

Abstract
Spinach (Spinacia oleracea) has cold tolerant but heat sensitive characteristics. The spinach variety ‘Island,’ is suitable for summer periods. There is lack molecular information available for spinach in response to heat stress. In this study, high throughput de novo transcriptome sequencing and gene expression analyses were carried out at different spinach variety ‘Island’ leaves (grown at 24 °C (control), exposed to 35 °C for 30 min (S1), and 5 h (S2)). A total of 133,200,898 clean reads were assembled into 59,413 unigenes (average size 1259.55 bp). 33,573 unigenes could match to public databases. The DEG of controls vs S1 was 986, the DEG of control vs S2 was 1741 and the DEG of S1 vs S2 was 1587. Gene Ontology (GO) and pathway enrichment analysis indicated that a great deal of heat-responsive genes and other stress-responsive genes were identified in these DEGs, suggesting that the heat stress may have induced an extensive abiotic stress effect. Comparative transcriptome analysis found 896 unique genes in spinach heat response transcript. The expression patterns of 13 selected genes were verified by RT-qPCR (quantitative real-time PCR). Our study found a series of candidate genes and pathways that may be related to heat resistance in spinach.
Agricultural production is threatened by abiotic stresses such as drought, extreme temperatures, flooding and chemical toxicity1. Heat stress can reduce crop production, and studies suggest that these reductions will become a major issue in coming years due to global warming2. Global warming predictions foretell that temperatures will rise further by 2–6 °C by the end of this century due to the rapid increase in atmospheric greenhouse gas concentrations3. Many studies reveal that heat resistance depends on species and genotype. However, there were some differences in response to heat stress among different species or genotype4. Therefore, it is essential to reveal molecular mechanism of crop plants in response to heat stress, which is helpful to breed heat-tolerant crop plants4.
Next-generation sequencing (NGS) technologies, such as Illumina/Solexa (San Diego, CA, USA), the SOLiD platform from Applied Biosystems, and Roche 454 sequencing, have revolutionized genomics because they allow faster and less expensive sequencing5. NGS-based RNA-Seq methods, which are used for transcriptome analysis, even allow simultaneous acquisition of sequences6. These fast sequencing methods are used to characterize genes, detect gene expression pattern and level, recognize and quantify rare transcripts without prior information of the particular gene or reference genome, and generate information on alternative splicing and sequence variations in identified genes6,7. In recent years, RNA-Seq has been successfully used to reveal the complex biotic and abiotic stress response mechanism in many plant species, such as grape (Vitis vinifera)8, cotton (Gossypium hirsutum)9, cucumber (Cucumis sativus)10, Ammopiptanthus mongolicus11, and Arabidopsis thaliana12. However, data on the molecular responses to heat stress in spinach leaves are very limited.
Spinach (Spinacia oleracea) belongs to the family Amaranthaceae and is an economically important vegetable crop that is grown worldwide. Spinach has cold tolerant but heat sensitive characteristics so that the crop may undergo heat stress throughout its entire life cycle. High temperatures affect spinach plant growth and significantly decrease yield and quality. Recently, some spinach varieties such as ‘Menorca’ and ‘Island,’ that originate from Seminis Vegetable Seeds Inc. have been shown to be suitable for summer periods. In the present study, the genome-wide analysis of gene expression during heat treatment was performed on the ‘Island’ summer spinach variety via a NGS-based Illumina paired-end sequencing platform. This study will aid in understanding the heat stress response in spinach at the molecular level, and enriches the genomic information for spinach.
Results and Discussion
Illumina sequencing and de novo assembly
Three cDNA libraries were generated with mRNA from three samples: control (grown at 24 °C), S1 (exposed at 35 °C for 30 min) and S2 (exposed at 35 °C for 5 h) spinach leaf. These cDNA libraries were then subjected to Illumina deep-sequencing. The raw reads were deposited in the Sequence Read Archive at GenBank databases ID: SRP051935. A total of 44,682,577, 42,290,681, and 46,227,640 clean reads with 4,349,881,212, 4,122,064,627, and 4,508,391,358 nucleotides were obtained from the control, S1, and S2, respectively. The clean reads were >12 gigabases (total length), which was equivalent to ~12-fold coverage of the genome of S. oleracea. All clean reads were de novo assembles with the Trinity method13 because S. oleracea does not have an appropriate reference genome sequence. Table 1 shows more than 80% of the reads could be mapped back to the assembled transcripts. Finally, a total of 133,200,898 clean reads were assembled into 59,413 unigenes with a total unigene length of 74,833,448 bp. The length of the unigenes ranged from 351–14,797 bp, with an average unigene size of 1259.55 bp. There were 26,684 unigenes (44.91%) in the length range of 401–1000 bp, 23,655 unigenes (39.81%) in the length range of 1001–3200 bp, and 3,233 unigenes (5.44%) with length >3200 bp (Fig. 1). The length distribution of Arabidopsis and rice (Oryza sativa) transcriptome sequences were obtained from publicly available datasets. Comparison analysis indicated that the length range of most unigenes was 400–3200 bp in these three species (Table 2).
Table 1. Total number of original and cleaned reads that were mapped to their respective assembled transcripts.
| Map to Genome | S1 sample |
Control sample |
S2 sample |
|||
|---|---|---|---|---|---|---|
| Number of reads | percentage | Number of reads | percentage | Number of reads | percentage | |
| Total reads | 44682577 | 100.00% | 42290681 | 100.00% | 46227640 | 100.00% |
| Total base pairs | 4349881212 | 100.00% | 4122064627 | 100.00% | 4508391358 | 100.00% |
| perfect match | 30160739 | 67.50% | 28842244 | 68.20% | 31120447 | 67.32% |
| < = 2bp mismatch | 5478084 | 12.26% | 4931093 | 11.66% | 5246837 | 11.35% |
| unique match | 18020483 | 40.33% | 17592923 | 41.60% | 18407846 | 39.82% |
| multi-position match | 19770071 | 42.35% | 17267285 | 40.83% | 20099778 | 43.48% |
| Total unmapped reads | 7694340 | 17.22% | 6884923 | 16.28% | 8390317 | 18.15% |
| Total mapped reads | 36988237 | 82.78% | 35405758 | 83.72% | 37837323 | 81.85% |
Figure 1. Length distribution of the assembled transcripts.
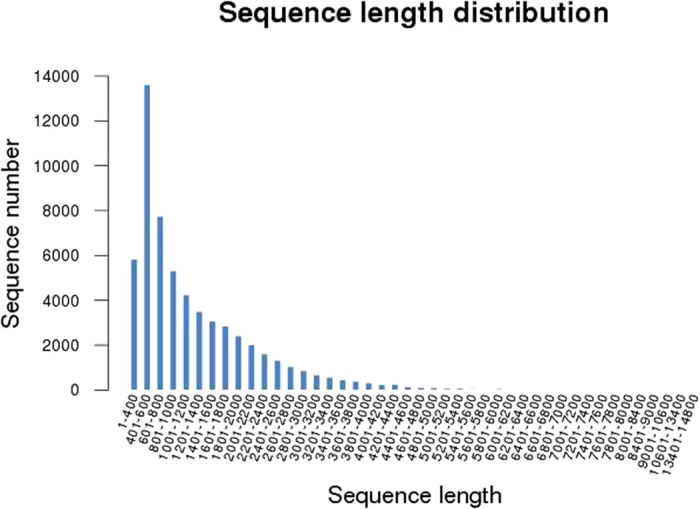
Table 2. The length distribution of spinach, rice and Arabidopsis.
| 0–400 bp | 401–1000 bp | 1001–3200 bp | >3200 bp | |
|---|---|---|---|---|
| Spinach | 5841 (9.83%) | 26684 (44.91%) | 23655 (39.81%) | 3233 (5.44%) |
| Rice | 6774 (10.21%) | 15277 (23.03%) | 36950 (55.7%) | 7337 (11.06%) |
| Arabidopsis | 3465 (8.32%) | 10137 (24.33%) | 25065 (60.15%) | 3004 (7.2%) |
ORF prediction analysis was made using get ORF from EMBOSS package. There were 35,805 (60.26%) unigenes were identified to have an ORF. These unigenes were compared with 31,282 sugar beet (Beta vulgaris) transcriptome sequences obtained from publicly available datasets which have an ORF (E-value <1e−10). The result show that 12,307 unigenes had significant matched to sugar beet transcripts, and there were 23,498 specific sequence in spinach compared to sugar beet transcripts.
The web-based tool ESTcal14 was used to assess the breadth and depth of all unigenes because S. oleracea does not have transcriptome profile for comparison. The average read-depth coverage was 31.65-fold, and 22.28% unigenes was greater than 20 fold (Fig. 2), which suggested that these unigenes have high contiguity and high coverage. So these unigenes could be used in further analyses.
Figure 2. A histogram of the average read-depth coverage for unigenes.

The x-axis indicates the number of unigenes, and the y-axis indicates the distribution of read-depth coverage.
Functional annotation and classification of spinach leaf transcriptome
Public databases (NR, NT, SwissProt, and KEGG) was used to annotated all unigenes with local BLAST programs (E value < 1.0E-5). The results showed that 33,573 (56.51% of 59,413) unigenes were matched to one or more of the databases. The large amount of unannotated unigenes generated in this study may be assembled uncorrectly or represent a specific gene pool for spinach leaf studies6. These unannotated unigenes may be useful for further study of heat stress response mechanisms and identification of novel genes in S. oleracea.
GO (http://www.geneontology.org/) is an international classification system for standardized gene functions, which have three GO categories: biological process, molecular function, and cellular component. Based on sequence homology, a total of 24,387 (41.05% of 59,413) unigenes were assigned to one or more GO term annotations (Fig. 3). Among which, ‘cell’ (15,020; 61.59% of 24,387), ‘cell part’ (15,020; 61.59% of 24,387), and ‘organelle’ (11,734; 44.12% of 24,387) were the terms that dominated in the cellular component category. ‘Cellular process’ (14,905; 61.11% of 24,387), ‘metabolic process’ (14,592; 59.84% of 24,387), and ‘response to stimulus’ (5731; 23.50% of 24,387) were the most representative terms in the biological process category. ‘Binding’ (12,444, 51.03% of 24,387) and ‘catalytic activity’ (13,374, 58.84% of 24,387) were the most abundant terms in the molecular function category. Only a few unigenes (less than 10) were clustered into the terms of ‘channel regulator activity,’ ‘protein tag,’ ‘metallochaperone activity,’ ‘translation regulator activity,’ ‘virion part,’ ‘extracellular matrix part,’ and ‘cell killing’. The GO terms in this study were compared with sugar beet GO terms which were obtained from publicly available datasets. Most GO terms of spinach were the same as sugar beet. In contrast to sugar beet, four GO terms were specific in spinach including ‘channel regulator activity’, ‘death’, ‘carbon utilization’ and ‘cell proliferation’. We speculate that the specific GO terms of spinach may result from heat stress or species differences.
Figure 3. Gene ontology classification of the S. oleracea transcriptome.

All unigenes were match with the Cluster of Orthologus Groups (COG database) for functional prediction and classification. A total of 9,760 (16.43% of 59,413) unigenes were assigned appropriate COG clusters, which could be classified into 25 functional categories (Fig. 4). Among them, the largest category was ‘General function prediction only’ (20.42%); followed by ‘transcription’ (10.26%); ‘Replication, recombination, and repair’ (10.07%); ‘Signal transduction mechanisms’ (9.0%); and ‘Posttranslational modification, protein turnover, and chaperones’ (6.98%).
Figure 4. COG functional classification of the S. oleracea transcriptome.

To identify biological pathways activated in spinach leaves under heat stress, all unigenes were annotated and mapped to the KEGG database (http://genome.jp/kegg/). Out of 33,573 total annotated unigenes, 10,143 unigenes were significantly matched to KEGG database and were assigned to 292 KEGG pathways (see Supplementary Table S1). The major pathways in this study were ‘metabolic pathways [ko01100]’(2,5525.22% of 10,143), ‘biosynthesis of secondary metabolites [ko01110]’(1,088, 10.73%8, of 10,143), ‘microbial metabolism in diverse environments [ko01120]’(439, 4.33% of 10,143), ‘spliceosome [ko03040]’(330, 3.25% of 10,143) and ‘ribosome [ko03010]’(272, 2.68% of 10,143). Furthermore, 975 unigenes were involved in the plant biotic/abiotic defense, which includes ‘MAPK signaling pathway’, ‘phosphatidylinositol’, ‘signaling system’, ‘ascorbate and aldarate metabolism’, ‘calcium signaling pathway’, ‘plant-pathogen interaction’, ‘glutathione’ metabolism’, ‘plant hormone signal transduction’, ‘ABC transporters’, and ‘phenylalanine metabolism’.
Differentially expressed genes (DEGs) involved in the heat stress response of spinach leaves
In order to found the differentially expressed genes in spinach leaves responding to heat stress among control, S1, and S2, clean reads of these libraries were respectively assigned to all unigenes using the RSEM (RNA-Seq by Expectation Maximization) software15, and the difference expression levels of unigenes were calculated with the FPKM method16. The DEGs were determined with the absolute change value of |log FC| > 2 using a greater statistical significance level (P < 0.05) and false discovery rates (FDR < 0.001). The DEG of controls vs S1 was 986, the DEG of controls vs S2 was 1741, and the DEG of S1 vs S2 was 1587. The number of up-regulated unigenes in the S1 and S2 samples compared with control was 550 and 1,131, respectively.
Validation of the differentially expressed genes (DEGs) via RT-qPCR
In order to experimentally confirm that the differentially expressed genes obtained in this study were credible, 13 DEGs from different gene families (including 8 up-regulated and 5 down-regulated in S1 or S2) were selected and their expression pattern was examined via RT-qPCR. The RT-qPCR results show that the expression of all these DEGs was similar to those obtained from the Illumina sequencing analysis (see Supplementary Table S2). Moreover, the fold-changes obtained by DEG were generally greater than those obtained by RT-qPCR, which was a universal phenomenon in other studies and attributed to the essentially different of algorithms and sensitivity between the two techniques17,18. These results indicated that the method used to determine DEGs in this study were valid.
GO functional enrichment and KEGG pathway enrichment analysis of DEGs
Goatools (https://github.com/tanghaibao/goatools) was used to identify GO terms that were remarkably enriched in DEGs. The remarkably enriched GO terms in DEGs was identified with a hyergeometric test (Bonferroni-correction P ≤ 0.05). According to GO functional enrichment analysis, 24 terms for the up-regulated DEGs and 19 terms for down-regulated DEGs were enriched in control vs. ein control vs. S2, and 35 terms for the up-regulated DEGs and 42 terms for down-regulated DEGs were enriched in S1 vs. S2 (see Supplementary Table S3). In the category of biological processes, five terms for up-regulated DEGs (including ‘response to abiotic stimulus,’ ‘response to heat,’ ‘response to temperature stimulus,’ ‘transmembrane transport,’ and ‘carbohydrate metabolic process’) as well as two terms for down-regulated DEGs (‘protein stabilization’ and ‘electron transport’) were enriched in both control vs. S1 and control vs. S2. This result suggested that genes involved in these biological processes may play important roles responding to heat stress. In the category of molecular function, four terms for up-regulated DEGs including ‘transcription regulator activity,’ ‘chaperone binding,’ ‘signal transducer activity,’ and ‘transmembrane transporter activity’ were enriched in both control vs. S1 and control vs. S2. As for the category of cellular component, ‘external encapsulating structure’ for up-regulated DEGs as well as ‘integral to membrane’ and ‘photosystem II’ for down-regulated DEGs were enriched in both control vs. S1 and control vs. S2. This indicates that genes related to photosynthesis may be seriously affected by heat stress. Additionally, ‘ubiquitin ligase complex,’ ‘phospholipid metabolic process,’ and ‘small molecule metabolic processes’ were also enriched for up-regulated DEGs in control vs. S1. ‘Ribonucleoprotein complex,’ ‘channel activity,’ ‘alcohol metabolic process,’ and ‘protein folding’ for up-regulated DEGs were enriched in control vs. S2. Therefore, genes involved in these biological processes may also participate in responding to heat stress. Furthermore, the terms ‘response to light stimulus,’ ‘response to water deprivation’, and ‘response to salt stress’ were also enriched for up-regulated DEGs in control vs. S2, suggesting that the heat stress may have caused an efficient abiotic stress.
The significantly enriched pathways for DEGs in this study were determined by the KEGG Orthology-Based Annotation System (KOBAS) (http://kobas.cbi.pku.edu.cn/home.do). According to the KEGG pathway analysis, 7, 17, and 11 pathways were observed as significantly enriched for DEGs of control vs. S1, control vs. S2, and S1 vs. S2, respectively (see Supplementary Table S4). The pathways enriched for control vs. S1 DEGs were ‘nucleotide excision repair’ [ko03420], ‘oxidative phosphorylation’ [ko00190], ‘ubiquitin mediated proteolysis’ [ko04120], ‘photosynthesis’ [ko00195], ‘protein processing in endoplasmic reticulum’ [ko04141], ‘calcium signaling pathway’ [ko04020], and ‘phenylalanine metabolism’ [ko00360], suggesting that genes in these pathways seem to be involved in early heat sense. The top five pathways enriched for control vs. S2 DEGs were ‘carbon fixation in photosynthetic organisms’ [ko00710], ‘fatty acid metabolism’ [ko00071], ‘phenylalanine metabolism’ [ko00360], ‘plant-pathogen metabolism’ [ko04626] and ‘glycine, serine and threonine metabolism’ [ko00260]. This indicates that metabolism processes play important roles in response to heat after 5 hours. Additionally, heat shock protein genes belonging to the ‘protein processing in endoplasmic reticulum’ [ko04141] pathway was enriched in both control vs. S1 and control vs. S2, which was consistent with previous studies that plants suffering from heat stress sharply accumulate heat shock proteins to enhance heat tolerance of the plant2.
Comparative analysis of genes response to heat stress in different plant species
In order to identify the similarities and differences of molecular mechanisms in response to heat stress between spinach and the other plant species, three other transcriptome profiles in response to heat stress from rice19, wheat20 and grape21 were compared with spinach. The comparison was carried out using publicly available datasets. In contrast to rice19, there were 1250 unique genes in spinach heat response transcript. In contrast to wheat20, there were 1340 unique genes in spinach heat response transcript. In contrast to grape21, there were 1701 unique genes in spinach heat response transcript. The total number of unique genes was 896 in spinach heat response transcript. Among these unique differentially expressed genes, 506 were up-regulated and 390 genes were down-regulated. GO enrichment analysis show that up regulated spinach specific genes belong to ‘response to stimulus’, ‘transmembrane transport’, ‘regulation of metabolic process’, ‘response to salt stress,’ ‘organic acid metabolic process,’ ‘channel activity’, ‘response to light stimulus’ ‘carbohydrate metabolic process’ ‘response to chemical stimulus’, ‘hydrolase activity’ and ‘regulation of transcription’. GO enrichment analysis show that down regulated spinach specific genes belong to ‘carbohydrate metabolic process’, ‘phosphorus metabolic process’, ‘metal ion binding’, ‘catalytic activity’, ‘transcription regulator activity’, ‘carotenoid metabolic process’, ‘protein kinase activity’ and ‘transferase activity’. Among these spinach unique genes, one gene related to violaxanthin was up regulated and three genes related to zeaxanthin were down-regulated during the heat treatment, which were not found in the other three species. However, it was report that xanthophylls (including violaxanthin, antheraxanthin, and zeaxanthin) could keep themostability of the membrane and lowers the harm of high temperatures22.
Twenty-six genes were commonly identified in all the four species. Interestingly, the expression patterns of these genes are very similar in the four plant species. Most of them were up-regulated, only four genes were down-regulated. The enriched GOs analysis indicated that these up regulated genes belong to protein folding, a common biochemical response to heat stress.
Common molecular response of spinach under heat stress
Analysis of the transcriptome profile in plant after heat treatment indicated that the HSP family plays a central role in responding to heat stress19,20,21. HSPs families, include HSP100, HSP90, HSP70, HSP60 and small HSPs (sHSPs), are involve in folding and assembling protein, keeping protein stabilization, activating protein, and degrading protein in many normal cellular processes and under stress conditions23. Thirty-three DEGs were identified in our study as candidate genes for membership in different HSP families. The genes of high and middle molecular weight HSPs included HSP101, HSP90, HSP83, HSP82, and HSP70. Low molecular HSPs genes included HSP18.1, HSP18.3, HSP20, HSP21.7, HSP25.3, HSP26.26, and HSP30. Most of the HSPs were significantly up-regulated during the heat treatment, and the expression level of some were sharply increased in the 30 min heat treatment but were decreased to low levels after the 5 h heat treatment (see Supplementary Table S5). In Arabidopsis, HSP101 homologue was involved in increasing chloroplasts thermotolerance during heat stress24, and HSP 90 was significantly induced by heat, salt, or heavy metal stresses25. In rice, over-expression of mitochondrial HSP70 inhibited programmed cell death (PCD) in protoplasts induced by heat- and H2O2-stress26. sHSPs, range from 15–42 kDa, have a conserved sequence at their C terminus, and it thought that chloroplast and mitochondrial sHSPs play a crucial role in heat tolerance27,28. In the present study, most sHSPs were significantly up-regulated under heat treatment, and chloroplast HSP25.3 and HSP26.26 increased by approximately 243-fold and 335-fold in the 30 min heat treatment. In Chenopodium album, chloroplast sHSPs are correlated with increased thermotolerance29. After expose to heat stress, wheat chloroplastic sHSP (HSP26) is highly expression in almost all the activated tissues and certain developmental growth stages30.
It is well-known that transcription factors (TFs) play an important role in biotic and abiotic stress responses (e.g., cold, high temperatures, high salinity, drought, and pathogen attacks)2. There were 87 DEGs including up- and down-regulated genes involved in seven TF families. These families include HSF (HSF30, A6b, A5, A1a, A8, and B2b), WRKY (WRKY4, 5, 7, 17, 24, 27, 31, 40, 41 and 57), DREB (DREB 2A and 2B), basic leucine zipper (bZIP9, 11, 12, and 37), NAC (NAC1 and 68), MYB family (MYB 1, 108, 306, and 811), and C2H2 (Table 3).
Table 3. The number of DEG identified as transcription factors in spinach leaves.
| Category | Total | S1a |
S2b |
||
|---|---|---|---|---|---|
| Up-regulated | Down-regulated | Up-regulated | Down-regulated | ||
| HSF | 16 | 12 | 1 | 6 | 1 |
| WRKY | 15 | 13 | 2 | 12 | 0 |
| DREB | 8 | 3 | 0 | 6 | 0 |
| bZIP | 12 | 9 | 0 | 9 | 3 |
| NAC | 9 | 5 | 3 | 4 | 1 |
| MYB | 16 | 7 | 9 | 3 | 4 |
| C2H2 | 11 | 9 | 3 | 9 | 0 |
aSpianch exposed to 35 °C for 30 min.
bSpinach exposed to 35 °C for 5 h.
HSFs have been shown to be involved in basal and acquired thermotolerance31. According to sequence homology and domain architecture, plants HSF proteins are classed into are three conserved categories: A, B, and C32. In the present study, sixteen DEG were similar to HSFs, including HSF30, HSFA6b, HSFA5, HSFA1a, HSFA8, and HSFB2b, and fourteen of these genes were induced during the heat treatment. It was reported that HSFA1a and HSF30 could govern the expression of some HSPs31,33,34,35. In Arabidopsis, HsfA4a and HsfA8 function as sensing elements of the reactive oxygen species (ROS) during the heat stress response36,37. More and more evidences indicate that several WRKY genes are involved in the regulation of various abiotic stress responses, including heat38. In Arabidopsis, WRKY7, 25 were regulated by heat shock39,40. In this study, thirteen DEGs similar to WRKY 7, 17, 27, 31, 40, and 57 were up-regulated in S1 and twelve DEGs similar to WRKY 7, 24, 31, and 41 were up-regulated in S2 compared to control sample. The dehydration-responsive element binding protein (DREB) family plays an important role in the responses to abiotic stress in plants41,42. In Arabidopsis, DREB2A and DREB2B are induced transiently within 1 h after heat stress, but DREB2B expression continued for 12 h after heat treatment41. It was reported that OsDREB2B improve the thermotolerance in rice42. In this study, DREB2A was up-regulated in S1 and DREB2B was up-regulated in S2. Basic leucine zipper (bZIP) transcription factors, plant-specific NAC family transcription factors, MYB transcription factors and C2H2-type zinc finger transcription factors have been implicated in various biological functions, including biotic and abiotic stress43,44,45,46. In the present study, twelve bZIP , nine NAC domain, sixteen MYB and eleven C2H2 genes were heat-regulated in spinach leaves, and the expression patterns were show in Table 3. These results should be helpful for explaining the important function of the bZIP, NAC, and C2H2 family genes in the heat response of spinach leaves.
When suffering from heat stress, plants could activate various stress-responsive signal transduction pathways to generate a series of innate defensive reactions47. Calcium signal is important in response to heat stress, and heat stress causes transient rise of Ca2+ levels in many plants48. In Arabidopsis, the calmodulin AtCaM3 is a required signaling element in heat stress49 and may activate different transcription factors, such as WRKY3950 and HSFs51. In wheat, a number of genes related to calcium signal pathways were heat regulated, including annexin, calcium-binding proteins (CBPs), calcium-dependent protein kinases (CDPKs), voltage-gated calcium channel activity, Ca2+ -binding protein EF hand, CBL (calcineurin B-like protein), and CIPK (CBL-interacting protein kinase)20. In this study, twenty DEGs were highly homolugous to calcium signaling genes including CaM, CDPK, CBK, and CBL, and sixteen of these genes were up-regulated in control vs.S1. Mitogen-activated protein kinase (MAPK) cascades are thought to act as useful role in many responses to abiotic stress47. The activation of HAMK in tobacco cells was observed together with HSP70 accumulation, suggesting that HAMK may induce a heat signaling cascade via HSP genes52. It was shown that the transcript abundance of the tomato SlMAPK increases after heat treatment53. In this study, fourteen DEGs were highly similar to MAPK genes, including MAPKKK2, MAPK2, MAPK6, and MPAK18. All the genes encoding MAPKs were up-regulated during the heat treatment compared with control samples, except those encoding MAPKKK2.
The differential expression analysis revealed that the genes related to secondary metabolism exhibited complex and significant expression changes under heat stress2. Phenolics, such as flavonoids, anthocyanins, lignins, play a variety of roles in response to abiotic stresses2. In rice, genes for PAL and 4-coumarate CoA ligase (4CL) were induced by heat stress54. In this study, seventeen DEGs were found to be involved with phenolics. Among them, one gene for PAL and three genes for 4CL were up-regulated during the heat treatment, two genes for caffeic acid 3-O-methyltransferase and two genes for dihydroflavonol 4-reductase were induced after the 5 h heat treatment, and the other genes were repressed. Genes involved in terpenoid metabolism have been investigated in response to various stresses and defense signals55. In this study, six DEGs were related to terpenoid biosynthesis after heat treatment. The genes encoding 3-hydroxy-3-methy lglutaryl-coenzyme A reductase, protein-S-isoprenylcysteine O-methyltransferase B, and geranylgeranyl hydrogenase were up-regulated after the 5 h heat treatment, and the other DEGs were repressed.
It was found that heat stress could induce some resistant protein involved with other abiotic or biotic stress2. Late embryogenesis abundant (LEA) proteins can protect the citrate synthase from dehydrating conditions, such as heat and drought stress56. In sugarcane leaves, three low-molecular-weight dehydrin proteins were induced in response to heat stress57. In this study, three DEGs for a NBS-LRR type resistance protein, disease resistance protein involved in disease stress, were up-regulated during the heat treatment. Seven DEGs for sugar transporter, LEA protein, salt tolerance-like protein, and water channel protein involved in osmotic stress were up-regulated after 5 h heat treatment. Oxidative stress is a kind of secondary stress during the heat stress response, which produces a large number of ROS2. ROS concentrations are regulated by a series of antioxidant enzymes, such as superoxide dismutases (SOD), ascorbate peroxidase (APX), dehydroascorbate reductase (DHAR), and catalase (CAT)38. In this study, eight DEGs related to oxidative stress were identified. Six DEGs encoding Cu/Zn superoxide dismutase (Cu/Zn-SOD), Fe superoxide dismutase (Fe-SOD), dehydroascorbate reductase (DHAR), and catalase (CAT) were found to be up-regulated and two DEGs for peroxidase (POD) were significantly repressed during the heat stress. It was reported that CAT primarily acts as a scavenger of ROS in plants under heat stress58. In grape, DHAR was up-regulated by 3.18-fold30. Therefore, these genes may play an important role in the heat stress response of spinach leaves.
Conclusion
Spinach varieties ‘Island’, come from Seminis Vegetable Seeds Inc, are suitable for summer periods. The spinach leaf de novo transcriptome under heat stress was analyzed using NGS-based RNA-Seq technology. Differentially expressed genes (DEGs) were determined with FPKM method. The DEG for controls vs S1 was 986, the DEG for controls vs S2 was 1741 and the DEG for S1 vs S2 was 1587. Compared with control, there were 550 and 1131 up-regulated unigenes in the S1 and S2 samples, respectively. In these identified heat-responsive genes, calcium signaling molecule and some transcription factors play an important role in response to early heat stress. Comparative transcriptome analysis found a number of unique genes in spinach heat response transcript. Among these unique differentially expressed genes, we speculate some genes involved in salt stress, organic acid metabolic and carotenoid metabolic deserve to pay more attention, because they represent special mechanism in spinach response to heat stress. In a word, these results were helpful for understanding the molecular adaptation mechanism of spinach during the heat stress.
Methods
Plant materials and heat treatment
S. oleracea L. var. Island was selected in this study for its high heat tolerance. The seeds were surface-sterilized and sown into soil in plastic pots, and then the seedlings were cultured in a growth chamber where they were maintained at 25 °C (14 h/10 h day/night cycle, relative humidity ranging from 50%–70%, and light intensity at 300 μmol m−2 s−1). Seedlings with four true leaves were then cultured in the same chamber with the same conditions except with the temperature at 35 °C. After 0 (control), 30 min (S1), and 5 h (S2) of heat treatment, the third true leaves were collected from all three different samples. The samples were immersed in liquid nitrogen and stored at −80 °C for RNA extraction and quantitative real-time PCR analysis.
RNA extraction, cDNA library construction, and Illumina sequencing
Total RNA from different samples was extracted using the TRIZOL reagent (Takara, China). Agilent 2100 Bioanalyzer was used to test the intergrity of RNA, which revealed that all RNA samples were integrate (integrity number value >8.0). Three libraries (control, S1 and S2) were prepared according to the manufacturer’s instructions of the TruseqTM RNA sample prep Kit (Illumina, Inc. San Diego, CA, USA). Firstly, mRNA was enriched using magnetic beads containing poly-T molecules. Subsequently, the enriched mRNA was broken into short fragments and then reverse transcribed into cDNA using PrimeScript 1st Strand cDNA Synthesis Kit (TaKaRa, China). Finally these cDNA fragments were carried out end repair and were ligated with Illumina adapters. Three libraries with an insert size of 200 bp were constructed and then sequenced using the Illumina HiSeqTM 2500.
Short reads de novo assembly
Firstly, the raw reads were cleaned by removing adaptor sequences, empty reads, and low quality (phred quality below 5). Then, transcriptome de novo assembly was carried out using Trinity (http://trinityrnaseq.sourceforge.net)59. The clean reads were first assembled to produce longer fragments named as contigs. In order to avoid obtain contigs from the same transcript, the paired-end reads were used to calculated the distance and relation among these contigs. Finally, contigs were connected until it cannot be extended on either end, and then these sequences need to perform a redundancy removal process with sequence clustering software to obtain non-redundant transcripts. These non-redundant transcripts were named as unigenes. Bowtie was used to map the clean reads back to the assembled transcripts.
Gene function annotations and classifications
The NCBI non-redundant (nr), Swiss-Prot, Cluster of Orthologous Groups (COG), and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases were used to annotated all the unigenes with local BLAST programs (E value <1.0E-5). For the nr annotation results, the Blast2GO program was applied to classify unigenes based on Gene Ontology (GO) terms, and then the WEGO tool was used to draw the GO tree. The COG database was also used to predict the functional classification of the unigenes. All unigenes were match with the KEGG pathway database to predict pathway unigenes participated with BLASTx (E value <1.0E-5).
Identification of differentially expressed genes (DEGs)
Differentially expressed genes (DEGs) of different libraries were analyzed using the FPKM (the fragments per kilobase of exon region in a given gene per million mapped fragments) method and DEGs were identified with edgeR package60. Briefly, FPKM was used to determine the number of mapping reads for every unigene, and also to assess the unigene expression levels. The trimmed mean of M-values (TMM) method was used to calculate normalization factors. The negative binomial distribution methods were applied to calculate p values. The Benjamini-Hochberg methods were chosen to adjust for multiple tests. Unigenes were determined to be significantly differentially expressed if they had a p-value <0.05 and a false discovery rate (FDR) <0.001. Besides, GO functional enrichment and KEGG pathway enrichment analysis of DEGs were carried out via comparison with the whole-transcriptome background using the formula described in previous studies (Bonferroni-corrected p-value ≤ 0.05)18.
Validation by real-time quantitative RT-PCR
Thirteen DEGs were chosen to confirm that they involve in responding heat stress using RT-qPCR method. Primer Premier 3.0 software was used to design gene-specific primers on the basis of the selected unigene sequences (see Supplementary Table S6). RT-qPCR was carried out on an ABI System using Fast Start Universal SYBR Green Master Mix (Roche). PCR amplifications included the following conditions: 95 °C for 3 min, followed by 40 cycles of 95 °C for 15 s, and then 60 °C for 30 s. A melting curve was obtained from 60 to 95 °C by increasing the temperature stepwise by 0.5 °C every 5 s. This was done to test the specificity of the amplified product. The expression levels of selected DEGs were normalized by comparing with an internal reference gene, 18SrRNA. Their relative expression level was calculated via the 2−ΔΔCt method61. All RT-qPCR were repeated in three biological and three technical replications.
Additional Information
How to cite this article: Yan, J. et al. De novo transcriptome sequencing and gene expression profiling of spinach (Spinacia oleracea L.) leaves under heat stress. Sci. Rep. 6, 19473; doi: 10.1038/srep19473 (2016).
Supplementary Material
Acknowledgments
We thank the reviewers for their critical reading and constructive suggestions. This work was funded by Green Leafy Vegetables Industrial Technology System of Shanghai (Hu nong ke chan zi NO. 2), the Yang Fan Plan Project (NO. 14YF1413100) and Excellent technical leaders plan in Shanghai (NO. 14XD1425100) supported by Science and Technology Commission of Shanghai in China. We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Footnotes
Author Contributions J.Y., L.Y. and W.Z. conceived and designed the study. J.Y. and L.Y. performed most of the experiments. S. L. bought the spinach variety. J.Y., J. X. and Y.L. processed and analyzed the data. J.Y., L.Y. and W.Z. wrote the manuscript. J. X. helps to revise the manuscript.
References
- Bita C. E. et al. Temperature stress differentially modulates transcription in meiotic anthers of heat-tolerant and heat-sensitive tomato plants. BMC Genomics 12, 384 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahid A., Gelani S., Ashraf M. & Foolad M. R. Heat tolerance in plants: An overview. Environmental and Experimental Botany 61, 199–223 (2007). [Google Scholar]
- Peck S. C. & Teisberg T. J. CETA: a model for carbon emissions trajectory assessment. Energy J 13, 55–77 (1992). [Google Scholar]
- Craita E. B. & Tom G. Plant tolerance to high temperature in a changing environment: scientific fundamentals and production of heat stress-tolerant crops. Frontiers in plant science 4, 273 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozova O. & Marra M. A. Applications of next-generation sequencing technologies in functional genomics. Genomics 92, 255–264 (2008). [DOI] [PubMed] [Google Scholar]
- Jia X. L. et al. De novo assembly, transcriptome characterization, lignin accumulation, and anatomic characteristics: novel insights into lignin biosynthesis during celery leaf development. Sci Rep 5, 8259 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickler S. R., Bombarely A. & Mueller L. A. Designing a transcriptome next generation sequencing project for a non model plant species. Am J Bot 99, 257–266 (2012). [DOI] [PubMed] [Google Scholar]
- Wu J. et al. Whole genome wide expression profiles of Vitis amurensis grape responding to downy mildew by using Solexa sequencing technology. BMC Plant Biology 10, 234 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G., Zhu Q., Meng Q. & Wu C. Transcript profiling during salt stress of young cotton (Gossypium hirsutum) seedlings via Solexa sequencing. Acta Physiol Plant 34, 107–115(2012). [Google Scholar]
- Qi X. H., Xu X. W., Lin X. J., Zhang W. J. & Chen X. H. Identification of differentially expressed genes in cucumber (Cucumis sativus L.) root under waterlogging stress by digital gene expression profile. Genomics, 99, 160–168 (2012). [DOI] [PubMed] [Google Scholar]
- Zhou Y., Gao F., Liu R., Feng J. & Li H. De novo sequencing and analysis of root transcriptome using 454 pyrosequencing to discover putative genes associated with drought tolerance in Ammopiptanthus mongolicus. BMC Genomics 13, 266 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuhiro H., Yozo O., Fumiyoshi M., Kazuo S. & Kazuki S. Landscape of the lipidome and transcriptome under heat stress in Arabidopsis thaliana. Sci Rep 5, 10533 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr M. G. et al. Full length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 29, 644–652 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall P. K. et al. Comparison of next generation sequencing technologies for transcriptome characterization. BMC Genomics 10, 347 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Trapnell C., Pop M. & Salzberg S. L. Ultra fast and memory efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Wei Z., Lam T. W. & Wang J. Next generation sequencing has lower sequence coverage and poorer SNP-detection capability in the regulatory regions. Sci Rep 1, 55 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P. et al. The developmental dynamic of the maize leaf transcriptome. Nat Genet 42, 1060–1067 (2010). [DOI] [PubMed] [Google Scholar]
- Shi T. et al. Identification of differentially- expressed genes associated with pistil abortion in Japanese apricot by genome-wide transcriptional analysis. PloS one 7, e47810 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. et al. Expression profile in rice panicle: insights into heat response mechanism at reproductive stage. Plos one 7, e49652 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin D. D. et al. Heat stress-responsive transcriptome analysis in heat susceptible and tolerant wheat (Triticum aestivum L.) by using Wheat Genome Array. BMC Genomics 9, 432 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G. T. et al. Transcriptomic analysis of grape (Vitis vinifera L.) leaves during and after recovery from heat stress. BMC plant Biology. 12, 174 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havaux M. Carotenoids as membrane stabilizers in chloroplasts. Trends Plant Sci. 3, 147–151 (1998). [Google Scholar]
- Huang B. & Xu C. Identification and characterization of proteins associated with plant tolerance to heat stress. J Integr Plant Biol. 50, 1230–1237 (2008). [DOI] [PubMed] [Google Scholar]
- Myouga F., Motohashi R., Kuromori T., Nagata N. & Shinozaki K. An Arabidopsis chloroplast-targeted Hsp101 homologue, APG6, has an essential role in chloroplast development as well as heat-stress response. Plant J. 48, 249–260 (2006). [DOI] [PubMed] [Google Scholar]
- Wang W. X., Vinocur B., Shoseyov O. & Altman A. Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends in Plant Science. 9(1), 244–252 (2004). [DOI] [PubMed] [Google Scholar]
- Qi Y. C. et al. Over-expression of mitochondrial heat shock protein 70 suppresses programmed cell death in rice. FEBS Letters. 585(1), 231–239 (2011). [DOI] [PubMed] [Google Scholar]
- Heckathorn S. A., Downs C. A., Sharkey T. D. & Coleman J. S. The small, methionine-rich chloroplast heat-shock protein protects photosystem II electron transport during heat stress. Plant Physiol. 116, 439–444 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanmiya K., Suzuki K., Egawa Y. & Shono M. Mitochondrial small heat-shock protein enhances thermotolerance in tobacco plants. FEBS Lett. 557, 265–268 (2004). [DOI] [PubMed] [Google Scholar]
- Samina S. et al. Ecotypic variation in chloroplast small heat-shock proteins and related thermotolerance in Chenopodium album. Plant Physiology and Biochemistry. 49(8), 898–908 (2011). [DOI] [PubMed] [Google Scholar]
- Harsh C. N. K., Aashima N., Jitendrap K. & Paramjit K. The wheat chloroplastic small heat shock protein (sHSP26) is involved in seed maturation and germination and imparts tolerance to heat stress. Plant, Cell & Environment. 35(11), 1912–1931 (2012). [DOI] [PubMed] [Google Scholar]
- Frank G. et al. Transcriptional profiling of maturing tomato (Solanum lycopersicum L.) microspores reveals the involvement of heat shock proteins, ROS scavengers, hormones, and sugars in the heat stress response. J Exp Bot. 60, 3891–3908 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjeev K. B. et al. Heat stress response in plants: a complex game with chaperones and more than twenty heat stress transcription factors. J. Biosci. 29, 471–487 (2004). [DOI] [PubMed] [Google Scholar]
- Klimecka M. & Muszynska G. Structure and functions of plant calcium dependent protein kinases. Acta Biochim Pol 54, 219–233 (2007). [PubMed] [Google Scholar]
- Chauhan H., Khurana N., Agarwal P. & Khurana P. Heat shock factors in rice (Oryza sativa L.): genome-wide expression analysis during reproductive development and abiotic stress. Mol Genet Genomics 286, 171–187 (2011). [DOI] [PubMed] [Google Scholar]
- Nishizawa A. et al. Arabidopsis heat shock transcription factor A2 as a key regulator in response to several types of environmental stress. Plant J. 48, 535–547 (2006). [DOI] [PubMed] [Google Scholar]
- Volkov R. A. et al. Heat stress-induced H2O2 is required for effective expression of heat shock genes in Arabidopsis. Plant Mol.Biol. 61, 733–746 (2006). [DOI] [PubMed] [Google Scholar]
- Chang Y. Y. et al. Heat inducible transcription factor, HsfA2, is required for extension of acquired thermotolerance in Arabidopsis. Plant Physiol. 143, 251–262 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller G., Shulaev V. & Mittler R. Reactive oxygen signaling and abiotic stress. Physiologia Plantarum. 133, 481–489 (2008) [DOI] [PubMed] [Google Scholar]
- Busch W., Wunderlich M. & Schoffl F. Identification of novel heat shock factor- dependent genes and biochemical pathways in Arabidopsis thaliana. Plant J 41, 1–14 (2005). [DOI] [PubMed] [Google Scholar]
- Li S. J., Fu Q. T., Huang W. D. & Yu D. Q. Functional analysis of an Arabidopsis transcription factor WRKY25 in heat stress. Plant Cell Rep. 28, 683–693 (2009). [DOI] [PubMed] [Google Scholar]
- Sakuma Y. et al. Dual function of an Arabidopsis transcription factor DREB2A in water-stress-responsive and heat-stress-responsive gene expression, Proc. Natl. Acad. Sci. USA 103, 18822–18827 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsukura S. et al. Comprehensive analysis of rice DREB2-type genes that encode transcription factors involved in the expression of abiotic stress-responsive genes. Mol Genet Genomics 283, 185–196 (2010). [DOI] [PubMed] [Google Scholar]
- Jakoby M., Weisshaar B. & Droge-Laser W. bZIP transcription factors in Arabidopsis. Trends Plant Sci. 7, 106–111 (2002). [DOI] [PubMed] [Google Scholar]
- Tran L. S. P. et al. Isolation and functional analysis of Arabidopsis stress-inducible NAC transcription factors that bind to a drought-responsive cis-element in the early responsive to dehydration stress 1 promoter. Plant Cell. 16, 2481–2498 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambawat S., Sharma P., Neelam R. Yadav. & Ram C. Yadav. MYB transcription factor genes as regulators for plant responses: an overview. Physiol Mol Biol Plants. 19(3), 307–321 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Wei X. L., Cheng L. J. & Tong Z. K. Identification of a C2H2-type zinc finger gene family from Eucalyptus grandis and its response to various abiotic stresses. Biologia Plantarum. 58(4), 385–390 (2014). [Google Scholar]
- Kaur N. & Gupta A. K. Signal transduction pathways under abiotic stresses in plants. Curr. Sci. 88, 1771–1780 (2005). [Google Scholar]
- Liu H. T. et al. Calmodulin is involved in heat shock signal transduction in wheat. Plant Physiol 132(3), 1186–1195 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W. et al. Molecular and genetic evidence for the key role of AtCaM3 in heat-shock signal transduction in Arabidopsis. Plant Physiol. 149, 1773–1784 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S. et al. Functional characterization of Arabidopsis thaliana WRKY39 in heat stress. Mol. Cells 29, 475–483 (2010). [DOI] [PubMed] [Google Scholar]
- Liu H. C. et al. The role of class A1 heat shock factors (HSFA1s) in response to heat and other stresses in Arabidopsis. Plant Cell Environ. 34, 738–751 (2011). [DOI] [PubMed] [Google Scholar]
- Suri S. S. & Dhindsa R. S. A heat-activated MAP kinase (HAMK) as a mediator of heat shock response in tobacco cells. Plant Cell Environ 31, 218–226 (2008). [DOI] [PubMed] [Google Scholar]
- Kong F. et al. Genome-wide analysis of the mitogen-activated protein kinase gene family in Solanum lycopersicum. Gene 499, 108–120 (2012). [DOI] [PubMed] [Google Scholar]
- Zhang X. W. et al. Transcriptome profile reveals heat response mechanism at molecular and metabolic level in rice flag leaf. Gene. 530(2), 185–192 (2013). [DOI] [PubMed] [Google Scholar]
- Irene P. & Angelos K. K. Stress and developmental responses of terpenoid biosynthetic genes in Cistus creticus subsp. Creticus. Plant Cell Rep. 29, 629–641 (2010). [DOI] [PubMed] [Google Scholar]
- Goyal K., Walton L. J. & Tunnacliffe A. LEA proteins prevent proteinaggregation due to water stress. Biochem. J. 388, 151–157 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahid A. & Close T. J. Expression of dehydrins under heat stress and their relationship with water relations of sugarcane leaves. Biol. Plant. 51, 104–109 (2007). [Google Scholar]
- Sarvajeet S. G. & Narendra T. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol. Biochem. 48, 909–930 (2010). [DOI] [PubMed] [Google Scholar]
- Grabherr M. G., Haas B. J., Yassour M., Levin J. Z. & Thompson D. A. Full length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 29, 644–652 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M. D., McCarthy D. J. & Smyth G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl M. W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2, 9 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.