

Abstract
Pixantrone is a new noncardiotoxic aza-anthracenedione anticancer drug structurally related to anthracyclines and anthracenediones, such as doxorubicin and mitoxantrone. Pixantrone is approved in the European Union for the treatment of relapsed or refractory aggressive B cell non-Hodgkin lymphoma. This study was undertaken to investigate both the mechanism(s) of its anticancer activity and its relative lack of cardiotoxicity. Pixantrone targeted DNA topoisomerase IIα as evidenced by its ability to inhibit kinetoplast DNA decatenation; to produce linear double-strand DNA in a pBR322 DNA cleavage assay; to produce DNA double-strand breaks in a cellular phospho-histone γH2AX assay; to form covalent topoisomerase II-DNA complexes in a cellular immunodetection of complex of enzyme-to-DNA assay; and to display cross-resistance in etoposide-resistant K562 cells. Pixantrone produced semiquinone free radicals in an enzymatic reducing system, although not in a cellular system, most likely due to low cellular uptake. Pixantrone was 10- to 12-fold less damaging to neonatal rat myocytes than doxorubicin or mitoxantrone, as measured by lactate dehydrogenase release. Three factors potentially contribute to the reduced cardiotoxicity of pixantrone. First, its lack of binding to iron(III) makes it unable to induce iron-based oxidative stress. Second, its low cellular uptake may limit its ability to produce semiquinone free radicals and redox cycle. Finally, because the β isoform of topoisomerase II predominates in postmitotic cardiomyocytes, and pixantrone is demonstrated in this study to be selective for topoisomerase IIα in stabilizing enzyme–DNA covalent complexes, the attenuated cardiotoxicity of this agent may also be due to its selectivity for targeting topoisomerase IIα over topoisomerase IIβ.

Introduction
Pixantrone (Fig. 1) is a DNA-intercalating aza-anthracenedione (Fig. 1) used for the treatment of aggressive non-Hodgkin lymphoma (NHL) that was designed to minimize cardiotoxicity (Mukherji and Pettengell, 2010; Boyle and Morschhauser, 2015). Pixantrone was granted conditional marketing approval in the European Union in May 2012 as a monotherapy for the treatment of adult patients with multiply relapsed or refractory aggressive B cell NHLs (Pean et al., 2013; Pettengell and Kaur, 2015). Anthracyclines such as doxorubicin are highly active against NHL, but their use in relapsed patients is compromised by their well-known total dose-limiting cardiotoxicity. A phase III clinical trial in heavily pretreated patients with relapsed or refractory aggressive NHL showed that pixantrone is efficacious and tolerable (Pean et al., 2013; Pettengell and Kaur, 2015). The National Institutes of Health clinical trials website (www.clinicaltrials.gov) currently lists 14 clinical trials involving pixantrone. Preclinical studies in mice showed that pixantrone displayed little or no cardiotoxicity compared with mitoxantrone (Beggiolin et al., 2001), or to doxorubicin (Cavalletti et al., 2007; Longo et al., 2014). In both doxorubicin-pretreated and naive mice, minimal cardiac changes were observed with repeated cycles of pixantrone, whereas repeated dosing with doxorubicin or mitoxantrone caused significant cardiotoxicity in naive mice or worsening of degenerative myopathy in doxorubicin-pretreated animals (Cavalletti et al., 2007).
Fig. 1.

Structure of pixantrone, mitoxantrone, and doxorubicin.
Both doxorubicin and mitoxantrone form a strong complex with Fe3+ (Herman et al., 1997). The cardiotoxicity of doxorubicin is thought to be due, at least in part, to iron-dependent oxygen free radical formation (Malisza and Hasinoff, 1995; Myers, 1998; Gewirtz, 1999). Pixantrone, which was designed not to bind iron, lacks the hydroquinone functionality of either doxorubicin or mitoxantrone (Fig. 1). Thus, the reduced cardiotoxicity of pixantrone, compared with doxorubicin or mitoxantrone, may be due to its lack of a quinone–hydroquinone functionality that can chelate iron. Theoretically, therefore, pixantrone would not directly participate in iron-mediated hydroxyl radical (HO·) formation through a Fenton reaction (Halliwell and Gutteridge, 1999).
In human myocardial strips, pixantrone lacked redox synergism with doxorubicin and formed neither 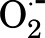 nor H2O2 (Salvatorelli et al., 2013). However, because pixantrone contains a quinone moiety, it can potentially be reductively activated to a semiquinone free radical like doxorubicin (Malisza and Hasinoff, 1996), which, in turn, could undergo aerobic oxidation and further redox cycling to induce cardiotoxic oxidative stress. Based on this consideration, electron paramagnetic resonance (EPR) experiments were carried out to determine the relative ability of pixantrone and doxorubicin to form the semiquinone free radical intermediate in both a hypoxic enzymatic reducing system and a cellular system.
nor H2O2 (Salvatorelli et al., 2013). However, because pixantrone contains a quinone moiety, it can potentially be reductively activated to a semiquinone free radical like doxorubicin (Malisza and Hasinoff, 1996), which, in turn, could undergo aerobic oxidation and further redox cycling to induce cardiotoxic oxidative stress. Based on this consideration, electron paramagnetic resonance (EPR) experiments were carried out to determine the relative ability of pixantrone and doxorubicin to form the semiquinone free radical intermediate in both a hypoxic enzymatic reducing system and a cellular system.
Prior cellular studies in HL-60 and cross-resistant HL-60/AMSA cells, containing a less sensitive topoisomerase II, demonstrated pixantrone-induced stabilization of a covalent topoisomerase II–DNA complex (Zwelling et al., 1993) and pixantrone-induced Simian virus 40 DNA cleavage mediated by topoisomerase II (De Isabella et al., 1995). Although the primary cytotoxic effect of pixantrone and its analogs were thought to be due to topoisomerase II-mediated effects, it was concluded that the pixantrone analogs exerted multiple effects at a cellular level (De Isabella et al., 1995), and the role of topoisomerase II as a target for pixantrone has been questioned (Hazlehurst et al., 1995a). More recently, it has been suggested that pixantrone induces a latent type of DNA damage that impairs mitosis (Beeharry et al., 2015). Evidence has been accumulating that anthracycline-induced cardiotoxicity may be due to topoisomerase IIβ-mediated responses to DNA damage as well as oxidative damage (Lyu et al., 2007; Zhang et al., 2012; Vejpongsa and Yeh, 2014). Importantly, the β isoform of topoisomerase II predominates in postmitotic cardiac cells (Capranico et al., 1992). It has also been suggested that targeting of topoisomerase IIβ is responsible for induction of anticancer drug-induced secondary malignancies produced by topoisomerase II-targeted anticancer drugs (Azarova et al., 2007; Cowell et al., 2012). Development of drugs such as NK314 (Toyoda et al., 2008) and etoposide analogs (Mariani et al., 2015) that specifically target the topoisomerase IIα isoform to reduce topoisomerase IIβ-mediated toxicity/carcinogenicity is being actively pursued (Vejpongsa and Yeh, 2014). Because preclinical studies with pixantrone showed little or no cardiotoxicity, this result suggested that pixantrone, unlike the anthracyclines, may be specifically targeting the topoisomerase IIα isoform. To further clarify pixantrone’s anticancer action targeting topoisomerase II isoforms as well as its relative lack of cardiac toxicity, a variety of cellular and in vitro experiments was implemented. Our well-validated neonatal rat myocyte model (Hasinoff and Patel, 2010; Herman et al., 2011; Hasinoff et al., 2013) was also used to compare the relative myocyte-damaging effects of doxorubicin, mitoxantrone, and pixantrone.
Materials and Methods
Materials, Cell Culture, Growth Inhibition Assays, and Drug Uptake in K562 Cells.
Pixantrone dimaleate was from LKT Laboratories (St. Paul, MN). The catenated kinetoplast DNA (kDNA) and the primary anti-topoisomerase I antibody were from TopoGEN (Port Orange, FL). A rabbit polyclonal topoisomerase IIα antibody obtained from Abcam (Cambridge, MA) was raised using a synthetic peptide from amino acids 14–27 of human topoisomerase IIα. The topoisomerase IIβ antibody obtained from Santa Cruz Biotechnology (Dallas, TX) is a mouse monoclonal raised against amino acids 1341–1626 from human topoisomerase IIβ. The secondary horseradish peroxidase-conjugated antibodies were from Jackson ImmunoResearch (West Grove, PA). Luminol/enhancer/peroxide solution and Immun-Star chemiluminescence reagent (Bio-Rad, Hercules, CA/Mississauga, Canada) were used for detection of immunoblots. Human leukemia K562 cells, obtained from the American Type Culture Collection (Manassas, VA), and the acquired etoposide-resistant K/VP.5 subline (containing decreased levels of topoisomerase IIα mRNA and protein) (Ritke and Yalowich, 1993; Ritke et al., 1994b) were maintained as suspension cultures in minimal essential medium α (Life Technologies, Burlington, Canada) containing 10% fetal calf serum and 20 mM HEPES (pH 7.2). The spectrophotometric 96-well plate cell (5 × 104 cell/ml, 0.1 ml/well) growth inhibition 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) CellTiter 96 AQueous One Solution Cell Proliferation assay (Promega, Madison, WI), which measures the ability of the cells to enzymatically reduce MTS after drug treatment, has been described previously (Hasinoff et al., 2014, 2015). Pixantrone was dissolved in 0.9% NaCl and doxorubicin in water for the myocyte experiments and in dimethylsulfoxide for the decatenation, cleavage, phospho-histone γH2AX, and cellular immunodetection of complex of enzyme-to-DNA (ICE) assays. The leukemia cells were incubated with the drugs for 72 hours and then assayed with MTS or evaluated directly for growth inhibition using a model ZBF Coulter counter. The effect of pixantrone on the 72-hour growth inhibition of the Madin Darby canine kidney (MDCK) and the multiple drug resistant (MDR) MDCK/MDR cell lines used to test for the role of an efflux transporter was assayed with a 3-[4,5-dimethylthiazol-2-yl]-2,5-tetrazolium bromide assay, as described (Hasinoff et al., 2015). The IC50 values for cell growth inhibition were measured by fitting the absorbance-drug concentration or cell count-drug concentration data to a four-parameter logistic equation, as we described (Yadav et al., 2014; Hasinoff et al., 2015).
The relative cellular uptake of pixantrone, doxorubicin, and mitoxantrone was measured in K562 cells, essentially as described (De Isabella et al., 1995). K562 cells in exponential growth were washed twice with Hanks’ balanced salt solution (HBSS) buffer (pH 7.4, with calcium, magnesium, and glucose) and resuspended in 1 ml HBSS buffer at a cell density of 10 × 106 cells/ml. The drugs (10 μM) were added and the samples were gently rocked at 37°C for 1 hour. The suspension was gently centrifuged, and the absorbance spectrum of the supernatant was measured. Cellular drug uptake was measured from the decrease in absorbance at the peak maxima (642, 495, and 611 nm for pixantrone, doxorubicin, and mitoxantrone, respectively), compared with drug controls without cells, and a control with cells and no added drug. MarvinSketch and its associated calculator plugins were used for calculating the nonionic partition coefficients (log P) and the distribution coefficients (log D) at pH 7.4 (Marvin version 6, 2013, ChemAxon, (http://www.chemaxon.com). Nonlinear least-squares curve fitting was done with SigmaPlot (Systat Software, San Jose, CA). Where significance is indicated (p < 0.05), an unpaired t test was used.
Topoisomerase IIα kDNA Decatenation, pBR322 DNA Relaxation, and Cleavage Assays.
A gel assay as previously described (Yadav et al., 2014; Hasinoff et al., 2015) was used to determine whether the drugs inhibited the catalytic decatenation activity of topoisomerase IIα. In this assay, kDNA, which consists of highly catenated networks of circular DNA, is decatenated by topoisomerase IIα in an ATP-dependent reaction to yield individual minicircles of DNA. Topoisomerase II-cleaved DNA covalent complexes produced by anticancer drugs may be trapped by rapidly denaturing the complexed enzyme with SDS (Burden et al., 2001; Yadav et al., 2014; Hasinoff et al., 2015). The drug-induced cleavage of double-strand closed circular plasmid pBR322 DNA to form linear DNA at 37°C was followed by separating the SDS-treated reaction products by ethidium bromide gel electrophoresis, essentially as described, except that all components of the assay mixture were assembled and mixed on ice prior to addition of the drug (Burden et al., 2001; Yadav et al., 2014; Hasinoff et al., 2015). The preparation of the full-length human topoisomerase IIα has been described in a previous publication (Hasinoff et al., 2005). The human topoisomerase IIβ, prepared as described (Smith et al., 2014), was a gift of N. Osheroff (Vanderbilt University School of Medicine, Nashville, TN).
γH2AX Assay for DNA Double-Strand Breaks in Pixantrone-Treated K562 Cells.
The γH2AX assay was carried out as described (Yadav et al., 2014; Hasinoff et al., 2015). K562 cells in growth medium (1.5 ml in a 24-well plate, 3.4 × 105 cells/ml) were incubated with drug or with dimethylsulfoxide vehicle control for 4 hours at 37°C. Cell lysates (70 μg protein) were subjected to SDS-PAGE on a 14% gel. Separated proteins were transferred to polyvinylidene fluoride membranes and then treated overnight with rabbit anti-γH2AX (Upstate, Charlottesville, VA) primary antibody diluted 1:1000. This was followed by incubation for 1 hour with both peroxidase-conjugated goat anti-rabbit secondary antibody (Cell Signaling Technology, Danvers, MA), diluted 1:2000, and anti-glyceraldehyde phosphate dehydrogenase (Cell Signaling Technology) primary antibody, diluted 1:1000. After incubation with luminol/enhancer/peroxide solution, chemiluminescence of the γH2AX and glyceraldehyde phosphate dehydrogenase bands was imaged on a Cell Biosciences (Santa Clara, CA) FluorChem FC2 imaging system equipped with a charge-coupled device camera.
Cellular ICE Assays for the Detection of Covalent DNA-Tpoisomerase IIα and Topoisomerase I.
The cellular ICE slot-blot assay for topoisomerase IIα covalently bound to DNA was carried out as described (Yadav et al., 2014; Hasinoff et al., 2015). The ICE assay used for the detection of covalent complexes of topoisomerase IIα bound to DNA was a modification of the original cesium chloride ultracentrifugation gradient assay used to isolate DNA (Subramanian et al., 2001), which instead employed the selective precipitation of genomic DNA using DNAzol (Invitrogen, Carlsbad, CA). Membranes were incubated overnight with either rabbit polyclonal antibody to human topoisomerase IIα diluted 1:500; or with a mouse monoclonal topoisomerase IIβ antibody diluted 1:200; or a monoclonal antibody to human topoisomerase I diluted 1:1000. The chemiluminescence bands on the nitrocellulose membranes were imaged on a ChemiDoc XRS+ imager Bio-Rad (Hercules, CA) or a FluorChem FC2 imager after incubation with appropriate secondary horseradish peroxidase-conjugated antibodies and chemiluminescent reagents.
EPR Experiments.
The EPR experiments were carried out essentially as we previously described on a Bruker EMX (Milton, Canada) spectrometer (Malisza and Hasinoff, 1996). A total of 15 μl hypoxanthine/xanthine oxidase reaction mixture or the K562 cell suspension containing the drugs indicated was added to gas-permeable Teflon tubing, which was then folded at both ends and inserted into a quartz EPR tube open at both ends, and placed in the EPR cavity. Thermostated prepurified nitrogen (400 l/h, 37°C) was passed over the sample in the EPR cavity to achieve hypoxic conditions. For the EPR experiments, five scans were averaged starting 8 minutes after the sample was loaded with the following instrument settings: microwave power 20 mW, modulation frequency 100 kHz, microwave frequency 9.3 GHz, modulation amplitude 2.0 G, 42-second scan time, 1024 data points/scan, magnetic field centered at 3310 G, and a 50 G scan range. The relative concentrations of the semiquinone free radical produced were measured by double integration of the EPR spectra.
Myocyte Isolation, Culture, and Lactate Dehydrogenase Determination.
Ventricular myocytes were isolated from 2- to 3-day-old Sprague-Dawley mixed-sex rats, as described (Hasinoff, 2010; Hasinoff et al., 2013). Briefly, minced ventricles were serially digested with collagenase and trypsin in Dulbecco’s phosphate-buffered saline (pH 7.4)/1% (wt/v) glucose at 37°C in the presence of deoxyribonuclease and preplated in large petri dishes to deplete fibroblasts. The preparation, which was typically >90% viable by trypan blue exclusion, yielded an almost confluent layer of uniformly beating cardiac myocytes by day 2. For the lactate dehydrogenase (LDH) release experiments, the myocyte-rich supernatant was plated on day 0 in 24-well plastic culture dishes (5 × 105 myocytes/well, 750 μl/well) in DF-15. On days 2 and 3, the medium was replaced with 750 μl fresh DF-10. To lower the background LDH levels, on day 4, 24 hours before the drug treatments, the medium was changed to DF-2 and again on day 5 just before the addition of drugs. The animal protocol was approved by the University of Manitoba Animal Care Committee.
Myocytes were treated with either pixantrone, mitoxantrone, or doxorubicin for the times indicated. Starting on day 6 after plating, samples (80 μl) of the myocyte supernatant were collected every 24 hours for 3 days after treatment. The samples were frozen at −80°C and analyzed within 1 week. After the last supernatant sample was taken, the myocytes were lysed with 250 μl 1% (v/v) Triton X-100/2 mM EDTA/1 mM dithiothreitol/0.1 M phosphate buffer (pH 7.8) for 20 minutes at room temperature. The total cellular LDH activity from which the percentage of LDH release was determined from the activity of the lysate plus the activity of the three 80 μl samples previously taken. The LDH activity was determined in quadruplicate in a spectrophotometric kinetic assay in 96-well plate format in a Molecular Devices (Menlo Park, CA) plate reader, as previously described (Hasinoff, 2010; Hasinoff et al., 2013).
Measurement of Mitochondrial Membrane Potential.
K562 cells were treated with various concentrations of pixantrone. The mitochondrial membrane potential sensing dye JC-1 (Life Technologies) (Reers et al., 1995; Hasinoff et al., 2013, 2015) was then loaded into suspended K562 cells (100,000 cells/well in 96-well black plates) by incubating cells with 8 μM JC-1 in HBSS buffer (pH 7.4 with 1.3/0.8 mM Ca2+/Mg2+) at 37°C for 20 minutes, as we previously described (Hasinoff et al., 2013, 2015). The cells were then gently washed with HBSS. The average ratio of the red fluorescence (λEx 544 nm, λEm 590 nm) to the green fluorescence (λEx 485 nm, λEm 520 nm), which is a measure of the mitochondrial membrane potential (Reers et al., 1995 Hasinoff et al., 2013), was determined for cells treated for 2 or 6 hours with various concentrations of pixantrone on a BMG Labtech (Cary, NC) Fluostar Galaxy fluorescence plate reader. The ionophore valinomycin (1 μM), which depolarizes the mitochondrial membrane, and doxorubicin (1.6 μM) were used as positive controls, as we previously described (Hasinoff et al., 2013, 2015). Measurement of the mitochondrial potential in attached myocytes was essentially the same as for K562 cells, except that myocytes were seeded at 125,000 cells/well and analyzed on day 6 after seeding.
Fluorometric Cellular Assay of Oxidation of 2′,7′-Dichlorofluorescin to 2′,7′-Dichlorofluorescein in K562 Cells.
The 2′,7′-dichlorofluorescin (DCFH) diacetate is a nonfluorescent ester compound that when taken up by cells is hydrolyzed by esterases to yield nonpermeable DCFH (O'Malley et al., 2004). Cellular or exogenous oxidants are then able to oxidize reduced DCFH to the fluorescent 2′,7′-dichlorofluorescein (DCF). The loading of K562 cells in HBSS with DCFH diacetate and the intracellular DCF assay (50,000 cells/well) was carried out, as we previously described (Hasinoff et al., 2014), on a Fluostar Galaxy fluorescence plate reader (excitation wavelength of 485 nm and emission wavelength of 520 nm, 30°C) equipped with excitation and emission probes directed to the bottom of the plate. The average rate of fluorescence increase was computed from data in six wells for 4 minutes after addition of the drug. H2O2, which rapidly enters cells and oxidizes DCFH to DCF, was used as a positive control (Hasinoff et al., 2014).
Thermal Denaturation of DNA Assay and Ethidium Bromide Displacement Assay.
Compounds that either intercalate into or bind in the minor groove of DNA stabilize the DNA double helix and increase the temperature at which the DNA denatures or unwinds (McGhee, 1976; Priebe et al., 2001; Zhang et al., 2011). The effect of pixantrone and doxorubicin on the melting temperature (ΔTm) of sonicated calf thymus DNA (6 μg/ml or 9.5 μM in base pairs) was measured in 10 mM Tris-HCl buffer (pH 7.4) in a Cary 1 (Varian, Mississauga, Canada) double-beam spectrophotometer by measuring the absorbance increase at 260 nm upon the application of a temperature ramp of 1°C/min, as we previously described (Zhang et al., 2010, 2011). The maximum of the first derivative of the absorbance-temperature curve was used to obtain the Tm. Doxorubicin, which is a strong DNA intercalator, was used for a comparison (Zhang et al., 2011). Linear least-squares calculated fits of the drug concentration versus ΔTm were used to obtain the slopes of the plots and a relative measure of the strength of their binding.
The fluorometric ethidium bromide displacement assay was carried out essentially as described (Jenkins, 1997; O'Hara et al., 2007) in Tris/NaCl/MgCl2 (10/100/20 mM, pH 7.0) buffer. Calf thymus DNA (16 μg/ml) and pixantrone were incubated for 5 minutes in the dark at room temperature in a 96-well plate, after which time ethidium bromide (1.6 μM) was added. The fluorescence of the solutions was immediately measured in a Fluostar Galaxy fluorescence plate reader using an excitation wavelength of 544 nm and an emission wavelength of 590 nm. The C50, which is the concentration of the drug that results in a 50% decrease in the fluorescence intensity of the ethidium bromide–DNA complex (Jenkins, 1997; O'Hara et al., 2007), was determined from a plot of pixantrone concentration versus fluorescence.
Results
Cell Growth-Inhibitory Effects of Pixantrone on Human Leukemia K562 Cells, Etoposide-Resistant K/VP.5 Cells with a Decreased Level of Topoisomerase IIα, and an ABCB1 (P-glycoprotein)-Overexpressing Efflux Transporter Cell Line.
Cancer cells can acquire resistance to topoisomerase II poisons by lowering their level and/or activity of topoisomerase II (Fattman et al., 1996). We have previously used (Hasinoff et al., 2012; Yadav et al., 2014, 2015) a clonal K562 cell line selected for resistance to etoposide as a screen to determine the ability of compounds to act as topoisomerase II poisons. The K/VP.5 cells were previously determined to be 26-fold resistant to etoposide, and to contain reduced levels of both topoisomerase IIα (sixfold) and topoisomerase IIβ (threefold) (Ritke et al., 1994a, 1994b). In addition, these cells are cross-resistant to other known topoisomerase II poisons, but are not cross-resistant to camptothecin and other nontopoisomerase IIα-targeted drugs (Ritke et al., 1994b). The cell growth-inhibitory effects of pixantrone on human leukemia K562 cells and the etoposide-resistant K/VP.5 cells (Ritke et al., 1994a, 1994b) were assessed by MTS assays (see Materials and Methods) and are shown in Fig. 2A. Pixantrone potently inhibited growth inhibition of K562 cells in the submicromolar range. The etoposide-resistant K/VP.5 cells were 5.7-fold cross-resistant to pixantrone. These results compare with 5.1- and 4.2-fold cross-resistance for doxorubicin and mitoxantrone, respectively, that we determined in a previous study (Hasinoff et al., 2008). The K562 IC50 value for pixantrone of 0.10 μM (Fig. 2A) compares to our previously determined IC50 values for doxorubicin and mitoxantrone of 0.08 and 0.42 μM, respectively (Hasinoff et al., 2008). In separate 72-hour growth-inhibitory assays using direct cell counting, the etoposide-resistant K/VP.5 cells were 21.6-fold cross-resistant to pixantrone, yielding IC50 values of 0.04 ± 0.01 and 0.85 ± 0.05 μM for K562 and K/VP.5 cells, respectively (mean ± S.E.M. from three experiments performed on separate days).
Fig. 2.

Comparison of the growth-inhibitory effects of pixantrone on K562 and K/VP.5 cells with reduced levels of topoisomerase IIα and on parental MDCK and efflux transporter overexpressing drug-resistant MDCK/MDR cell lines. (A) K562 (Δ) and K/VP.5 (○) cells were treated with pixantrone for 72 hours prior to the assessment of growth inhibition. Curve fitting yielded IC50 values of 0.10 μM and 0.56 μM, respectively. (B) MDCK (Δ) and MDCK/MDR (○) cells were treated with pixantrone for 72 hours prior to the assessment of growth inhibition. Curve fitting of absorbance values (n = 3) yielded IC50 values of 0.058 μM and 4.5 μM, respectively. The curved lines were calculated from nonlinear least-squares fits to 4-parameter logistic equations.
To assess whether pixantrone was a substrate for a common drug efflux transporter, the IC50 values for pixantrone were determined in ABCB1 [P-glycoprotein (Pgp)]-overexpressing cell line and in its parental cell line. The pair studied was MDCK cells and its ABCB1-transfected MDCK/MDR derivative (Pastan et al., 1988) (maintained in 0.2 μM colchicine). The growth inhibition curves (Fig. 2B) showed that the MDCK/MDR cells were 77-fold resistant to pixantrone compared with parental MDCK cells. These results are consistent with an earlier study that had likewise shown that pixantrone and mitoxantrone were both highly cross-resistant (50- and 65-fold, respectively) to S180/A10 MDR (Pgp) doxorubicin-resistant cells (Chou et al., 2002).
Relative Cellular Uptake of Pixantrone, Doxorubicin, and Mitoxantrone in K562 Cells.
The relative ability of pixantrone, doxorubicin, and mitoxantrone to be taken up by K562 cells was measured, as described (De Isabella et al., 1995). K562 cells (10 × 106 cells in 1 ml) were incubated for 1 hour at 37°C in a final concentration of 10 μM (10 nmol) each drug, and total accumulation was assessed. Total pixantrone uptake (1.5 nmol) was reduced compared with doxorubicin (6.5 nmol) and mitoxantrone (8.1 nmol) under these experimental conditions. Their relative uptake after 1 hour can be correlated with their calculated distribution coefficients D at pH 7.4 with log D values of −3.2, −0.28, and −1.2 for pixantrone, doxorubicin, and mitoxantrone, respectively. The relatively lower uptake of pixantrone can be partially explained by its highly negative log D value in comparison with those for doxorubicin and mitoxantrone. At pH 7.4, the major pixantrone microspecies is essentially all dicationic, making it less cell permeable compared with monocationic doxorubicin. In addition, although both pixantrone and mitoxantrone are dicationic at pH 7.4, the calculated nonionic partition coefficient log P for pixantrone is 0.0 compared with 1.4 for mitoxantrone. Thus, the larger uptake of mitoxantrone in K562 cells, compared with pixantrone, can be partly explained by mitoxantrone having a more hydrophobic core structure, even though both are present as dicationic species. Also, the decreased cellular accumulation of pixantrone compared with mitoxantrone in K562 cells (5.4-fold) reported in this work compares well to a previous study that found pixantrone uptake in NCI-H187 cells was 12-fold less than for mitoxantrone (De Isabella et al., 1995).
Pixantrone Inhibited Topoisomerase IIα and Topoisomerase IIβ Decatenation Activity, Acted as Topoisomerase IIα and Topoisomerase IIβ Poisons, and Induced DNA Double-Strand Breaks in Cells.
The torsional stress that occurs in DNA during replication and transcription and daughter-strand separation during mitosis can be relieved by topoisomerase II. Topoisomerase II alters DNA topology by catalyzing the passage of an intact DNA double helix through a transient double-strand break made in a second helix (Deweese and Osheroff, 2009; Nitiss, 2009; Pommier and Marchand, 2012; Pommier, 2013). As shown in Fig. 3A, pixantrone, doxorubicin, and etoposide all inhibited the decatenation of kDNA by topoisomerase IIα (upper figure) and topoisomerase IIβ (lower figure). Both pixantrone and doxorubicin were similar in their ability to inhibit the decatenation activity of topoisomerase IIα and topoisomerase IIβ. In these experiments, topoisomerase IIα and topoisomerase IIβ-alone controls were of equal decatenation activity, which was determined by measuring the decatenation activity as a function of the enzyme concentration (results not shown). The decatenation assay is a measure of the ability of compounds to inhibit the catalytic activity only, and is not a measure of whether they acted as topoisomerase II poisons.
Fig. 3.

Effect of pixantrone on the inhibition of the decatenation activity of topoisomerase IIα and topoisomerase IIβ and the topoisomerase IIα- and topoisomerase IIβ-mediated relaxation and cleavage of supercoiled pBR322 plasmid DNA to produce linear DNA. (A) The fluorescent images of the ethidium bromide–containing gel show that in the absence of added drug topoisomerase IIα (upper figure) and topoisomerase IIβ (lower figure) decatenated kDNA to its open circular (OC) and nicked circular (NC) forms. ORI is the gel origin. Topoisomerase IIα or topoisomerase IIβ, as indicated, was present in the reaction mixture for all lanes, except the lane marked kDNA. The 20 μl reaction mixture contained 40 ng kDNA and either 45 ng topoisomerase IIα or 4.5 ng topoisomerase IIβ, as indicated. The separation was carried out on a 1.2% agarose gel containing ethidium bromide only in the gel at 80 V for 0.5 hour. (B) These fluorescent images of ethidium bromide–stained gels show that topoisomerase IIα and topoisomerase IIβ, as indicated, converted supercoiled (SC) pBR322 DNA to relaxed (RLX) DNA. In this assay, the supercoiled DNA runs slightly ahead of the nicked circular (NC) DNA because of the separation conditions. The pixantrone concentration-dependent shift of the relaxed band (RLX) was most likely due to binding of pixantrone to the different forms of DNA and inhibition of catalytic topoisomerase II-mediated strand passage. Topoisomerase IIα or topoisomerase IIβ, as indicated, was present in the reaction mixture in all but the lane marked pBR322. The 20 μl reaction mixture contained 40 ng pBR322 DNA and 60 ng topoisomerase IIα or 6 ng topoisomerase IIβ, as indicated, to achieve equal enzyme activities. The separation was carried out on a 1.2% agarose gel containing ethidium bromide in both the running buffer and the gel at 10 V for 18 hours. The etoposide-positive control produced detectable amounts of linear DNA (LIN) with both topoisomerase IIα and topoisomerase IIβ. Pixantrone treatment induced detectable amounts of topoisomerase IIα-mediated linear DNA. Likewise, pixantrone treatment also produced detectable amounts of topoisomerase IIβ-mediated linear DNA, although to a lesser extent. A small amount of nicked circular DNA (NC), which may arise from strand breakage during isolation, is normally present in pBR322 DNA. The results were typical of experiments carried out in gels either with or without ethidium bromide in the running buffer on 5 different days. (See also Supplemental Figs. 1 and 2.)
Stabilization of the covalent complex by a topoisomerase II-targeted drug can lead to double-strand DNA breaks that are toxic to the cell. Thus, DNA cleavage assay experiments (Burden et al., 2001; Hasinoff et al., 2012; Yadav et al., 2014) were carried out to determine whether pixantrone stabilized the cleavable complex induced by topoisomerase IIα and topoisomerase IIβ. As shown in Fig. 3B, the addition of the etoposide-positive control to reaction mixtures containing supercoiled pBR322 DNA and either topoisomerase IIα or topoisomerase IIβ induced formation of linear pBR322 DNA. Linear DNA was identified by comparison with linear pBR322 DNA produced by action of the restriction enzyme HindIII acting on a single site on pBR322 DNA (data not shown). Pixantrone, acting on topoisomerase IIα, induced a concentration-dependent formation of linear DNA in the 0.01– 0.2 μM concentration range, which indicated that it acted as a topoisomerase IIα poison. Pixantrone, acting on topoisomerase IIβ, also detectably increased formation of linear DNA, although to a lesser extent than with topoisomerase IIα. Pixantrone at 1 and 5 μM almost completely inhibited relaxation of supercoiled DNA for both topoisomerase IIα and topoisomerase IIβ. Under the electrophoresis conditions used (ethidium bromide in both the gel and the running buffer), pBR322-supercoiled DNA ran slightly slower than the linear DNA band and much slower than fully relaxed DNA. In the experiment shown in Supplemental Fig. 1, also indicating less pixantrone-induced DNA cleavage with topoisomerase IIβ compared with topoisomerase IIα, ethidium bromide was included only in the agarose gel. Under these conditions, the supercoiled DNA ran ahead of the linear DNA, but again slower than relaxed DNA. These cleavage experiments were carried out with equal activities of the two enzyme isoforms, determined as described above for the decatenation experiments. Results from additional confirmatory pixantrone-induced specific topoisomerase IIα cleavage experiments run under slightly different separation and reaction conditions are also shown in Supplemental Fig. 2. With increasing pixantrone concentrations (Fig. 3B; Supplemental Figs. 1 and 2), there was a shift from relaxed to supercoiled DNA, indicating an inhibition of catalytic topoisomerase II–induced DNA strand passage, most likely due to pixantrone binding to DNA. Additionally, at higher pixantrone concentrations, there was a decrease in the amount of linear DNA produced (Supplemental Figs. 1 and 2). This most likely occurred because pixantrone bound strongly to DNA and limited formation of topoisomerase II–DNA cleavable complexes, as has been proposed for intercalative drugs such as doxorubicin (Tewey et al., 1984a, 1984b), and as we have seen for strongly DNA-intercalating anthrapyrazoles (Liang et al., 2006). It has also been previously shown that topoisomerase II-mediated pixantrone-induced Simian virus 40 DNA cleavage was reduced above 1 μM drug (De Isabella et al., 1995) and that DNA–protein covalent complexes in HL-60 cells and with purified topoisomerase IIα were reduced at higher pixantrone concentrations (Zwelling et al., 1993).
Because the results of Fig. 3B suggested that pixantrone was more selective in inducing linear DNA formation by acting on topoisomerase IIα than on topoisomerase IIβ, a cellular ICE assay was also used to determine the effects of pixantrone and mitoxantrone for comparison on production of topoisomerase IIα– and topoisomerase IIβ–DNA covalent complexes in K562 cells. Experiments, carried out as we previously described (Hasinoff et al., 2012, 2015; Yadav et al., 2014), showed that pixantrone and mitoxantrone induced concentration-dependent increases in the amount of topoisomerase IIα and topoisomerase IIβ covalently bound to DNA (Fig. 4, A and B). Etoposide, used as a positive control, also increased both topoisomerase IIα– and topoisomerase IIβ–DNA covalent complexes. Etoposide (2 and 5 µM) effects were similar at the level of both topoisomerase II isoforms. Interestingly, pixantrone, relative to its vehicle control, was much less effective in producing topoisomerase IIβ– compared with topoisomerase IIα–DNA covalent complexes (Fig. 4, A and B). Mitoxantrone (1 µM) also was relatively less effective inducing topoisomerase IIβ– compared with topoisomerase IIα–DNA covalent complexes. At higher mitoxantrone concentrations (2, 5, and 10 µM), effects at the level of topoisomerase IIβ were similar to or greater than effects on topoisomerase IIα (Fig. 4, A and B). Taken together, these results, and those in Fig. 3B, indicate that pixantrone selectively targets topoisomerase II and is relatively more specific in stabilizing topoisomerase IIα– compared with topoisomerase IIβ–DNA covalent complexes. Pixantrone did not induce formation of topoisomerase I-covalent complexes (Fig. 4D).
Fig. 4.

Cell-based assays of the ability of various topoisomerase inhibitors to stabilize topoisomerase IIα–, topoisomerase IIβ–DNA, and topoisomerase I–DNA covalent complexes, and to produce DNA double-strand breaks. (A) Chemiluminescent images of Western slot-blot determinations of pixantrone-, mitoxantrone-, and etoposide-induced cellular covalent topoisomerase IIα– and topoisomerase IIβ–DNA cleavage complexes produced in K562 cells determined using an ICE assay. In these experiments, K562 cells were treated with the control vehicle, or with the indicated concentrations of drug for 1 hour, after which isolated genomic DNA was isolated and 2.5 µg DNA from each experimental condition was applied to a slot-blot apparatus for immunoblot analysis (see Materials and Methods). Two representative experiments are shown (experiments 1 and 2). (B) Quantified image analyses of the fold increase (over control) of topoisomerase II–DNA covalent complexes from replicate experiments. Bars from the pixantrone and mitoxantrone results represent the mean from four to five determinations from five replicate experiments performed on separate days. Bars from the etoposide results represent the mean from three to four determinations from four replicate experiments performed on separate days. Error bars represent the S.E.M. * and **, Indicate statistically significant differences in drug activity comparing effects on topoisomerase IIα versus topoisomerase IIβ using an unpaired t test (p < 0.05 and p < 0.01, respectively). (C) Induction of double-strand DNA breaks in K562 cells by pixantrone and the etoposide-positive control, as indicated by formation of γH2AX. K562 cells were treated with etoposide or the concentrations of pixantrone indicated for 4 hours in growth medium, lysed, and subjected to SDS-PAGE and Western blotting. The blots were probed with antibodies to γH2AX and glyceraldehyde phosphate dehydrogenase as a loading control and a chemiluminescent-inducing horseradish peroxidase-conjugated secondary antibody. Results are typical of those found on two different days. (D) Chemiluminescent image of a Western slot-blot determination of cellular covalent topoisomerase I–DNA cleavage complexes produced in K562 cells determined using an ICE assay. In these experiments, K562 cells were treated either with control vehicle, pixantrone, or the camptothecin-positive control for 1 hour.
H2AX is a variant of an H2A core histone that becomes phosphorylated to produce γH2AX in the vicinity of double-strand DNA breaks upon treatment of cells with drugs or ionizing radiation (Pilch et al., 2003). This phosphorylation becomes amplified over megabases of DNA surrounding the break, thus acting as a scaffold for the recruitment of key signaling and repair proteins, and, thus, is an accepted marker of double-strand breaks (Pilch et al., 2003). To determine whether pixantrone could induce double-strand breaks in intact K562 cells, the level of γH2AX was determined by Western blotting with etoposide as the positive control (Burden et al., 2001). Experiments carried out as we previously described (Hasinoff et al., 2012, 2015), and shown in Fig. 4C, indicate that pixantrone and the etoposide-positive control increased the levels of γH2AX in K562 cells. The results of Fig. 4C also show that the γH2AX levels decrease with an increase in the pixantrone concentration, suggesting that intercalation interferes with topoisomerase II-mediated DNA double-strand breaks, as has been demonstrated previously with other intercalating topoisomerase II poisons (Tewey et al., 1984a, 1984b).
Binding of Pixantrone to DNA as Evaluated by the Thermal Denaturation of DNA and in the Ethidium Bromide Displacement Assay.
Because pixantrone is a DNA intercalator (Hazlehurst et al., 1995b), and at higher concentrations limits induction of γH2AX (Fig. 4C) and topoisomerase II-mediated DNA cleavage (Fig. 3B), there is a suggestion of avid DNA binding. The strength of pixantrone–DNA binding, therefore, was determined and compared with that of doxorubicin. The ΔTm of sonicated calf thymus DNA with increasing concentrations of pixantrone and doxorubicin are compared in Fig. 5A. The linear least-squares calculated slope was 3.3-fold larger for pixantrone than it was for doxorubicin, indicating stronger pixantrone DNA binding compared with doxorubicin. We previously showed that mitoxantrone also binds DNA stronger than doxorubicin, as evidenced by a ΔTm at 2 μM that is 1.4-fold higher than for doxorubicin (Liang et al., 2006).
Fig. 5.

Pixantrone strongly binds to DNA, as shown by an increase in the DNA ΔTm, and by the fluorometric ethidium bromide displacement assay. (A) Concentration dependence of ΔTm for pixantrone and doxorubicin binding to DNA. The assay buffer (pH 7.5) contained 10 mM Tris. The straight lines were linear least-squares calculated fits to the data (n = 2–3). Error bars are average deviations where n = 2. (B) Average changes (n = 4) in the fluorescence at 590 nm ethidium bromide bound to calf thymus DNA are plotted as a function of the pixantrone concentration. The reaction mixture contained calf thymus DNA (16 μg/ml) and ethidium bromide (1.6 μM) at 20°C. The assay buffer (pH 7.2) contained 10 mM Tris, 100 mM NaCl, and 20 mM MgCl2.
To determine the apparent association constant Kapp for pixantrone binding to DNA, an ethidium bromide displacement assay was used, as described (Jenkins, 1997; O'Hara et al., 2007). Ethidium bromide fluoresces strongly when bound to DNA, but only weakly fluoresces when free. Thus, the displacement of ethidium bromide by pixantrone was measured fluorometrically. As shown in Fig. 5B, pixantrone displaced ethidium bromide from DNA and caused a 50% reduction in fluorescence at a concentration of 1.1 μM. The apparent association constant, Kapp, for pixantrone binding to DNA was calculated from the formula Kapp = KEtBrCEtBr/C50 (Jenkins, 1997) to be 1.4 × 107 M−1, which is some 2.6-fold larger than the value of 5.2 × 106 M−1 for doxorubicin under comparable conditions (Chaires et al., 1996). In this formula, CEtB is the concentration of ethidium bromide (1.6 µM) and C50 is the concentration of pixantrone that reduced the fluorescence of ethidium bromide by 50% (1.1 µM); and KEtBr is the association constant for ethidium bromide binding to DNA (9.5 × 106 M−1) (Jenkins, 1997).
Pixantrone Semiquinone Radical Formation in a Xanthine Oxidase/Hypoxanthine-Reducing System and in a K562 Cell Suspension.
The xanthine oxidase/hypoxanthine-reducing system has been previously used to generate and detect anthracycline semiquinone free radical species under hypoxic conditions (Kalyanaraman et al., 1991; Malisza and Hasinoff, 1996). The results in Fig. 6A show that both pixantrone and doxorubicin produced semiquinone free radical species in this reducing system, whereas mitoxantrone did not. The peak-to-peak line widths (ΔHpp) of the spectra in Fig. 6A were 3.2 and 7.2 G for doxorubicin and pixantrone, respectively. The ΔHpp for doxorubicin can be compared with previously measured values of 3.25 G (Kalyanaraman et al., 1991; Malisza and Hasinoff, 1996). Double integration of the pixantrone and doxorubicin spectra was carried out to determine the relative concentrations of the semiquinones produced and showed that pixantrone produced 1.8-fold more semiquinone than did doxorubicin. The lack of semiquinone formation by mitoxantrone, relative to pixantrone and doxorubicin, using a xanthine oxidase/hypoxanthine-reducing system (Fig. 6A), can be explained by the relatively less negative half-wave reduction potentials, E1/2 of −0.54 V and −0.6 V for pixantrone and doxorubicin, respectively (Nguyen and Gutierrez, 1990; De Isabella et al., 1995), compared with an E1/2 of −0.74 V for mitoxantrone (Nguyen and Gutierrez, 1990), which makes mitoxantrone more difficult to reduce with biologic reductants.
Fig. 6.

EPR spectra obtained under hypoxic conditions showing the reduction of pixantrone and doxorubicin to their semiquinone free radical species in a hypoxanthine/xanthine oxidase-reducing system (A) and in a K562 cell suspension (B). (a–d) EPR spectra produced by the control, pixantrone, mitoxantrone, and doxorubicin, respectively, in the reaction system containing the xanthine oxidase/hypoxanthine-reducing system. The complete reaction system contained the drug indicated (1 mM), xanthine oxidase (0.1 U/ml), and hypoxanthine (400 μM) in 50 mM Tris buffer (pH 7.4) at 37°C. (e–h) EPR spectra produced by the control, pixantrone, mitoxantrone, and doxorubicin, respectively. The K562 cell suspension (4 × 108 cell/ml) contained the drugs indicated (1 mM) in Dulbecco’s phosphate-buffered saline (pH 7.4)/1% glucose buffer. The spectra are an average of five scans recorded over 3.5 minutes shortly after the components were assembled.
We previously measured semiquinone formation by doxorubicin and other anthracyclines in a suspension of Chinese hamster ovary (CHO) cells under hypoxic conditions (Malisza and Hasinoff, 1996). We also previously showed that mitoxantrone did not produce a semiquinone in a CHO cell suspension (Malisza and Hasinoff, 1996). As shown in Fig. 6B, the addition of either pixantrone (1 mM) or mitoxantrone (1 mM) to a K562 cell suspension did not produce an EPR signal compared with control cells. In contrast, the addition of doxorubicin (1 mM) to the K562 cell suspension did produce a free radical EPR signal under hypoxic conditions. The doxorubicin-derived EPR signal showed appreciable asymmetry, as we showed before in CHO cells, and is suggestive of a partly immobilized free radical species within the cell (Malisza and Hasinoff, 1996).
Effect of Pixantrone on the Oxidation of DCFH to DCF, and on the Mitochondrial Membrane Potential in K562 Cells.
To investigate whether pixantrone and doxorubicin produced oxidative stress in K562 cells, the oxidation of DCFH loaded into K562 cells was followed in a fluorescence plate reader. We previously showed that 10 μM doxorubicin increased DCF fluorescence in neonatal rat myocytes (Hasinoff et al., 2003). As shown in Fig. 7A, treatment of K562 cells with pixantrone did not significantly increase the rate of oxidation of DCFH to DCF relative to the control. However, doxorubicin did significantly increase the rate of DCF formation, but only at the highest concentration tested (10 μM).
Fig. 7.

Effect of pixantrone and doxorubicin treatment on oxidation of DCFH to DCF in K562 cells and in myocytes and the effect of pixantrone on the mitochondrial membrane potential in K562 cells. (A) As measured by the increase in the DCF fluorescence doxorubicin only slightly increased the rate of oxidation to DCF in K562 cells at the highest concentration tested. The H2O2 positive control-treated K562 cells all showed significant increases in DCF fluorescence. The rate of change in DCF fluorescence was measured for 4 minutes directly after the addition of either pixantrone or doxorubicin or H2O2. The results are an average of six wells. (B) Effect of pixantrone treatment on the mitochondrial membrane potential of K562 cells that were loaded with the mitochondrial membrane potential sensing dye JC-1 measured 2 hours and 6 hours after drug treatment. Valinomycin (Val, 1 μM, 3 hours) and doxorubicin (Dox, 1.6 μM, 3 hours) were used as positive controls. Both valinomycin and doxorubicin significantly, and strongly, reduced the mitochondrial membrane potential. Treatment with pixantrone progressively and significantly reduced the mitochondrial membrane potential at 2 hours and 6 hours. The results are an average of eight wells and are typical of experiments carried out on 2 different days. (C) Effect of pixantrone and doxorubicin treatment on the mitochondrial membrane potential of attached cardiac myocytes 6 hours after drug treatment. Treatment with pixantrone and doxorubicin both progressively decreased the mitochondrial membrane potential. The JC-1 results are an average of eight wells and were typical of experiments carried out on 2 different days. The mitochondrial membrane potential was measured by the ratio of the red fluorescence (λEx 544 nm, λEm 590 nm) to the green fluorescence (λEx 485 nm, λEm 520 nm).
To determine whether pixantrone treatment resulted in mitochondrial damage to K562 cells, the effect on mitochondrial membrane potential was determined using the ratiometric mitochondrial membrane potential sensing dye JC-1 (Reers et al., 1995; Hasinoff et al., 2013). K562 cells were treated with pixantrone for either 2 or 6 hours. Figure 7B results show that pixantrone significantly decreased the mitochondrial membrane potential relative to the control in a concentration-dependent manner after a 6-hour treatment. A 2-hour treatment was relatively ineffective, except at the highest pixantrone concentration (5 μM). Pixantrone was much less effective in reducing the mitochondrial membrane potential than was doxorubicin (1.6 µM, 3 hours) or the valinomycin (1 µM, 3 hours) positive control (Fig. 7B).
Comparison of the Effect of Pixantrone and Doxorubicin on the Mitochondrial Membrane Potential in Myocytes.
We previously showed that doxorubicin reduced the mitochondrial membrane potential in myocytes, which may be an important mechanism for its cardiotoxicity (Hasinoff et al., 2003; Wallace, 2003; Ichikawa et al., 2014). Cationic compounds may be preferentially taken up by mitochondria because of their negative mitochondrial membrane potential. Figure 7C results demonstrate that, after 6 hours, both pixantrone and doxorubicin significantly, and in a concentration-dependent manner, reduced the myocyte mitochondrial membrane potential relative to the control, with doxorubicin eliciting greater effects than pixantrone. As can be seen in Fig. 7C, a concentration of 0.2 µM doxorubicin was approximately equipotent to 5 μM pixantrone in reducing the mitochondrial membrane potential. A t test comparing the effect on the mitochondrial membrane potential at these two concentrations showed that they were not significantly different. A t test carried out comparing 2 μM pixantrone and a 2 μM doxorubicin treatment, and 5 μM pixantrone and a 5 μM doxorubicin treatment showed that the mitochondrial membrane potential was significantly reduced (p = 0.0002 and 0.0005, respectively) in both cases. This experiment was also repeated at the same drug concentrations, but with a 2-hour treatment. These 2-hour results (data not shown) indicated no significant reduction in mitochondrial membrane potential for pixantrone at any concentration, and a significant effect of doxorubicin only at 1 and 5 μM.
Comparison of Pixantrone-, Mitoxantrone-, and Doxorubicin-Induced Damage to Neonatal Cardiac Myocytes as Measured by Cumulative Percentage of LDH Release.
As previously described (Hasinoff and Patel, 2010; Hasinoff et al., 2013), we used percentage of LDH release to measure myocyte damage, which is a widely used measure of drug-induced damage to myocytes (Adderley and Fitzgerald, 1999; Schroeder et al., 2008). Using this LDH release assay, we compared the ability of continuous treatment with pixantrone or doxorubicin to damage myocytes starting 5 days after isolation (Fig. 8) by which time the myocytes would be essentially nonproliferating (Li et al., 1996). Pixantrone showed a dramatic shift to the right of the concentration-response curve compared with doxorubicin and mitoxantrone, indicating much less myocyte toxicity consistent with published preclinical and clinical literature (Beggiolin et al., 2001; Cavalletti et al., 2007; Mukherji and Pettengell, 2010; Longo et al., 2014; Boyle and Morschhauser, 2015). In these experiments, the doxorubicin concentrations that induced myocyte damage were less than doxorubicin plasma concentrations (12 μM) seen clinically at the end of a 60 mg/m2 infusion period (Hochster et al., 1992). For comparison, maximum pixantrone plasma concentrations of 1.2 μM are seen after a 37.5 mg/m2 infusion (Faivre et al., 2001).
Fig. 8.

Comparison of pixantrone-, mitoxantrone-, and doxorubicin-induced damage to neonatal rat cardiac myocytes as measured by cumulative percentage of LDH release. Effect on percentage of LDH release from myocytes treated at the concentrations of pixantrone (○), mitoxantrone (Δ), and doxorubicin (●) indicated 72 hours after drug treatment. Data at 24 and 48 hours were also obtained, but were not plotted for clarity. The doxorubicin and mitoxantrone treatments significantly increased percentage of LDH release over untreated controls at all concentrations 0.2 μM and greater. The pixantrone treatment significantly increased percentage of LDH release over untreated controls only at all concentrations 10 μM and greater. Doxorubicin, mitoxantrone, and pixantrone are equipotent (∼15% LDH release) in inducing myocyte damage at 0.8, 1.0, and 10 μM, respectively. The results are an average of percentage of LDH release in a minimum of four wells. Where error bars are not seen, they are smaller than the size of the symbol. The results are typical of experiments carried out on two separate myocyte isolations.
Spectrophotometric Titration of Pixantrone with Fe3+ and Cu2+.
Spectrophotometric titration experiments, as we described (Martin et al., 2009), were carried out to determine whether pixantrone could bind either Fe3+ or Cu2+. Spectral scanning (250–800 nm) of solutions containing FeCl3 (5–250 μM final concentrations) and pixantrone (30 μM final concentration) added to Tris/KCl buffer (50 mM/150 mM, pH 7.4, 25°C) showed no significant spectral changes (data not shown). This result suggested that Fe3+ did not bind pixantrone, at least over this concentration range. However, when pixantrone was titrated with CuCl2, changes in the absorbance spectrum were seen that were consistent with Cu2+ forming a complex with this agent (Fig. 9A). As shown in Fig. 9B, the absorbance at the 642 nm peak maximum systematically decreased with the addition of Cu2+. A least-squares fit of the two linear segments of the plot of Cu2+ concentration versus absorbance (Fig. 9B) intersected at 40 ± 4 μM or at a Cu2+:pixantrone ratio of approximately 1.33:1. This result suggests that more than one Cu2+ may bind to pixantrone. The fact that the plot had significant curvature in the equivalence region was consistent with the formation of a relatively weak complex, and the lack of well-defined isosbestic points suggests that there were more than two species present in solution. Thus, the stoichiometry of the Cu(II) –pixantrone complex or complexes formed could not be reliably determined. Cu2+ typically forms strong complexes with polyamino-containing compounds such as triethylenetetramine (Crisponi et al., 2010), and thus, it is not unexpected that the polyamine-containing pixantrone formed a complex with Cu2+. The formation of a weak complex between Cu2+ and pixantrone is unlikely to be pharmacologically significant, however, as the concentration of free or loosely bound copper in plasma or in cells is tightly controlled because of its potential toxicity. Because Cu2+ binds pixantrone with relatively low affinity, it would be unlikely to be able to displace copper from high-affinity (subfemtomolar) copper–protein complexes (Crisponi et al., 2010) and form a potentially redox-active copper complex.
Fig. 9.

Spectrophotometric titration of pixantrone by CuCl2. (A) UV-vis spectral changes observed when microliter amounts of CuCl2 were added to 30 μM pixantrone in Tris/KCl (50/150 mM, pH 7.4) buffer at 25°C in a 1-cm spectrophotometer cell. The peaks at 597 and 642 nm progressively decreased with increasing concentrations of CuCl2 added (0, 10, 20, 30, 40, 50, 60, 70, 80, 90, 110, and 130 μM, respectively), indicating complex formation. (B) Spectrophotometric titration of pixantrone by CuCl2 at 642 nm. The intersection of the least-squares calculated straight lines occurred at 40 ± 4 μM, which was consistent with complex formation between Cu2+ and pixantrone. The curvature in the plot indicates that a relatively weak complex was formed.
Discussion
Growth inhibition experiments shown in Fig. 2A, demonstrating pixantrone cross-resistance in the etoposide-resistant K/VP.5 cell line, which contains reduced cellular topoisomerase II, strongly suggest that it targets this enzyme. In a cleavage assay with topoisomerase IIα and topoisomerase IIβ, pixantrone induced linear DNA formation (Fig. 3B), consistent with its acting as a topoisomerase II poison. Pixantrone also induced DNA double-strand breaks in K562 cells, as determined by the increase in γH2AX levels (Fig. 4C). This result was also consistent with its targeting topoisomerase II, although it was not as potent as etoposide at a nearly equivalent concentration. The decrease in γH2AX levels with increasing pixantrone concentration is also consistent with DNA intercalation limiting cleavable complex formation (Tewey et al., 1984a, 1984b), and corresponds to results in Fig. 3B, in which higher concentrations of pixantrone limited DNA cleavage activity.
The cellular ICE assay in K562 cells (Fig. 4, A and B), which express similar levels of both topoisomerase IIα and topoisomerase IIβ (Padget et al., 2000), showed that pixantrone, mitoxantrone, and etoposide all increased topoisomerase IIα and topoisomerase IIβ covalently bound to DNA. Consistent with these results, etoposide and mitoxantrone have been shown to target both topoisomerase IIα and topoisomerase IIβ (Willmore et al., 1998; Errington et al., 2004; Mariani et al., 2015). An earlier study had suggested topoisomerase IIβ as an important target for mitoxantrone (Errington et al., 1999). Whereas these three drugs targeted both topoisomerase II isoforms, pixantrone’s effects (1–10 μM) after a 1-hour incubation were more selective than either mitoxantrone or etoposide for topoisomerase IIα compared with topoisomerase IIβ. However, whereas mitoxantrone showed good selectivity for topoisomerase IIα compared with topoisomerase IIβ, this selectivity was lost at higher mitoxantrone concentrations and, in fact, exhibited greater activity against topoisomerase IIβ compared with topoisomerase IIα at 10 µM, consistent with evidence that secondary malignancies, known to be associated with use of mitoxantrone, are mediated by targeting topoisomerase IIβ (Pendleton et al., 2014). Pixantrone, by comparison, showed good selectivity for topoisomerase IIα over topoisomerase IIα over all concentrations used. Consequently, the reduced cardiotoxicity of pixantrone may be due to pixantrone’s attenuated effects at the level of topoisomerase IIβ, in accord with the suggestion that anthracycline-induced cardiotoxicity is due to topoisomerase IIβ-mediated responses to DNA damage as well as oxidative damage (Zhang et al., 2012; Vejpongsa and Yeh, 2014). In addition, it has been shown that topoisomerase IIβ levels are much higher than topoisomerase IIα levels in the adult nonproliferating murine heart (Capranico et al., 1992), supporting the idea that topoisomerase IIβ is the targeted isoform responsible for cardiotoxicity.
Pixantrone was shown to be a stronger DNA-intercalating agent than doxorubicin, as indicated by its ability to increase ΔTm (Fig. 5A) and from its ability to displace ethidium bromide from DNA (Fig. 5B). The X-ray structure of the pixantrone analogs mitoxantrone (Protein Data Bank ID: 4G0V) and ametantrone (Protein Data Bank ID: 4G0W) complexed to a cleaved DNA–topoisomerase IIβ ternary complex has recently been determined (Wu et al., 2013). In these structures, two molecules of mitoxantrone and ametantrone intercalate into the cleaved DNA separated by four base pairs, similar to the analogous X-ray structure of etoposide (Protein Data Bank ID: 3QX3) in the ternary complex (Wu et al., 2011). Given the structural similarity of pixantrone to mitoxantrone and ametantrone, it is likely that pixantrone exerts its anticancer activity through stabilization of a similar ternary complex, but more selectively with topoisomerase IIα. The high-fold cross-resistance that pixantrone displayed to an ABCB1-transfected overexpressing MDCK/MDR (Pgp) cell line (Fig. 2B) suggests that the clinical use of pixantrone might be expected to result in transport-related resistance.
We also showed that pixantrone is capable of producing semiquinone free radicals in an enzymatic reducing system, and thus has the potential to produce damaging reactive oxygen species such as H2O2. However, in a K562 cell suspension, pixantrone produced no detectable semiquinone (Fig. 6B), whereas doxorubicin generated a distinct semiquinone radical signal. The reason for this lack of semiquinone formation in K562 cells, compared with doxorubicin, was most likely due, in part, to decreased (4.3-fold) pixantrone cell accumulation over the short duration of these experiments. These results are consistent with the much decreased uptake of dicationic pixantrone into cells compared with monocationic doxorubicin. Our EPR results, which were obtained under hypoxic conditions, to enhance our ability to observe EPR semiquinone signals, are at odds with a study in a more complex human myocardial strip model in which it was concluded that pixantrone was virtually resistant to one-electron reduction (Salvatorelli et al., 2013). Our EPR results in cells are also consistent with our previous study of the comparative semiquinone formation of doxorubicin, epirubicin, idarubicin, and daunorubicin in CHO cell suspensions, and their fluorescence-determined uptake in CHO cells, in which we showed that anthracycline uptake paralleled formation of the semiquinone signal (Malisza and Hasinoff, 1996).
Compared with doxorubicin, pixantrone was not effective in producing reactive oxygen species in K562 cells, as measured by oxidation of DCFH to DCF (Fig. 7A). These results are consistent with the EPR results that showed that it produced no detectable semiquinone in K562 cells (Fig. 6B). Again, these results are consistent with a decreased uptake of pixantrone, limiting its ability to produce oxidizing species such as H2O2 through redox cycling of the semiquinone. Although pixantrone decreased the mitochondrial membrane potential of K562 cells in a concentration- and time-dependent manner, it was much less effective than doxorubicin in this regard (Fig. 7B).
The ability of pixantrone and doxorubicin to reduce the mitochondrial membrane potential was compared in myocytes (Fig. 7C), as we and others previously showed that this may be a mechanism by which doxorubicin exerts its cardiotoxicity (Hasinoff et al., 2003; Wallace, 2003; Ichikawa et al., 2014). Whereas both drugs reduced the mitochondrial membrane potential in a concentration-dependent manner, pixantrone was much less effective than doxorubicin.
A comparison of the ability of doxorubicin, mitoxantrone, and pixantrone to damage myocytes as evaluated by percentage of LDH release (Fig. 8) clearly shows that pixantrone was much less toxic toward myocytes than either doxorubicin or mitoxantrone, a result in line with its reduced preclinical cardiotoxicity in mice (Beggiolin et al., 2001; Cavalletti et al., 2007; Longo et al., 2014).
No spectrophotometric evidence for the formation of a complex between Fe3+ and pixantrone was detected. The inability of pixantrone to bind Fe3+, unlike doxorubicin and mitoxantrone (Herman et al., 1997), was probably due to its lack of a hydroquinone functionality (Fig. 1). Thus, the relatively low myocyte toxicity displayed by pixantrone (Fig. 8) may be due, in part, to its inability to generate HO· and other reactive oxygen species through iron-based Fenton chemistry (Malisza and Hasinoff, 1995; Halliwell and Gutteridge, 1999). In an EPR spin-trapping study with a xanthine oxidase/hypoxanthine-reducing enzymatic system, we previously showed that both Fe3+–doxorubicin and Fe3+–mitoxantrone complexes produced HO·, although Fe3+–mitoxantrone was eightfold less potent than the Fe3+–doxorubicin complex in this regard (Malisza and Hasinoff, 1995). This decreased HO· formation from the Fe3+–mitoxantrone complex compared with the Fe3+–doxorubicin complex is in accord with the reduced ability of mitoxantrone to damage myocytes compared with doxorubicin (Fig. 8). These previous results with mitoxantrone and doxorubicin strengthen the contention that the lack of pixantrone binding with Fe3+ and its inability to generate reactive oxygen species are also responsible, in part, for its low cardiomyocyte toxicity.
In summary, the results of this study have shown that pixantrone most likely exerted its cancer cell growth-inhibitory (and cytotoxic) effects through targeting of topoisomerase II. Both the in vitro cleavage and the cellular ICE assay results were consistent with pixantrone selectively targeting topoisomerase IIα over topoisomerase IIβ. The ability of pixantrone to induce linear DNA formation in a cleavage assay; to induce topoisomerase II-covalent complexes in an ICE assay; to induce DNA double-strand breaks in K562 cells; and to show cross-resistance in K562 cells with reduced levels of topoisomerase II isoforms are all consistent with this conclusion.
Additionally, this study has shown that pixantrone was much less potent than doxorubicin in damaging cardiac myocytes, as measured by its ability to both reduce the myocyte mitochondrial membrane potential and induce LDH release. Pixantrone may, in part, be less damaging than either doxorubicin or mitoxantrone due to its reduced cellular uptake, thereby limiting its ability to form redox-active semiquinone radicals and generate damaging reactive oxygen species. In addition, our results also suggest that another important reason for the lack of cardiotoxicity of pixantrone may be that, unlike doxorubicin and mitoxantrone, pixantrone did not form a complex with Fe3+. Thus, pixantrone would not have the potential to directly generate damaging HO· through a Fenton reaction (Malisza and Hasinoff, 1995; Halliwell and Gutteridge, 1999). Lastly, pixantrone may also exhibit reduced cardiotoxicity because it selectively targets topoisomerase IIα over topoisomerase IIβ, the latter form of which predominates in nonproliferating heart cells. Continuing studies will focus on characterizing the DNA-damaging mechanisms of pixantrone and its cellular pharmacokinetic and pharmacodynamic properties that impact on its clinical efficacy.
Supplementary Material
Abbreviations
- CHO
Chinese hamster ovary
- DCF
2′,7′-dichlorofluorescein
- DCFH
2′,7′-dichlorofluorescin
- EPR
electron paramagnetic resonance
- HBSS
Hanks’ balanced salt solution
- HO·
hydroxyl radical
- ICE
cellular immunodetection of complex of enzyme-to-DNA assay
- kDNA
kinetoplast DNA
- LDH
lactate dehydrogenase
- MDCK
Madin Darby canine kidney
- MTS
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- NHL
non-Hodgkin lymphoma
- Pgp
P-glycoprotein
- ΔTm
melting temperature
Authorship Contributions
Participated in research design: Hasinoff, Wu, Patel, Kanagasabai, Yalowich.
Conducted experiments: Wu, Patel, Karmahapatra, Kanagasabai.
Performed data analysis: Hasinoff, Wu, Kanagasabai, Karmahapatra, Yalowich.
Wrote or contributed to the writing of the manuscript: Hasinoff, Kanagasabai, Yalowich.
Footnotes
This work was supported by the Canadian Institutes of Health Research [Grant MOP13748], a Canada Research Chairs Program grant, and a Canada Research Chair in Drug Development grant to (B.B.H.), and National Institutes of Health [Grant CA090787; to J.C.Y.].
The authors declare no competing financial interests. The funding sources had no involvement in the study design; in the collection, analysis, and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
 This article has supplemental material available at (jpet.aspetjournals.org).
This article has supplemental material available at (jpet.aspetjournals.org).
References
- Adderley SR, Fitzgerald DJ. (1999) Oxidative damage of cardiomyocytes is limited by extracellular regulated kinases 1/2-mediated induction of cyclooxygenase-2. J Biol Chem 274:5038–5046. [DOI] [PubMed] [Google Scholar]
- Azarova AM, Lyu YL, Lin CP, Tsai YC, Lau JY, Wang JC, Liu LF. (2007) Roles of DNA topoisomerase II isozymes in chemotherapy and secondary malignancies. Proc Natl Acad Sci USA 104:11014–11019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeharry N, Di Rora AG, Smith MR, Yen TJ. (2015) Pixantrone induces cell death through mitotic perturbations and subsequent aberrant cell divisions. Cancer Biol Ther 16:1397–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beggiolin G, Crippa L, Menta E, Manzotti C, Cavalletti E, Pezzoni G, Torriani D, Randisi E, Cavagnoli R, Sala F, et al. (2001) Bbr 2778, an aza-anthracenedione endowed with preclinical anticancer activity and lack of delayed cardiotoxicity. Tumori 87:407–416. [DOI] [PubMed] [Google Scholar]
- Boyle EM, Morschhauser F. (2015) Pixantrone: a novel anthracycline-like drug for the treatment of non-Hodgkin lymphoma. Expert Opin Drug Saf 14:601–607. [DOI] [PubMed] [Google Scholar]
- Burden DA, Froelich-Ammon SJ, Osheroff N. (2001) Topoisomerase II-mediated cleavage of plasmid DNA. Methods Mol Biol 95:283–289. [DOI] [PubMed] [Google Scholar]
- Capranico G, Tinelli S, Austin CA, Fisher ML, Zunino F. (1992) Different patterns of gene expression of topoisomerase II isoforms in differentiated tissues during murine development. Biochim Biophys Acta 1132:43–48. [DOI] [PubMed] [Google Scholar]
- Cavalletti E, Crippa L, Mainardi P, Oggioni N, Cavagnoli R, Bellini O, Sala F. (2007) Pixantrone (BBR 2778) has reduced cardiotoxic potential in mice pretreated with doxorubicin: comparative studies against doxorubicin and mitoxantrone. Invest New Drugs 25:187–195. [DOI] [PubMed] [Google Scholar]
- Chaires JB, Satyanarayana S, Suh D, Fokt I, Przewloka T, Priebe W. (1996) Parsing the free energy of anthracycline antibiotic binding to DNA. Biochemistry 35:2047–2053. [DOI] [PubMed] [Google Scholar]
- Chou KM, Krapcho AP, Horn D, Hacker M. (2002) Characterization of anthracenediones and their photoaffinity analogs. Biochem Pharmacol 63:1143–1147. [DOI] [PubMed] [Google Scholar]
- Cowell IG, Sondka Z, Smith K, Lee KC, Manville CM, Sidorczuk-Lesthuruge M, Rance HA, Padget K, Jackson GH, Adachi N, et al. (2012) Model for MLL translocations in therapy-related leukemia involving topoisomerase IIβ-mediated DNA strand breaks and gene proximity. Proc Natl Acad Sci USA 109:8989–8994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisponi G, Nurchi VM, Fanni D, Gerosa C, Nemolato S, Faa G. (2010) Copper-related diseases: from chemistry to molecular pathology. Coord Chem Rev 254:876–889. [Google Scholar]
- De Isabella P, Palumbo M, Sissi C, Capranico G, Carenini N, Menta E, Oliva A, Spinelli S, Krapcho AP, Giuliani FC, et al. (1995) Topoisomerase II DNA cleavage stimulation, DNA binding activity, cytotoxicity, and physico-chemical properties of 2-aza- and 2-aza-oxide-anthracenedione derivatives. Mol Pharmacol 48:30–38. [PubMed] [Google Scholar]
- Deweese JE, Osheroff N. (2009) The DNA cleavage reaction of topoisomerase II: wolf in sheep’s clothing. Nucleic Acids Res 37:738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errington F, Willmore E, Leontiou C, Tilby MJ, Austin CA. (2004) Differences in the longevity of topo IIα and topo IIβ drug-stabilized cleavable complexes and the relationship to drug sensitivity. Cancer Chemother Pharmacol 53:155–162. [DOI] [PubMed] [Google Scholar]
- Errington F, Willmore E, Tilby MJ, Li L, Li G, Li W, Baguley BC, Austin CA. (1999) Murine transgenic cells lacking DNA topoisomerase IIβ are resistant to acridines and mitoxantrone: analysis of cytotoxicity and cleavable complex formation. Mol Pharmacol 56:1309–1316. [DOI] [PubMed] [Google Scholar]
- Faivre S, Raymond E, Boige V, Gatineau M, Buthaut X, Rixe O, Bernareggi A, Camboni G, Armand JP. (2001) A phase I and pharmacokinetic study of the novel aza-anthracenedione compound BBR 2778 in patients with advanced solid malignancies. Clin Cancer Res 7:43–50. [PubMed] [Google Scholar]
- Fattman CL, Allan WP, Hasinoff BB, Yalowich JC. (1996) Collateral sensitivity to the bisdioxopiperazine dexrazoxane (ICRF-187) in etoposide (VP-16)-resistant human leukemia K562 cells. Biochem Pharmacol 52:635–642. [DOI] [PubMed] [Google Scholar]
- Gewirtz DA. (1999) A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol 57:727–741. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JMC. (1999) Free Radicals in Biology and Medicine, Clarendon Press, Oxford. [Google Scholar]
- Hasinoff BB. (2010) The cardiotoxicity and myocyte damage caused by small molecule anticancer tyrosine kinase inhibitors is correlated with lack of target specificity. Toxicol Appl Pharmacol 244:190–195. [DOI] [PubMed] [Google Scholar]
- Hasinoff BB, Liang H, Wu X, Guziec LJ, Guziec FS, Jr, Marshall K, Yalowich JC. (2008) The structure-based design, synthesis and biological evaluation of DNA-binding bisintercalating bisanthrapyrazole anticancer compounds. Bioorg Med Chem 16:3959–3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasinoff BB, Patel D. (2010) The lack of target specificity of small molecule anticancer kinase inhibitors is correlated with their ability to damage myocytes in vitro. Toxicol Appl Pharmacol 249:132–139. [DOI] [PubMed] [Google Scholar]
- Hasinoff BB, Patel D, Wu X. (2013) The dual-targeted HER1/HER2 tyrosine kinase inhibitor lapatinib strongly potentiates the cardiac myocyte-damaging effects of doxorubicin. Cardiovasc Toxicol 13:33–47. [DOI] [PubMed] [Google Scholar]
- Hasinoff BB, Schnabl KL, Marusak RA, Patel D, Huebner E. (2003) Dexrazoxane (ICRF-187) protects cardiac myocytes against doxorubicin by preventing damage to mitochondria. Cardiovasc Toxicol 3:89–99. [DOI] [PubMed] [Google Scholar]
- Hasinoff BB, Wu X, Krokhin OV, Ens W, Standing KG, Nitiss JL, Sivaram T, Giorgianni A, Yang S, Jiang Y, et al. (2005) Biochemical and proteomics approaches to characterize topoisomerase IIα cysteines and DNA as targets responsible for cisplatin-induced inhibition of topoisomerase IIα. Mol Pharmacol 67:937–947. [DOI] [PubMed] [Google Scholar]
- Hasinoff BB, Wu X, Nitiss JL, Kanagasabai R, Yalowich JC. (2012) The anticancer multi-kinase inhibitor dovitinib also targets topoisomerase I and topoisomerase II. Biochem Pharmacol 84:1617–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasinoff BB, Wu X, Yadav AA, Patel D, Zhang H, Wang D-S, Chen Z-S, Yalowich JC. (2015) Cellular mechanisms of the cytotoxicity of the anticancer drug elesclomol and its complex with Cu(II). Biochem Pharmacol 93:266–276. [DOI] [PubMed] [Google Scholar]
- Hasinoff BB, Yadav AA, Patel D, Wu X. (2014) The cytotoxicity of the anticancer drug elesclomol is due to oxidative stress indirectly mediated through its complex with Cu(II). J Inorg Biochem 137:22–30. [DOI] [PubMed] [Google Scholar]
- Hazlehurst LA, Krapcho AP, Hacker MP. (1995a) Comparison of aza-anthracenedione-induced DNA damage and cytotoxicity in experimental tumor cells. Biochem Pharmacol 50:1087–1094. [DOI] [PubMed] [Google Scholar]
- Hazlehurst LA, Krapcho AP, Hacker MP. (1995b) Correlation of DNA reactivity and cytotoxicity of a new class of anticancer agents: aza-anthracenediones. Cancer Lett 91:115–124. [DOI] [PubMed] [Google Scholar]
- Herman EH, Knapton A, Rosen E, Thompson K, Rosenzweig B, Estis J, Agee S, Lu QA, Todd JA, Lipshultz S, et al. (2011) A multifaceted evaluation of imatinib-induced cardiotoxicity in the rat. Toxicol Pathol 39:1091–1106. [DOI] [PubMed] [Google Scholar]
- Herman EH, Zhang J, Hasinoff BB, Clark JRJ, Jr, Ferrans VJ. (1997) Comparison of the structural changes induced by doxorubicin and mitoxantrone in the heart, kidney and intestine and characterization of the Fe(III)-mitoxantrone complex. J Mol Cell Cardiol 29:2415–2430. [DOI] [PubMed] [Google Scholar]
- Hochster H, Liebes L, Wadler S, Oratz R, Wernz JC, Meyers M, Green M, Blum RH, Speyer JL. (1992) Pharmacokinetics of the cardioprotector ADR-529 (ICRF-187) in escalating doses combined with fixed-dose doxorubicin. J Natl Cancer Inst 84:1725–1730. [DOI] [PubMed] [Google Scholar]
- Ichikawa Y, Ghanefar M, Bayeva M, Wu R, Khechaduri A, Naga Prasad SV, Mutharasan RK, Naik TJ, Ardehali H. (2014) Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J Clin Invest 124:617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins TC. (1997) Optical absorbance and fluorescence techniques for measuring DNA-drug interactions. Methods Mol Biol 90:195–218. [DOI] [PubMed] [Google Scholar]
- Kalyanaraman B, Morehouse KM, Mason RP. (1991) An electron paramagnetic resonance study of the interactions between the adriamycin semiquinone, hydrogen peroxide, iron-chelators, and radical scavengers. Arch Biochem Biophys 286:164–170. [DOI] [PubMed] [Google Scholar]
- Li F, Wang X, Capasso JM, Gerdes AM. (1996) Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J Mol Cell Cardiol 28:1737–1746. [DOI] [PubMed] [Google Scholar]
- Liang H, Wu X, Guziec LJ, Guziec FS, Jr, Larson KK, Lang J, Yalowich JC, Hasinoff BB. (2006) A structure-based 3D-QSAR study of anthrapyrazole analogues of the anticancer agents losoxantrone and piroxantrone. J Chem Inf Model 46:1827–1835. [DOI] [PubMed] [Google Scholar]
- Longo M, Della Torre P, Allievi C, Morisetti A, Al-Fayoumi S, Singer JW. (2014) Tolerability and toxicological profile of pixantrone (Pixuvri®) in juvenile mice: comparative study with doxorubicin. Reprod Toxicol 46:20–30. [DOI] [PubMed] [Google Scholar]
- Lyu YL, Kerrigan JE, Lin CP, Azarova AM, Tsai YC, Ban Y, Liu LF. (2007) Topoisomerase IIβ mediated DNA double-strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res 67:8839–8846. [DOI] [PubMed] [Google Scholar]
- Malisza KL, Hasinoff BB. (1995) Production of hydroxyl radical by iron(III)-anthraquinone complexes through self-reduction and through reductive activation by the xanthine oxidase/hypoxanthine system. Arch Biochem Biophys 321:51–60. [DOI] [PubMed] [Google Scholar]
- Malisza KL, Hasinoff BB. (1996) Inhibition of anthracycline semiquinone formation by ICRF-187 (dexrazoxane) in cells. Free Radic Biol Med 20:905–914. [DOI] [PubMed] [Google Scholar]
- Mariani A, Bartoli A, Atwal M, Lee KC, Austin CA, Rodriguez R. (2015) Differential targeting of human topoisomerase II isoforms with small molecules. J Med Chem 58:4851–4856. [DOI] [PubMed] [Google Scholar]
- Martin E, Thougaard AV, Grauslund M, Jensen PB, Bjorkling F, Hasinoff BB, Tjørnelund J, Sehested M, Jensen LH. (2009) Evaluation of the topoisomerase II-inactive bisdioxopiperazine ICRF-161 as a protectant against doxorubicin-induced cardiomyopathy. Toxicology 255:72–79. [DOI] [PubMed] [Google Scholar]
- McGhee JD. (1976) Theoretical calculations of the helix-coil transition of DNA in the presence of large, cooperatively binding ligands. Biopolymers 15:1345–1375. [DOI] [PubMed] [Google Scholar]
- Mukherji D, Pettengell R. (2010) Pixantrone for the treatment of aggressive non-Hodgkin lymphoma. Expert Opin Pharmacother 11:1915–1923. [DOI] [PubMed] [Google Scholar]
- Myers C. (1998) The role of iron in doxorubicin-induced cardiomyopathy. Semin Oncol 25(Suppl 10):10–14. [PubMed] [Google Scholar]
- Nguyen B, Gutierrez PL. (1990) Mechanism(s) for the metabolism of mitoxantrone: electron spin resonance and electrochemical studies. Chem Biol Interact 74:139–162. [DOI] [PubMed] [Google Scholar]
- Nitiss JL. (2009) Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer 9:338–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hara KA, Wu X, Patel D, Liang H, Yalowich JC, Chen N, Goodfellow V, Adedayo O, Dmitrienko GI, Hasinoff BB. (2007) Mechanism of the cytotoxicity of the diazoparaquinone antitumor antibiotic kinamycin F. Free Radic Biol Med 43:1132–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Malley YQ, Reszka KJ, Britigan BE. (2004) Direct oxidation of 2′,7′-dichlorodihydrofluorescein by pyocyanin and other redox-active compounds independent of reactive oxygen species production. Free Radic Biol Med 36:90–100. [DOI] [PubMed] [Google Scholar]
- Padget K, Pearson AD, Austin CA. (2000) Quantitation of DNA topoisomerase IIα and β in human leukaemia cells by immunoblotting. Leukemia 14:1997–2005. [DOI] [PubMed] [Google Scholar]
- Pastan I, Gottesman MM, Ueda K, Lovelace E, Rutherford AV, Willingham MC. (1988) A retrovirus carrying an MDR1 cDNA confers multidrug resistance and polarized expression of P-glycoprotein in MDCK cells. Proc Natl Acad Sci USA 85:4486–4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Péan E, Flores B, Hudson I, Sjöberg J, Dunder K, Salmonson T, Gisselbrecht C, Laane E, Pignatti F. (2013) The European Medicines Agency review of pixantrone for the treatment of adult patients with multiply relapsed or refractory aggressive non-Hodgkin’s B-cell lymphomas: summary of the scientific assessment of the committee for medicinal products for human use. Oncologist 18:625–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendleton M, Lindsey RH, Jr, Felix CA, Grimwade D, Osheroff N. (2014) Topoisomerase II and leukemia. Ann N Y Acad Sci 1310:98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettengell R, Kaur J. (2015) Pixantrone dimaleate for treating non-Hodgkin’s lymphoma. Expert Opin Orphan Drugs 3:747–757. [Google Scholar]
- Pilch DR, Sedelnikova OA, Redon C, Celeste A, Nussenzweig A, Bonner WM. (2003) Characteristics of γ-H2AX foci at DNA double-strand breaks sites. Biochem Cell Biol 81:123–129. [DOI] [PubMed] [Google Scholar]
- Pommier Y. (2013) Drugging topoisomerases: lessons and challenges. ACS Chem Biol 8:82–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y, Marchand C. (2012) Interfacial inhibitors: targeting macromolecular complexes. Nat Rev Drug Discov 11:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priebe W, Fokt I, Przewloka T, Chaires JB, Portugal J, Trent JO. (2001) Exploiting anthracycline scaffold for designing DNA-targeting agents. Methods Enzymol 340:529–555. [DOI] [PubMed] [Google Scholar]
- Reers M, Smiley ST, Mottola-Hartshorn C, Chen A, Lin M, Chen LB. (1995) Mitochondrial membrane potential monitored by JC-1 dye. Methods Enzymol 260:406–417. [DOI] [PubMed] [Google Scholar]
- Ritke MK, Allan WP, Fattman C, Gunduz NN, Yalowich JC. (1994a) Reduced phosphorylation of topoisomerase II in etoposide-resistant human leukemia K562 cells. Mol Pharmacol 46:58–66. [PubMed] [Google Scholar]
- Ritke MK, Roberts D, Allan WP, Raymond J, Bergoltz VV, Yalowich JC. (1994b) Altered stability of etoposide-induced topoisomerase II-DNA complexes in resistant human leukaemia K562 cells. Br J Cancer 69:687–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritke MK, Yalowich JC. (1993) Altered gene expression in human leukemia K562 cells selected for resistance to etoposide. Biochem Pharmacol 46:2007–2020. [DOI] [PubMed] [Google Scholar]
- Salvatorelli E, Menna P, Paz OG, Chello M, Covino E, Singer JW, Minotti G. (2013) The novel anthracenedione, pixantrone, lacks redox activity and inhibits doxorubicinol formation in human myocardium: insight to explain the cardiac safety of pixantrone in doxorubicin-treated patients. J Pharmacol Exp Ther 344:467–478. [DOI] [PubMed] [Google Scholar]
- Schroeder PE, Patel D, Hasinoff BB. (2008) The dihydroorotase inhibitor 5-aminoorotic acid inhibits the metabolism in the rat of the cardioprotective drug dexrazoxane and its one-ring open metabolites. Drug Metab Dispos 36:1780–1785. [DOI] [PubMed] [Google Scholar]
- Smith NA, Byl JA, Mercer SL, Deweese JE, Osheroff N. (2014) Etoposide quinone is a covalent poison of human topoisomerase IIβ. Biochemistry 53:3229–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian D, Furbee CS, Muller MT. (2001) ICE bioassay: isolating in vivo complexes of enzyme to DNA. Methods Mol Biol 95:137–147. [DOI] [PubMed] [Google Scholar]
- Tewey KM, Chen GL, Nelson EM, Liu LF. (1984a) Intercalative antitumor drugs interfere with the breakage-reunion reaction of mammalian DNA topoisomerase II. J Biol Chem 259:9182–9187. [PubMed] [Google Scholar]
- Tewey KM, Rowe TC, Yang L, Halligan BD, Liu LF. (1984b) Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science 226:466–468. [DOI] [PubMed] [Google Scholar]
- Toyoda E, Kagaya S, Cowell IG, Kurosawa A, Kamoshita K, Nishikawa K, Iiizumi S, Koyama H, Austin CA, Adachi N. (2008) NK314, a topoisomerase II inhibitor that specifically targets the α isoform. J Biol Chem 283:23711–23720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vejpongsa P, Yeh ET. (2014) Topoisomerase 2β: a promising molecular target for primary prevention of anthracycline-induced cardiotoxicity. Clin Pharmacol Ther 95:45–52. [DOI] [PubMed] [Google Scholar]
- Wallace KB. (2003) Doxorubicin-induced cardiac mitochondrionopathy. Pharmacol Toxicol 93:105–115. [DOI] [PubMed] [Google Scholar]
- Willmore E, Frank AJ, Padget K, Tilby MJ, Austin CA. (1998) Etoposide targets topoisomerase IIα and IIβ in leukemic cells: isoform-specific cleavable complexes visualized and quantified in situ by a novel immunofluorescence technique. Mol Pharmacol 54:78–85. [DOI] [PubMed] [Google Scholar]
- Wu CC, Li TK, Farh L, Lin LY, Lin TS, Yu YJ, Yen TJ, Chiang CW, Chan NL. (2011) Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 333:459–462. [DOI] [PubMed] [Google Scholar]
- Wu CC, Li YC, Wang YR, Li TK, Chan NL. (2013) On the structural basis and design guidelines for type II topoisomerase-targeting anticancer drugs. Nucleic Acids Res 41:10630–10640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav AA, Chee G-L, Wu X, Patel D, Yalowich JC, Hasinoff BB. (2015) Structure-based design, synthesis and biological testing of piperazine-linked bis-epipodophyllotoxin etoposide analogs. Bioorg Med Chem 23:3542–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav AA, Wu X, Patel D, Yalowich JC, Hasinoff BB. (2014) Structure-based design, synthesis and biological testing of etoposide analog epipodophyllotoxin-N-mustard hybrid compounds designed to covalently bind to topoisomerase II and DNA. Bioorg Med Chem 22:5935–5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Wu X, Guziec LJ, Guziec FS, Jr, Chee G-L, Yalowich JC, Hasinoff BB. (2010) Design, synthesis and biological evaluation of a novel series of anthrapyrazoles linked with netropsin-like oligopyrrole carboxamides as anticancer agents. Bioorg Med Chem 18:3974–3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Wu X, Yalowich JC, Hasinoff BB. (2011) Design, synthesis, and biological evaluation of a novel series of bisintercalating DNA-binding piperazine-linked bisanthrapyrazole compounds as anticancer agents. Bioorg Med Chem 19:7023–7032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu YL, Liu LF, Yeh ET. (2012) Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med 18:1639–1642. [DOI] [PubMed] [Google Scholar]
- Zwelling LA, Mayes J, Altschuler E, Satitpunwaycha P, Tritton TR, Hacker MP. (1993) Activity of two novel anthracene-9,10-diones against human leukemia cells containing intercalator-sensitive or -resistant forms of topoisomerase II. Biochem Pharmacol 46:265–271. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.