

Abstract
Background
Repositioning approved drug and small molecules in novel therapeutic areas is of key interest to the pharmaceutical industry. A number of promising computational techniques have been developed to aid in repositioning, however, the majority of available methodologies require highly specific data inputs that preclude the use of many datasets and databases. There is a clear unmet need for a generalized methodology that enables the integration of multiple types of both gene expression data and database schema.
Results
ksRepo eliminates the need for a single microarray platform as input and allows for the use of a variety of drug and chemical exposure databases. We tested ksRepo’s performance on a set of five prostate cancer datasets using the Comparative Toxicogenomics Database (CTD) as our database of gene-compound interactions. ksRepo successfully predicted significance for five frontline prostate cancer therapies, representing a significant enrichment from over 7000 CTD compounds, and achieved specificity similar to other repositioning methods.
Conclusions
We present ksRepo, which enables investigators to use any data inputs for computational drug repositioning. ksRepo is implemented in a series of four functions in the R statistical environment under a BSD3 license. Source code is freely available at http://github.com/adam-sam-brown/ksRepo. A vignette is provided to aid users in performing ksRepo analysis.
Keywords: Repositioning, Drug discovery, Prostate cancer, Gene expression
Background
Repositioning of previously approved drugs is a promising methodology because it reduces the cost and duration of the drug development pipeline and reduces the likelihood of adverse events [1–4]. High-throughput repositioning efforts are especially appealing given their ability to yield many potential development opportunities [5–7]. A major goal in repositioning is the development of in silico tools that reduce the number of potential candidate molecules to be screened while also suggesting unlikely and novel possibilities. To this end, a number of groups have developed computational approaches that display high degrees of both sensitivity and specificity [8]. Many successful computational repositioning methodologies have relied on comparing individual disease RNA-level expression profiles to large databases of pre-generated multi-drug exposure profiles or known gene-drug interactions [9–12]. Unfortunately, the majority of these methodologies are hindered by their need for specific data types and formats, including requirements for detailed genomic or phenotypic annotations [9, 10], expression levels from a single microarray platform [12], and pre-determined databases of drug-gene interactions [13]. These limitations prevent investigators from utilizing newer profiling technologies, such as RNA-seq, and from utilizing alternative or proprietary compound exposure profiles. Despite these drawbacks, numerous successes using these techniques, including the highly cited Connectivity Map (Broad), suggest the utility of a pipeline capable of surpassing these hindrances [12, 14–17].
To address these limitations, a universally applicable computational repositioning tool should have flexibility in the types of data sets and databases that can be used. Specifically, we envision such a tool having 1) the ability to interrogate any case/control disease study-derived expression profile, 2) the ability to use any compound database, including those with limited numbers of gene-drug interactions, and 3) an extensible, open-source distribution. Here, we propose a generalized tool for computational repositioning that builds on the successes of previous expression-based repositioning tools while allowing greater flexibility for the investigator called ksRepo. Our methodology modifies the Kolmogorov-Smirnov (KS) enrichment approach used by the Broad Connectivity Map to enable the use of any expression-level disease study with any database containing, at minimum, gene-drug interactions from any source (with or without information about the directionality of association) [12, 13]. The only requirement of our methodology is that there is a common identifier system to which the information from both the disease and exposure databases can be converted (see Fig. 1). Unlike many popular repositioning tools (e.g. [9–13]), we provide source code for ksRepo that enables investigators to extend our methodology as new datatypes become available. We demonstrated our methodology using five independent freely available Prostate Cancer datasets [18–22] downloaded from the Gene Expression Omnibus (GEO) [23] and the open-source gene-drug interaction database, the Comparative Toxicogenomics Database (CTD) [24].
Fig. 1.
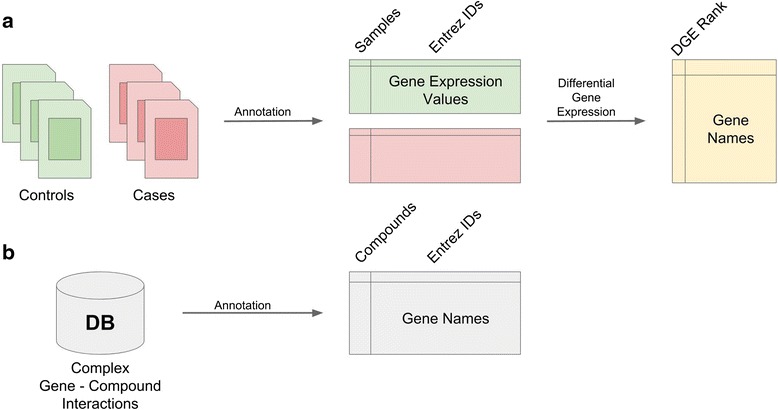
ksRepo can use any input data types. a Preparation of arbitrary expression data is accomplished by conversion to Entrez ID using manufacturer or online annotation tools followed by the user’s preferred algorithm for determining differential gene expression. b Preparation of an arbitrary database only required conversion to Entrez ID, which is often provided by the database directly
Methods
GEO dataset processing
All GEO datasets were accessed through the NCBI GEO portal and analyzed using the integrated GEO2R tool [23]. As input for GEO2R, we classified each sample within a GEO series as either normal tissue or tumor tissue. GEO2R provides a list of all probes (and corresponding gene aliases) ranked according to their degree of differential expression. We imported all of the results from GEO2R into R [25] and converted all gene aliases into EntrezGene Identifiers using the org.Hs.eg.db R package [26]. The prostate cancer datasets used in this study are GSE3868, GSE12348, GSE45016, GSE55945, and GSE6919 [18–22]. The five prostate cancer datasets were chosen on the basis of three criteria: (1) the expression profiles were derived from primary prostate cancer cells, as opposed to cell lines or short-term cultures, (2) there were healthy prostate tissue controls included in the study, and (3) tissue samples were from fresh-frozen biopsies, and not preserved (e.g. by FFPE).
CTD database construction
To generate a ksRepo-compatible database, we first downloaded the entirety of the CTD and imported the database into R (downloaded February, 2014). The CTD contains manually curated compound-gene interactions collected from the primary literature by trained experts [24]. We filtered the full database for literature-supported interactions between compounds and human genes or gene products (e.g. transcripts, proteins, or peptides). Following filtering, the resulting database contained interactions between 7170 compounds and 18,768 unique human genes. Of these 7170 compounds, 1660 are drugs approved by the FDA. A script for converting the downloadable files from CTD to a ksRepo compatible format is available in the ksRepo GitHub repository (CTDget.R script).
Kolmogorov-Smirnov enrichment score calculations
Our modified method is analogous to an “inverse” version of the Connectivity Map implementation in that we compare a single instance (complete gene expression profile) to a number of signatures (short compound-gene interaction lists) rather than comparing a single signature to a number of instances. In addition, we focus on interaction without directionality to accommodate compound exposure databases with no regulatory component or conflicting regulatory information. In addition, we consider all genes in the ranked instance gene list regardless of significance to ensure overlap between the instance and signatures. KS enrichment scores for our method are calculated as follows.
Let n be the number of genes in the instance and t be the number of genes in a given signature. Order all n genes in the instance by their differential expression. Construct a vector V of the position (∈ {1,…,n}) of each signature gene in the instance ordered gene list and sort these components in ascending order such that V(j) is the position of gene j, where j ∈ {1,…,t}. Calculate the following values:
| 1 |
| 2 |
and set KS = a if a > b. Else set KS = −b.
Both a and b quantify differences in the expected distribution of gene ranks (∈ {1,…,n}) and the observed sample of ranks in the signature. The value a ∈ U(0,1) and scales inversely with the meansignature rank (mean V(j)), with deviations proportional to the standard deviation of signature rank; the value b is the inverse of a. If a > b, then the mean signature rank is low, corresponding to enrichment, and we assign a as the KS score. If b > a, we assign -b as the KS score. In this way, signatures with highly enriched gene sets are assigned highly positive KS scores, while signatures with unenriched or inversely enriched (e.g. very high meansignature rank) are assigned KS scores near zero or negative KS scores respectively.
Because our KS test statistic has no empirical distribution, we calculated significance by bootstrapping as follows. Construct a vector L of the number of genes in each signature. For each unique ℓ in L, generate 10,000 independent resamples of the instance gene list of length ℓ and calculate KS scores for each resample. For each signature, compare the observed signatureKS score to the corresponding resample with the same number of genes. Set the p value of that signature as the proportion of resample KS scores that exceed the signatureKS score and FDR adjust to correct for multiple hypothesis testing [27].
ksRepo implementation
We implemented ksRepo testing in R as a series of four functions. The core testing function, ks_single, performs KS enrichment testing between the ranked instance gene list and one unranked signature gene list (see Fig. 2). Bootstrapped P-value calculation is accomplished by the functions boot_ks and boot_p. The final function included in the implementation, repo, is a wrapper function which calls the other three functions and formats the output. All four functions are available for non-commercial use from GitHub (http://github.com/adam-sam-brown/ksRepo). In addition, we provide a comprehensive vignette that demonstrates the use of ksRepo using one of the prostate cancer datasets, GSE6919, described above.
Fig. 2.
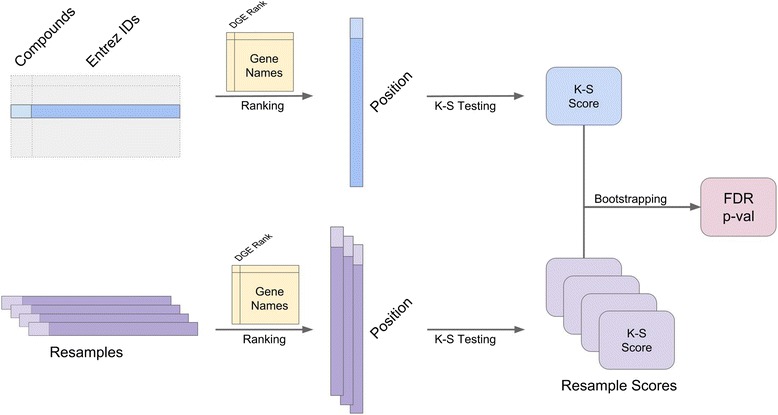
ksRepo employs a generalized K-S enrichment test and bootstrapping to determine candidate repositioning targets. See Methods for a detailed description
Results and discussion
We implemented ksRepo, an expression-based, generalized tool for computational drug repositioning. ksRepo avoids the requirements of currently available methods for specific data inputs [9–13]. Our methodology is capable of utilizing any pair of disease expression dataset and compound exposure database with the simple constraint that they be mappable to a single, common identifier system. Once an investigator has chosen the two inputs, our method straightforwardly expands the methods of Lamb and colleagues [12] to allow for varying numbers and types of gene-compound associations in the exposure database. We then compute K-S enrichment scores for each compound and report bootstrapped and FDR-corrected p-values for ease of interpretation.
To demonstrate ksRepo’s applicability, we applied our method to five independent prostate cancer datasets (GSE3868, GSE12348, GSE45016, GSE55945, and GSE6919) from three distinct microarray platforms, and attempted to detect signal from FDA-approved prostate cancer therapies from the CTD. The CTD reports expert curated gene-compound interactions from the primary literature. Unlike currently available methods (e.g. [9–13]), in which a full profile is necessary, ksRepo is able to analyze the CTD and databases like it. We also note that the use of three distinct microarray platforms precludes the use of some methods (e.g. [12, 13]), but is possible with ksRepo.
We first identified all FDA-approved prostate cancer therapies using DrugBank [28] and then determined that out of 11 small-molecule therapies, seven (Bicalutamide, Nilutamide, Leuprolide, Zoledronic Acid, Docetaxel, Aminoglutethimide, and Estropipate) were also included in the CTD. We then applied ksRepo to the five GEO prostate cancer datasets and determined the FDR-corrected p-values for each of the seven annotated therapies in the CTD. ksRepo predicted on average approximately 300 compounds (median 319 compounds, FDR-corrected p-value <0.05) corresponding to a specificity around 5 %, which is similar to reported specificities for other repositioning strategies [9–13].
For each of the five prostate cancer datasets we were able to detect significance for between one and three FDA-approved therapies at a FDR-corrected p-value less than 0.05. In each case, this represented a significant enrichment for approved therapies (Hypergeometric Test, p < 0.027, expected number of drugs μ = 0.029). Among compounds, significant prostate cancer therapies ranked on average in the 3.5th percentile and of the seven therapies, five were significant for at least one of the five datasets. We did not detect significance for two therapies, Aminoglutethimide and Estropipate; we hypothesize that due to the nature of the microarray datasets we included (tissue from primary, non-metastatic tumors), it is unlikely that we would detect secondary hormone modulatory treatments, which are typically used in treatment refractory patients with metastases [29].
These results suggest that ksRepo is a generalized methodology for computational drug repositioning. Even after intentionally reducing the information content presented to our methodology by using a database with a modest number of gene interactors by compound (as annotated in the CTD), we were still able to recover many of the FDA-approved drugs for prostate cancer. In addition, we have enabled the use of any microarray platform as input, bypassing an impediment to using a popular repositioning tool, the Broad Connectivity Map. By allowing investigators to choose any expression study and drug exposure database we hope to spur the analysis of as-yet unexplored diseases and databases. Furthermore, because ksRepo is flexible and generalized, we hope to apply it to a variety of future projects, including the incorporation of other exposure databases such as Drugbank [28] and PharmGKB [30], as well as new input types such as mRNA-seq and epigenomic information.
Conclusions
Here, we have described ksRepo, a generalized, expression-level tool for computational drug repositioning. Our implementation enables investigators to choose any case/control disease study and exposure database to suit their experimental needs. To validate our method, we applied ksRepo to five distinct prostate cancer datasets and the Comparative Toxicogenomics Database (CTD) and ksRepo successfully detected significance for a majority of FDA-approved prostate cancer therapies and significantly enriched for these compounds from the CTD. Our methodology is implemented in an open-source GitHub repository for free use. Future work with ksRepo will focus on exploring as-yet under utilized databases and the possibility of incorporating novel expression and genomic information.
Availability and requirements
Project Name: ksRepo
Project Home Page: http://github.com/adam-sam-brown/ksRepo
Operating System: Platform Independent
Programming Language: R
License: BSD-3
Acknowledgements
ASB was funded by the National Institutes of Health (NIH) Training Grant T32 GM007306-39. CJP is funded by a NIH National Institute of Environmental Health Sciences (NIEHS) K99/R00 Pathway to Independence Award (K99ES023504), a Pharmaceutical Researchers and Manufacturers Association (PhRMA) foundation fellowship, and a NIH Common Fund grant (U54HG007963).
Abbreviations
- FDA
Food and Drug Administration of the United States of America
- CTD
comparative toxicology database
- K-S
Kolmogorov-Smirnov
- GEO
gene expression omnibus
- FDR
false discovery rate
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Conceived and designed the methods: ASB, SWK, IK, CJP. Analyzed and interpreted the data: ASB, CJP; Drafted the manuscript: ASB, CJP. All authors read and approved the final version of the manuscript.
Contributor Information
Adam S. Brown, Email: adambrown@fas.harvard.edu
Sek Won Kong, Email: sekwon.kong@childrens.harvard.edu.
Isaac S. Kohane, Email: isaac_kohane@harvard.edu
Chirag J. Patel, Email: Chirag_Patel@hms.harvard.edu
References
- 1.Readhead B, Dudley J. Translational bioinformatics approaches to drug development. Adv Wound Care (New Rochelle) 2013;2(9):470–89. doi: 10.1089/wound.2012.0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jin G, Wong STC. Toward better drug repositioning: prioritizing and integrating existing methods into efficient pipelines. Drug Discov Today. 2014;19(5):637–44. doi: 10.1016/j.drudis.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shim JS, Liu JO. Recent advances in drug repositioning for the discovery of new anticancer drugs. Int J Biol Sci. 2014;10(7):654–63. doi: 10.7150/ijbs.9224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stenvang J, Kümler I, Nygård SB, Smith DH, Nielsen D, Brünner N, et al. Biomarker-guided repurposing of chemotherapeutic drugs for cancer therapy: a novel strategy in drug development. Front Oncol. 2013;3:313. doi: 10.3389/fonc.2013.00313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Munson J, Bonner M, Fried L, Hofmekler J, Arbiser J, Bellamkonda R. Identifying new small molecule anti-invasive compounds for glioma treatment. Cell Cycle. 2013;12(14):2200–9. doi: 10.4161/cc.25334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bellera CL, Sbaraglini ML, Balcazar DE, Fraccaroli L, Vanrell MC, Casassa AF, et al. High-throughput drug repositioning for the discovery of new treatments for Chagas disease. Mini Rev Med Chem. 2015;15(3):182–93. doi: 10.2174/138955751503150312120208. [DOI] [PubMed] [Google Scholar]
- 7.Shu C-W, Liu P-F, Huang C-M. High throughput screening for drug discovery of autophagy modulators. Comb Chem High Throughput Screen. 2012;15(9):721–9. doi: 10.2174/138620712803519734. [DOI] [PubMed] [Google Scholar]
- 8.Li J, Zheng S, Chen B, Butte AJ, Swamidass SJ, Lu Z. A survey of current trends in computational drug repositioning. Brief Bioinform. 2016 Jan;17(1):2-12. [DOI] [PMC free article] [PubMed]
- 9.Menden MP, Iorio F, Garnett M, McDermott U, Benes CH, Ballester PJ, et al. Machine learning prediction of cancer cell sensitivity to drugs based on genomic and chemical properties. PLoS One. 2013;8(4) doi: 10.1371/journal.pone.0061318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gottlieb A, Stein GY, Ruppin E, Sharan R. PREDICT: a method for inferring novel drug indications with application to personalized medicine. Mol Syst Biol. 2011;7:496. doi: 10.1038/msb.2011.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grover MP, Ballouz S, Mohanasundaram KA, George RA, Sherman CDH, Crowley TM, et al. Identification of novel therapeutics for complex diseases from genome-wide association data. BMC Med Genomics. 2014;7 Suppl 1:S8. doi: 10.1186/1755-8794-7-S1-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313(5795):1929–35. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 13.Huang H, Nguyen T, Ibrahim S, Shantharam S, Yue Z, Chen JY. DMAP: a connectivity map database to enable identification of novel drug repositioning candidates. BMC Bioinformatics. 2015;16 Suppl 13:S4. doi: 10.1186/1471-2105-16-S13-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu J, Putcha P, Silva JM. Recovering drug-induced apoptosis subnetwork from Connectivity Map data. Biomed Res Int. 2015;2015:708563. doi: 10.1155/2015/708563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lv J, Zhu B, Zhang L, Xie Q, Zhuo W. Detection and screening of small molecule agents for overcoming Sorafenib resistance of hepatocellular carcinoma: a bioinformatics study. Int J Clin Exp Med. 2015;8(2):2317–25. [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang M, Luo H, Xi Z, Rogaeva E. Drug repositioning for diabetes based on “omics” data mining. PLoS One. 2015;10(5) doi: 10.1371/journal.pone.0126082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manzotti G, Parenti S, Ferrari-Amorotti G, Soliera AR, Cattelani S, Montanari M, et al. Monocyte-macrophage differentiation of acute myeloid leukemia cell lines by small molecules identified through interrogation of the Connectivity Map database. Cell Cycle. 2015 Jun 23;0 [DOI] [PMC free article] [PubMed]
- 18.Yu YP, Landsittel D, Jing L, Nelson J, Ren B, Liu L, et al. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J Clin Oncol. 2004;22(14):2790–9. doi: 10.1200/JCO.2004.05.158. [DOI] [PubMed] [Google Scholar]
- 19.Arredouani MS, Lu B, Bhasin M, Eljanne M, Yue W, Mosquera J-M, et al. Identification of the transcription factor single-minded homologue 2 as a potential biomarker and immunotherapy target in prostate cancer. Clin Cancer Res. 2009;15(18):5794–802. doi: 10.1158/1078-0432.CCR-09-0911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Satake H, Tamura K, Furihata M, Anchi T, Sakoda H, Kawada C, et al. The ubiquitin-like molecule interferon-stimulated gene 15 is overexpressed in human prostate cancer. Oncol Rep. 2010;23(1):11–6. [PubMed] [Google Scholar]
- 21.Yegnasubramanian S, Haffner MC, Zhang Y, Gurel B, Cornish TC, Wu Z, et al. DNA hypomethylation arises later in prostate cancer progression than CpG island hypermethylation and contributes to metastatic tumor heterogeneity. Cancer Res. 2008;68(21):8954–67. doi: 10.1158/0008-5472.CAN-07-6088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nanni S, Priolo C, Grasselli A, D’Eletto M, Merola R, Moretti F, et al. Epithelial-restricted gene profile of primary cultures from human prostate tumors: a molecular approach to predict clinical behavior of prostate cancer. Mol Cancer Res. 2006;4(2):79–92. doi: 10.1158/1541-7786.MCR-05-0098. [DOI] [PubMed] [Google Scholar]
- 23.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: archive for functional genomics data sets—update. Nucl Acids Res [Internet]. 2013 1 [cited 2015 Jun 26];41(D1):D991–D995. [DOI] [PMC free article] [PubMed]
- 24.Davis AP, Grondin CJ, Lennon-Hopkins K, Saraceni-Richards C, Sciaky D, King BL, et al. The Comparative Toxicogenomics Database’s 10th year anniversary: update 2015. Nucleic Acids Res. 2015;43(Database issue):D914–20. doi: 10.1093/nar/gku935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.The R team . R: a language and environment for statistical computing. R foundation for statistical computing, Vienna, Austria. 2008. [Google Scholar]
- 26.Carlson M. org.Hs.eg.db: Genome wide annotation for Human. R package version 3.1.2 2015.
- 27.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995;57:289–300. [Google Scholar]
- 28.Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34(Database issue):D668–72. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lam JS, Leppert JT, Vemulapali SN, Shvarts O, Belldegrun AS. Secondary hormonal therapy for advanced prostate cancer. J Urol. 2006;175(1):27–34. doi: 10.1016/S0022-5347(05)00034-0. [DOI] [PubMed] [Google Scholar]
- 30.Thorn CF, Klein TE, Altman RB. PharmGKB: the pharmacogenetics and pharmacogenomics knowledge base. Methods Mol Biol. 2005;311:179–91. doi: 10.1385/1-59259-957-5:179. [DOI] [PubMed] [Google Scholar]