/or radiation therapy, combined with PARP inhibitors, remembering the toxicity reported in the present study.
Implications for Practice:
Microsatellite instability (MSI-H) colorectal tumors exhibit hypermethylation in tumor mismatch repair genes, or have mutations in one or more of these genes resulting from a germ-line defect (Lynch syndrome). PARP inhibitors such as olaparib are most effective in tumors associated with inability to repair DNA damage. However, in this trial, single agent olaparib failed to elicit responses in patients with MSI-H colorectal tumors, and in those with microsatellite-stable tumors. It is possible that by adding olaparib to radiation therapy, or to a systemic DNA damaging agent, tumor lethality could be obtained. However, the price would be increased toxicity.
Introduction
Systemic therapy has improved overall survival (OS) for patients with disseminated colorectal cancer (CRC). First-line chemotherapy combined with a targeted agent has led to expected response rates of approximately 60%, with OS exceeding 2 years [1, 2]. Although standard-of-care therapy is available after failure of first-line therapy, the response rates have generally been <20%, and progression-free survival (PFS) decreases with subsequent systemic treatments [3]. Furthermore, we now recognize that the heterogeneous molecular biology of CRC has an important role in the choice of appropriate therapy [4].
Although patients with stage II high-level microsatellite instability (MSI-H) colon cancer have improved survival compared with those with tumors without mismatch repair gene abnormalities (microsatellite-stable [MSS]), MSI-H status alone does not influence OS for patients with stage III CRC or disseminated CRC [5, 6]. An analysis of four phase III trials that included 3,063 patients with disseminated colorectal cancer revealed that patients exhibiting deficient mismatch repair (MSI-H) had statistically inferior survival compared with those with proficient mismatch repair (MSS). In the cohort of MSI-H patients, BRAF mutations were found in 34.6%. In contrast, only 6.8% of patients who were MSS exhibited a BRAF mutation. The investigators concluded that both MSI-H and BRAF mutational status confer a statistically significant inferior survival compared with MSS and BRAF wild-type status. Although BRAF mutations clearly influence the biologic behavior of MSI-H patients, the investigators “. . . caution against a firm conclusion on this issue since . . . [the] trial was not sufficiently powered to test this interaction” [7]. The need for improved therapy for MSI-H CRC patients is all the more compelling, because in the setting of adjuvant treatment, the data suggest that MSI-H patients treated with single-agent 5-fluorouracil might have worse outcomes than untreated MSI-H patients [8].
BRCA1 and BRCA2 encode proteins that repair double-strand DNA breaks. In contrast, PARP genes repair single-strand DNA breaks [9, 10]. Inhibition of PARP in the presence of dysfunctional BRCA genes leads to contextual synthetic lethality [11]. Furthermore, loss of PARP-1 activity increases chemo- and radiosensitivity [12]. Tumor cells with BRCA mutations are susceptible to PARP inhibitors such as olaparib (Lynparza; AZ2281) [13–15]. CRC has been linked to mutations in DNA repair proteins that repair double-strand breaks [16, 17]. McCabe et al. have linked the sensitivity of PARP inhibitors to deficiencies in the protein complex consisting of MRE11, Rad50, and Nbs1 (MRN) [18]. MRE11, a component of the MRN complex, is mutated in approximately 80% of MSI-H tumors and leads to aberrant splicing and a truncated protein [16]. We hypothesized that targeting aberrant DNA repair with a PARP inhibitor could be a successful strategy for patients with stage IV MSI-H CRC and that patients with MSH-H CRC tumors would benefit more from a PARP inhibitor than those with non-MSI-H tumors.
In a phase I study of patients with advanced solid tumors, the first clinical response was noted with olaparib 100 mg twice daily. Dose escalations to twice-daily doses of 200 mg and 400 mg were associated with response; grade 1 or 2 nausea was the most frequently reported adverse event (AE). In an expanded cohort at 400 mg twice daily, 9 of 19 patients responded [19]. Thus, we designed an open-label trial testing single-agent olaparib 400 mg twice daily for patients with measurable disease for whom all standard therapies for disseminated CRC had failed. To test our hypothesis, the patients were stratified into two groups: microsatellite stable (MSS)—a group that also included MSI-low tumors—and those exhibiting a high degree of microsatellite instability (MSI-H).
The primary objective of the present trial (ClinicalTrials.gov identifier, NCT00912743) was to determine the response rate for olaparib in patients with progression or failure with all standard therapy. The secondary objectives included the determination of safety, tolerability, and toxicities of olaparib for patients previously treated for advanced CRC and the estimation of PFS and OS for patients treated with olaparib.
Methods
The eligibility criteria included patients >18 years of age, Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0 to 1, histologically proven disseminated CRC, and measurable lesions (≥10 mm) on spiral computed tomography scans. Patients were required to show progression of metastatic cancer after receiving at least two regimens, including a fluoropyrimidine, irinotecan, oxaliplatin, and appropriate targeted agents. Before therapy, submission of a pathology specimen from the primary tumor or metastatic lesion was required for central analysis of microsatellite instability (MSI) status with polymerase chain reaction-based methodology, as reported previously [20]. Standard parameters for adequate bone marrow, liver, and renal function were required. All patients enrolled had to have the ability to understand and sign an informed consent document approved by the treating investigator’s institutional review board. The exclusion criteria included previous PARP inhibitor treatment, symptomatic uncontrolled brain metastases, active seizures, antiepileptic medications, gastrointestinal disorders potentially interfering with oral drug absorption, a positive pregnancy test, active breast feeding, or an unwillingness to use effective contraception during the trial.
Study Design
The present study was a multicenter, open-label, phase II trial conducted by the Academic Gastrointestinal Cancer Consortium (AGICC). Registration was performed centrally by AGICC Operations in Los Angeles, California. AstraZeneca provided the study drug and supported our study as in Investigator Initiated Trial. AstraZeneca employees reviewed and contributed to the final draft of the protocol and to the final draft of this manuscript. The starting dose of olaparib (capsule formulation) for all patients was 400 mg p.o. b.i.d. The patients were instructed to take olaparib at least 1 hour after the last intake of food and to refrain from food intake for 2 hours after taking olaparib. The treatment was patient-administered until progression, unacceptable toxicity, or withdrawal of consent. Treatment interruptions were built into the protocol for grade 3 and 4 toxicities, according to the National Cancer Institute-Common Terminology Criteria for Adverse Events, version 3, and were allowed, at the investigator’s discretion, for any toxicity grade, for a maximum of 28 days. Any interruption lasting longer than 28 days necessitated withdrawal of the patient from the trial. Olaparib could be restarted after grade 3 or 4 toxicity had returned to grade 1 or baseline. Dose reduction to 200 mg p.o. b.i.d. was mandatory for grade ≥3 toxicity. A second dose reduction to 100 mg p.o. b.i.d. was allowed if a grade 3 or 4 AE recurred after the first dose reduction. Study subjects were withdrawn for a third occurrence of grade ≥3 toxicity. A cycle was defined as 28 days (4 weeks). Evaluation of tumor progression or regression was scheduled after completion of every two cycles.
Statistical Analysis
The primary endpoint was the tumor response as assessed using RECIST, version 1.0. The secondary endpoints were safety/toxicity and PFS, calculated as the time from the start of treatment with olaparib until disease progression or death. Patients alive with stable disease were censored at the last follow-up visit. OS was defined as the time from the start of treatment until death from any cause.
The trial used the first stage of the Gehan phase II design [21] for the MSI-H cohort, and a Simon two-stage design [22] for the MSS cohort. The maximum target accrual was 74 patients, with 59 and 15 patients in the MSS and MSI-H groups, respectively. If no responses were recorded in the 15 MSI-H patients enrolled, it would be fair to conclude that the response rate would be less than 12%, and cohort enrollment would be discontinued. In the MSS cohort, if at least 1 of the first 20 patients had an objective response, 39 more patients would be enrolled. If none of the first 20 patients responded, the enrollment into the MSS cohort would be discontinued. With this design, the probability of falsely declaring a regimen with a 2% response rate as warranting further study was 0.10 (α), and the probability of correctly declaring a regimen with a 10% response rate as warranting further study was 0.85 (power = 1 − β).
Toxicity was assessed and reported for all patients who received olaparib. A safety monitoring boundary (i.e., a modified sequential probability ratio test to detect an excessive number of patients removed from study because of toxicity during the first cycle) was established to trigger trial suspension and possible amendment if crossed [23].
Results
Patient Demographics and Tumor Biology
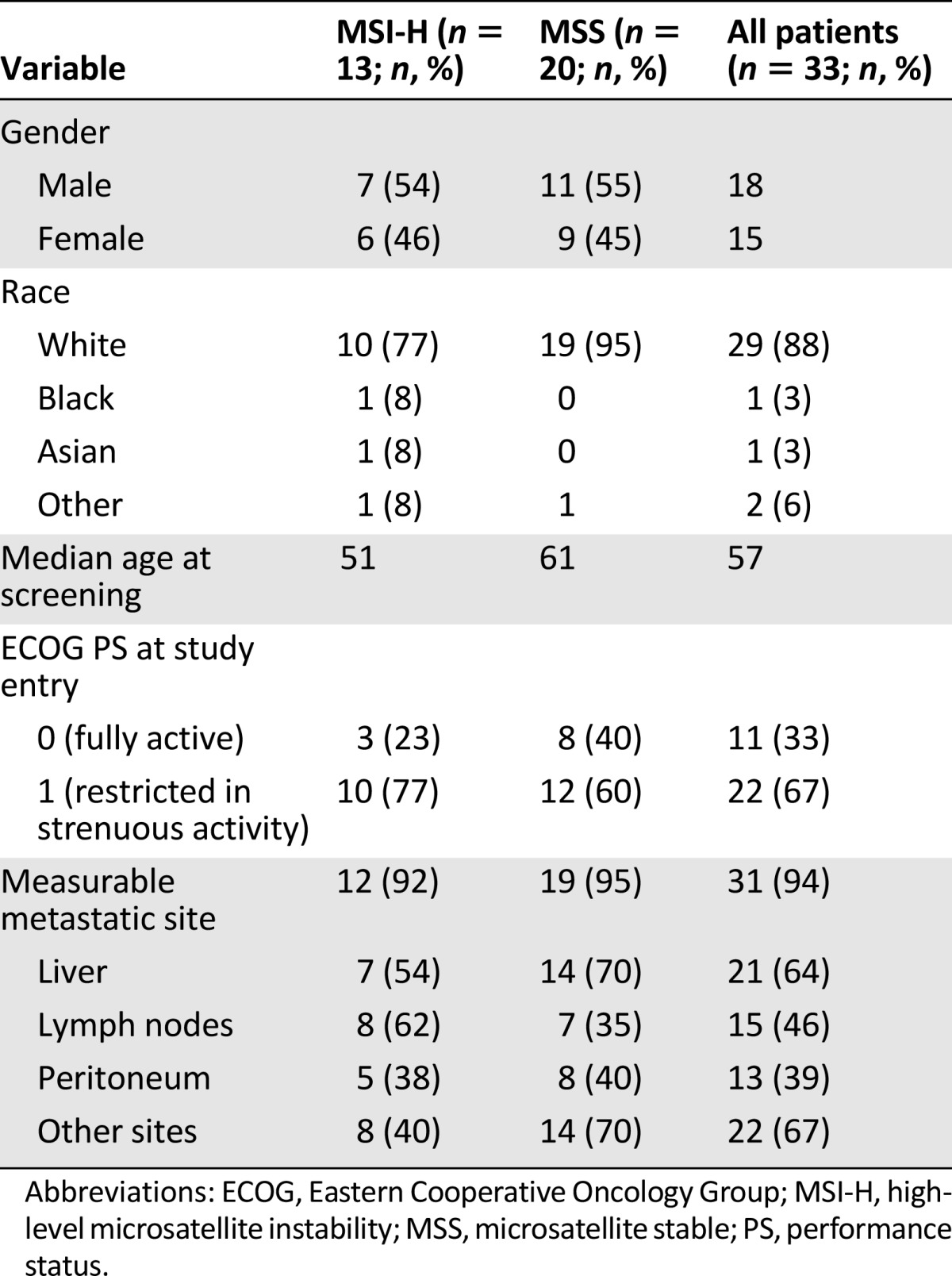
A total of 33 patients from eight AGICC institutions provided written informed consent and were enrolled in the present study, which opened May 30, 2009, and closed with the last patient visit on December 1, 2010 (Table 1). Enrollment included 20 patients with MSS colorectal cancer and 13 with MSI-H tumors. Of the 33 patients, 18 were men (7 with MSI-H) and 15 were women (6 with MSI-H). The median age for all patients was 57 years. For MSI-H patients and MSS patients, the median age was 51 and 61 years, respectively. Sixty-seven percent of the patients had an ECOG PS of 1. Nine patients (6 MSS and 3 MSI-H) had received previous radiation therapy. The liver (64%) and lymph nodes (46%) were the predominant sites of measurable lesions.
Table 1.
Summary of demographic data
Efficacy Evaluation
At a median follow-up period of 31.5 months (range, 3.5–33.5), 28 patients had died and 5 were alive. Two patients, both with MSI-H, died of disease progression before disease assessment at the end of cycle 2; all others were assessed for response. All 33 patients were included in all analyses. No responses were noted in either cohort. Although most had progression at the first efficacy assessment, 5 patients had stable disease for >8 weeks (2 with MSS and 3 with MSI-H tumors). The difference in the median PFS was small: 1.81 months (95% confidence interval [CI], 1.61–1.84) versus 2.00 months (95% CI, 1.81–3.25) in MSS and MSI-H patients, respectively (Fig. 1). At the time of study closure, 18 (90%) of the MSS and 10 (77%) of the MSI-H patients had died. No difference in OS between MSS and MSI-H patients was observed: 9.3 months (95% CI, 6.4–14.1) versus 8.1 months (95% CI, 4.0–11.8; Fig. 2).
Figure 1.

Kaplan-Meier plot of progression-free survival: Evaluable set.
Abbreviations: MSI-H, high-level microsatellite instability; MSS, microsatellite stable.
Figure 2.

Kaplan-Meier plot of overall survival: Evaluable set.
Abbreviations: MSI-H, high-level microsatellite instability; MSS, microsatellite stable.
Safety and Tolerability
The median treatment duration of 1.9 months (range, 0.70–31) was identical for both non-MSI-H and MSI-H patients. Fifteen patients (45.5%) had ≥1 dose delays, with 14 of these attributed to an AE. Twenty-four (73%) needed ≥1 dose reductions. The most commonly reported all-grade AEs were 31 (94%) gastrointestinal disorders, mainly nausea and vomiting, 16 (48%) cytopenias, and 13 (39%) fatigue. The AE distribution was similar in MSS and MSI-H patients. The AEs occurring in ≥10% of patients are summarized in Table 2. Although fatigue is always difficult to treat, in almost all cases, nausea and vomiting were well-controlled with appropriate antiemetic medications. Six MSI-H (46%) and 10 MSS (50%) patients experienced any AE grade ≥3. The most frequently reported grade ≥3 AEs were anemia in 5, neutropenia in 3, and small intestinal obstruction in 2. The study drug was discontinued in 9 patients (27%) because of an AE; in 6 because of treatment-related AEs. In the MSI-H group, these were neutropenia and anemia. In the MSS group, these were neutropenia, fatigue, thrombocytopenia, and diarrhea. No drug-related deaths occurred, with all deaths during the study attributable to disease progression.
Table 2.
Summary of adverse events (any grade) occurring in at least 10% of patients in either treatment group, arranged by system organ class

Discussion
The present multicenter, open-label, phase II trial was based on the hypothesis that olaparib, a PARP-1 inhibitor, would be more effective for CRC patients with defective DNA mismatch repair and resulting microsatellite instability. However, as a single agent delivered after failure of standard systemic therapies, olaparib did not demonstrate promising activity. Although designed for a total of 74 patients, the early stopping rule necessitated study closure after 20 MSS patients had been treated without a response. The accrual of 15 MSI-H patients proved difficult owing to the low frequency of MSI-H in advanced disease, and the study was halted after 13 MSI-H patients were registered and treated without a response. Overall, the toxicities, AEs, and severe AEs were similar for both cohorts. It is possible that a randomized comparison of olaparib and a placebo as third-line therapy, restricted to the MSI-H CRC population, would have more clearly defined the role of single-agent olaparib in this population. However, a prospective trial in this cohort would take years to complete, and our results do not suggest that undertaking such a trial would be worthwhile.
The median OS for the patients in the present trial was longer for the MSS patients than for the MSI-H patients: 9.3 versus 8.1 months. This suggests that although MSI-H status might inform for improved survival for patients with stage II colon cancer, it does not do so for patients with disseminated disease. The role of the higher rate of BRAF mutations in the MSI-H CRC patients in determining these seemingly paradoxical results needs further analysis [7]. In a review of the survival influence of BRAF mutations for patients with disseminated CRC, Tran et al. found those with MSI-H tumors had worse survival than those with MSS tumors (11.1 vs. 22.1 months; p = .017) [6]. We did not require knowledge of BRAF status for entry into the protocol. However, at this time, we are not able to identify those in our MSI-H population who had BRAF mutations.
In a trial without clinical responses in our target population of MSI-H CRC patients, it is reasonable to review our central hypothesis: a PARP inhibitor would cause synthetic lethality in tumors harboring a high rate of MRE11 mutations. Support for this hypothesis can be found in a report by Vilar et al., in which they noted that a deficiency in MRE11 sensitizes colorectal cells to PARP-1 inhibition. This group tested 17 colorectal cell lines and 46 primary tumors and found that MRE11 deficiency increases sensitivity to poly(ADP-ribose) polymerase inhibition in MSI-H colorectal cancers exhibiting biallelic mutation in MRE11 [24]. Because biallelic mutations represent 36% of all MRE11 mutations, if we had honed our target of MSI-H patients to those with MRE11 biallelic mutations, it is possible we would have had a better outcome. An alternative hypothesis was presented by Regal et al., who demonstrated that malfunction of the MRN complex binds double-strand breaks and initiates damage-induced signaling cascades via activation of the ataxia-telangiectasia mutated (ATM) and ataxia-telangiectasia- and rad3-related kinases. Thus, targeting the ATM pathway for patients with CRC might have been a better strategy than targeting MRE11 with a PARP inhibitor [25].
We can also speculate that combination therapy with olaparib and cytotoxic agents, including radiation therapy, could have been a better strategy as a trial design for MSI-H CRC patients. It has been shown that PARP inhibitors might function as sensitizing agents for chemotherapy and radiotherapy that cause DNA damage [26]. Palma et al. have shown what they called “potent antitumor efficacy” of the PARP inhibitor, ABT-888 with temozolomide (TMZ) in orthotopic and metastatic implantation models across a spectrum of histologic types. Their results are worth noting, because the efficacy was independent of TMZ activity and overcame both inherent and acquired TMZ resistance [27]. Japanese investigators found that adding olaparib to SN-38 or irinotecan potentiated S-phase double-strand breaks, producing a synergistic effect in cells that were MSI-H and non-MSI-H [28]. In a prospective, randomized study of second-line therapy for Korean gastric cancer patients, Bang et al. randomized their patients to receive olaparib 100 mg p.o. b.i.d., combined with weekly paclitaxel on a 28-day cycle or paclitaxel plus a placebo. In their report, the OS was 13.1 months for those receiving olaparib with paclitaxel versus 8.3 months for those receiving paclitaxel with placebo (p = .010) [29]. Additionally, Oza et al. reported the results of a randomized phase II trial in which patients with recurrent platinum-sensitive ovarian cancer (38% BRCA-mutated) were introduced to olaparib with chemotherapy or chemotherapy alone and found a longer PFS for those receiving olaparib plus chemotherapy (12.2 vs. 9.6 months; p = .0012) [30].
Finally, a clinical trial reported by Le et al. might point the way to an entirely new approach to the treatment of MSI-H CRC. Noting that in reports of programmed death (PD-1) checkpoint inhibitors, the solitary CRC responder harbored an MSI-H tumor, they conducted a phase II trial testing pembrolizumab, an anti-PD-1 immune check point inhibitor, in 41 patients with or without mismatch-repair deficiency. Although no immune-related responses were noted for 18 CRC patients who were MSS, 4 of 10 patients who were MSI-H had objective responses to pembrolizumab. They hypothesized that the sensitivity of MSI-H tumors to the immune check point inhibitor is related to the prominent lymphocytic infiltrates uniformly found in MSI-H patients and to high mutational rate (some of which might be recognized by the patient’s immune system) found in MSI-H CRC patients compared with MSS patients [31]. These results open a promising avenue for therapy for patients with MSI-H colon cancers. Whether PARP inhibitors will have a role, combined with either standard chemotherapy or immune check point inhibitors, in patients with MSI-H CRC remains a subject for further clinical evaluation.
Acknowledgment
We gratefully acknowledge the editorial assistance of Katherine Vandris, B.A., GI Cancer Program Coordinator, New York University.
Author Contributions
Conception/design: Lawrence Leichman, Bert H. O’Neil, Wells Messersmith, Jordan Berlin, Cynthia G. Leichman, Steven J. Cohen, Deirdre Cohen, Heinz-Josef Lenz, Philip Gold, Bruce Boman, Stan R. Hamilton, Howard S. Hochster
Provision of study material or patients: Lawrence Leichman, Bert H. O’Neil, Wells Messersmith, Jordan Berlin, Emily Chan, Cynthia G. Leichman, Steven J. Cohen, Deirdre Cohen, Heinz-Josef Lenz, Philip Gold, Bruce Boman, Stan R. Hamilton, Howard S. Hochster
Collection and/or assembly of data: Lawrence Leichman, Susan Groshen, Bert H. O’Neil, Wells Messersmith, Jordan Berlin, Cynthia G. Leichman, Steven J. Cohen, Deirdre Cohen, Heinz-Josef Lenz, Philip Gold, Bruce Boman, Anitra Fielding, Gershon Locker, Ronald C. Cason, Stan R. Hamilton, Howard S. Hochster
Data analysis and interpretation: Lawrence Leichman, Susan Groshen, Bert H. O’Neil, Wells Messersmith, Jordan Berlin, Cynthia G. Leichman, Steven J. Cohen, Deirdre Cohen, Heinz-Josef Lenz, Philip Gold, Bruce Boman, Anitra Fielding, Gershon Locker, Stan R. Hamilton, Howard S. Hochster
Manuscript writing: Lawrence Leichman, Susan Groshen, Bert H. O’Neil, Wells Messersmith, Jordan Berlin, Cynthia G. Leichman, Steven J. Cohen, Deirdre Cohen, Heinz-Josef Lenz, Philip Gold, Bruce Boman, Anitra Fielding, Gershon Locker, Stan R. Hamilton, Howard S. Hochster
Final approval of manuscript: Lawrence Leichman, Susan Groshen, Bert H. O’Neil, Wells Messersmith, Jordan Berlin, Cynthia G. Leichman, Steven J. Cohen, Deirdre Cohen, Heinz-Josef Lenz, Philip Gold, Bruce Boman, Anitra Fielding, Gershon Locker, Stan R. Hamilton, Howard S. Hochster
Disclosures
Jordan Berlin: AstraZeneca (RF); Anitra Fielding: AstraZeneca (E, OI); Gershon Locker: AstraZeneca (E); Stan R. Hamilton: Intervention Insights, LLC, Amgen, Inc., Palmetto GBA, Medical Research Council (C/A); Howard S. Hochster: AstraZeneca (H). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.Falcone A, Ricci S, Brunetti I, et al. Phase III trial of infusional fluorouracil, leucovorin, oxaliplatin, and irinotecan (FOLFOXIRI) compared with infusional fluorouracil, leucovorin, and irinotecan (FOLFIRI) as first-line treatment for metastatic colorectal cancer: The Gruppo Oncologico Nord Ovest. J Clin Oncol. 2007;25:1670–1676. doi: 10.1200/JCO.2006.09.0928. [DOI] [PubMed] [Google Scholar]
- 2.Heinemann V, von Weikersthal LF, Decker T, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): A randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15:1065–1075. doi: 10.1016/S1470-2045(14)70330-4. [DOI] [PubMed] [Google Scholar]
- 3.Tournigand C, André T, Achille E, et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: A randomized GERCOR study. J Clin Oncol. 2004;22:229–237. doi: 10.1200/JCO.2004.05.113. [DOI] [PubMed] [Google Scholar]
- 4.Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–1765. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 5.Sinicrope FA, Mahoney MR, Smyrk TC, et al. Prognostic impact of deficient DNA mismatch repair in patients with stage III colon cancer from a randomized trial of FOLFOX-based adjuvant chemotherapy. J Clin Oncol. 2013;31:3664–3672. doi: 10.1200/JCO.2013.48.9591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tran B, Kopetz S, Tie J, et al. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer. 2011;117:4623–4632. doi: 10.1002/cncr.26086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Venderbosch S, Nagtegaal ID, Maughan TS, et al. Mismatch repair status and BRAF mutation status in metastatic colorectal cancer patients: A pooled analysis of the CAIRO, CAIRO2, COIN, and FOCUS studies. Clin Cancer Res. 2014;20:5322–5330. doi: 10.1158/1078-0432.CCR-14-0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349:247–257. doi: 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 10.Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72:5588–5599. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 12.Nguewa PA, Fuertes MA, Valladares B, et al. Poly (ADP-ribose) polymerases: Homology, structural domains and functions: Novel therapeutic applications. Prog Biophys Mol Biol. 2005;88:143–172. doi: 10.1016/j.pbiomolbio.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 13.Tutt A, Robson M, Garber JE, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet. 2010;376:235–244. doi: 10.1016/S0140-6736(10)60892-6. [DOI] [PubMed] [Google Scholar]
- 14.Audeh MW, Carmichael J, Penson RT, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: A proof-of-concept trial. Lancet. 2010;376:245–251. doi: 10.1016/S0140-6736(10)60893-8. [DOI] [PubMed] [Google Scholar]
- 15.Kaufman B, Shapira-Frommer R, Schmutzler RK, et al. Olaparib monotherapy in patients advanced cancer and germline BRACA1/2 mutation. J Clin Oncol. 2014:32. doi: 10.1200/JCO.2014.56.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giannini G, Ristori E, Cerignoli F, et al. Human MRE11 is inactivated in mismatch repair-deficient cancers. EMBO Rep. 2002;3:248–254. doi: 10.1093/embo-reports/kvf044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ottini L, Falchetti M, Saieva C, et al. MRE11 expression is impaired in gastric cancer with microsatellite instability. Carcinogenesis. 2004;25:2337–2343. doi: 10.1093/carcin/bgh257. [DOI] [PubMed] [Google Scholar]
- 18.McCabe N, Turner NC, Lord CJ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66:8109–8115. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]
- 19.Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 20.Earle JSL, Luthra R, Romans A, et al. Association of microRNA expression with microsatellite instability status in colorectal adenocarcinoma. J Mol Diagn. 2010;12:433–440. doi: 10.2353/jmoldx.2010.090154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gehan EA. The determination of patients required in a follow-up of a new chemotherapeutic agent. J Chronic Dis. 1961;13:346–353. doi: 10.1016/0021-9681(61)90060-1. [DOI] [PubMed] [Google Scholar]
- 22.Simon R. Optimal two-stage designs for phase II clinical trials. Control Clin Trials. 1989;10:1–10. doi: 10.1016/0197-2456(89)90015-9. [DOI] [PubMed] [Google Scholar]
- 23.Wald A. Sequential tests of statistical hypotheses. Ann Math Stat. 1945;16:117–186. [Google Scholar]
- 24.Vilar E, Bartnik CM, Stenzel SL, et al. MRE11 deficiency increases sensitivity to poly(ADP-ribose) polymerase inhibition in microsatellite unstable colorectal cancers. Cancer Res. 2011;71:2632–2642. doi: 10.1158/0008-5472.CAN-10-1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Regal JA, Festerling TA, Buis JM, et al. Disease-associated MRE11 mutants impact ATM/ATR DNA damage signaling by distinct mechanisms. Hum Mol Genet. 2013;22:5146–5159. doi: 10.1093/hmg/ddt368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reiss KA, Herman JM, Zahurak M, et al. A phase I study of veliparib (ABT-888) in combination with low-dose fractionated whole abdominal radiation therapy in patients with advanced solid malignancies and peritoneal carcinomatosis. Clin Cancer Res. 2015;21:68–76. doi: 10.1158/1078-0432.CCR-14-1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palma JP, Wang YC, Rodriguez LE, et al. ABT-888 confers broad in vivo activity in combination with temozolomide in diverse tumors. Clin Cancer Res. 2009;15:7277–7290. doi: 10.1158/1078-0432.CCR-09-1245. [DOI] [PubMed] [Google Scholar]
- 28.Tahara M, Inoue T, Sato F, et al. The use of Olaparib (AZD2281) potentiates SN-38 cytotoxicity in colon cancer cells by indirect inhibition of Rad51-mediated repair of DNA double-strand breaks. Mol Cancer Ther. 2014;13:1170–1180. doi: 10.1158/1535-7163.MCT-13-0683. [DOI] [PubMed] [Google Scholar]
- 29.Bang Y-J, Im SA, Lee KW, et al. Olaparib plus paclitaxel in patients with recurrent or metastatic gastric cancer: A randomized double-blind phase II study. J Clin Oncol. 2013;31(suppl):40130a. doi: 10.1200/JCO.2014.60.0320. [DOI] [PubMed] [Google Scholar]
- 30.Oza AM, Cibula D, Benzaquen AO, et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: A randomised phase 2 trial. Lancet Oncol. 2015;16:87–97. doi: 10.1016/S1470-2045(14)71135-0. [DOI] [PubMed] [Google Scholar]
- 31.Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]