

Abstract
Coagulation factor VIII and von Willebrand factor (VWF) are key proteins in procoagulant activation. Higher FVIII coagulant activity (FVIII:C) and VWF antigen (VWF:Ag) are risk factors for cardiovascular disease and venous thromboembolism. Beyond associations with ABO blood group, genetic determinants of FVIII and VWF are not well understood, especially in non European-American populations. We performed a genetic association study of FVIII:C and VWF:Ag that assessed 50,000 gene-centric single nucleotide polymorphisms (SNPs) in 18,556 European Americans (EAs) and 5,047 African Americans (AAs) from five population-based cohorts. Previously unreported associations for FVIII:C were identified in both AAs and EAs with KNG1 (most significantly associated SNP rs710446, Ile581Thr, P=5.10 × 10−7 in EAs and P=3.88 × 10−3 in AAs) and VWF rs7962217 (Gly2705Arg, P=6.30 × 10−9 in EAs and P=2.98 × 10−2 in AAs). Significant associations for FVIII:C were also observed with F8/TMLHE region SNP rs12557310 in EAs (P=8.02 × 10−10), with VWF rs1800380 in AAs (P=5.62 × 10−11), and with MAT1A rs2236568 in AAs (P=1.69 × 10−6). We replicated previously reported associations of FVIII:C and VWF:Ag with the ABO blood group, VWF rs1063856 (Thr789Ala), rs216321 (Ala852Gln), and VWF rs2229446 (Arg2185Gln). Findings from this study expand our understanding of genetic influences for FVIII:C and VWF:Ag in both EAs and AAs.
Introduction
Coagulation factor VIII (FVIII) and von Willebrand factor (VWF) provide critical functions in hemostasis. In the circulation, 95% of FVIII is bound to VWF as an inactive complex that stabilizes FVIII [1,2]. Activated FVIII, after its release from VWF, acts as a cofactor for factor IXa-mediated activation of factor X and the subsequent conversion of prothrombin to its active form, thrombin [1]. VWF, an adhesive glycoprotein, also promotes platelet adhesion and aggregation [3]. Levels of FVIII coagulant activity (FVIII:C) and VWF antigen (VWF:Ag) in the top 25% of the population distribution are risk factors for venous thromboembolism and arterial vascular events [3,4].
In family studies, a significant genetic contribution has been documented for variation in FVIII:C and VWF:Ag, with estimated heritability ranging between 0.31 and 0.75 [5–7]. Shared genetic effects of FVIII:C and VWF:Ag on their risk to thrombosis have also been documented [8]. There are few reports of candidate gene studies focused on the FVIII, VWF, and ABO structural genes [9–14] and two genome-wide association studies (GWAS) [15,16] for FVIII:C or VWF:Ag traits. The ABO locus is a major contributor to their levels [10,15,16] and ABO alleles are also associated with the risk of venous thrombosis [17–19]. The first GWAS study, which included both FVIII:C and VWF:Ag in individuals of European ancestry (EA), reported associations at additional loci including STXBP5, SCARA5, VWF, STAB2, STX2, TC2N, and CLEC4M. These loci, including ABO, explained only 10.0-12.8% of the variation in FVIII:C and VWF:Ag [15]. The second GWAS, for VWF:Ag based on two young and healthy cohorts of EA ancestry and mixed ancestry, respectively, confirmed the associations at ABO and VWF and identified a new linkage region at chromosome 2q12–2p13 [16]. Data from other ethnic populations, such as African Americans (AAs), are limited to one report that sequenced only coding regions of the VWF gene [14].
In this study, we investigated the associations of circulating levels of FVIII:C and VWF:Ag with single nucleotide polymorphisms (SNPs) on the gene-centric 50K SNP ITMAT-Broad-CARe (IBC) genotyping array. The CARe IBC chip was designed to include a greater SNP marker density, including more non-synonymous variants, for more than 2,000 cardiovascular candidate regions, compared with the genome-wide arrays used in the published GWAS study [20]. The analyses were based upon 18,556 European Americans (EAs) and 5,047 AAs from five population-based cohorts in the Candidate Gene Association Resource (CARe) Consortium [21].
Material and Methods
Study population
This analysis included five participating cohorts in the CARe Consortium [21] that had assayed FVIII:C and/or VWF:Ag: the Atherosclerosis Risk in Communities (ARIC) study [22], the Coronary Artery Risk Development in Young Adults (CARDIA) study [23], the Cardiovascular Health Study (CHS) [24,25], the Framingham Heart Study (FHS) [26], and the Multi-Ethnic Study of Atherosclerosis (MESA) [27] (Table I). All participating institutions gave institutional review board approval for this study and all participants gave written informed consent. Details on the participating cohorts are provided in Supporting Information Methods.
Table I. Description of Basic Characteristics in the CARe Cohorts: Mean ± Standard Deviation or Percentage Unless Otherwise Mentioned.
| Phenotypes | ARIC EA | ARIC AA | CARDIA EA | CARDIA AA | CHS EA | CHS AA | MESA EA | MESA AA | FHS |
|---|---|---|---|---|---|---|---|---|---|
| Total Na | 9,050 | 2,800 | 801 | 484 | 3,830 | 175 | 2,270 | 1,588 | 2,605 |
| Age, years | 54.1 ± 5.7 | 53.2 ± 5.8 | 25.8 ± 3.2b | 24.4 ± 4.0b | 72.8 ± 5.6 | 72.7 ± 5.6 | 62.7 ± 10.3 | 62.2 ± 10.1 | 54.4 ± 9.7 |
| Age range | 44–66 | 44–66 | 18–32b | 17–34b | 65–100 | 65–88 | 44–84 | 45–84 | 26–82 |
| Female (%) | 54.9 | 63.4 | 52.4 | 59.5 | 56.4 | 64.0 | 52.3 | 54.2 | 54.6 |
| FVIII:C (%) (N) | 125 ± 34 (9,041) | 146 ± 46 (2,799) | 93 ± 28 (780) | 106 ± 38 (458) | 121 ± 37 (3,830) | 140 ± 44 (175) | 95 ± 36 (2,270) | 108 ± 43 (1,588) | NA |
| Median FVIII: C, % (IQR) | 121 (101–143) | 140 (114–170) | 90 (74–109) | 101 (78–129) | 116 (95–141) | 134 (106–166) | 90 (70–114) | 101 (77–132) | NA |
| VWF: Ag, % (N) | 111 ± 42 (9,045) | 133 ± 55 (2,799) | 97 ± 32 (799) | 111 ± 44 (483) | NA | NA | 134 ± 54 (414) | 157 ± 63 (178) | 126 ± 45 (2,605) |
| Median VWF: Ag, % (IQR) | 104 (81–133) | 124 (92–164) | 94 (74–115) | 102 (80–134) | NA | NA | 123 (95–166) | 147 (115–199) | 120 (91-156) |
EA = European American, AA = African American, NA = phenotype not available; IQR = interquartle range.
overall N available for the analysis of FVIII:C or VWF:Ag.
Age at baseline.
Phenotype measurement
Plasma FVIII:C and/or VWF:Ag was measured in the entire cohort at baseline for ARIC (both FVIII:C and VWF:Ag) [28,29], CHS (FVIIIc) [30], and MESA (FVIIIc) [31]. In CARDIA, participants attending two field centers were measured at the years 5 and 7 exams for plasma FVIII:C and at the years 2, 5, and 7 exams for VWF:Ag [32]. Additionally, plasma VWF:Ag was measured in the FHS Offspring cohort during the fifth examination cycle [33] and in a random sample of 1,000 participants in MESA [31]. In each cohort, FVIII:C was measured using standard clinical clotting time-based assays and VWF:Ag was measured by ELISA or immunoturbidometric assays. Details on the laboratory assays are provided in Supporting Information Table SI. Information on clinical, demographic, lifestyle, and anthropometric characteristics were obtained in each cohort at the same visit as the measurements for FVIII:C and VWF:Ag.
Genotyping, imputation, and quality control
All samples were genotyped at the Broad Institute using the IBC Illumina iSELECT array [20]. The 50,000 SNPs on the IBC array are distributed across ∼2,100 genes selected to cover a range of cardiovascular, metabolic, lung, blood, sleep, and inflammatory pathways [20]. The SNP selection, designed to capture maximal genetic information specific to European and African ancestries excluding rare variants, was informed by the HapMap data and supplemented by the SeattleSNPs and Environmental Genome Project resequencing data [20]. All non-synonymous variants of minor allele frequency (MAF) > 0.01 were included to supplement tagging SNPs. Imputation to the phase 2 HapMap data at gene regions for un-genotyped SNPs was conducted using MACH 1.0.16 [34], with the CEU founders of the HapMap2 as the reference panel for the CARe EA samples and the combined CEU+YRI samples as the reference panel for the CARe AA samples. SNPs with imputation quality score < 0.6 were excluded. Details on genotyping, imputation, and quality control (QC) in these five participating cohorts are provided in Supporting Information Methods.
Statistical analysis
Participants who were taking anticoagulants or had FVIII:C or VWF:Ag values more than 6 standard deviations from the mean were excluded. Untransformed measurements for FVIII:C and VWF:Ag were regressed on age, sex, and study site (where appropriate) using linear regression models stratified by cohort and ethnic group to create cohort and ethnic-specific residuals. These residuals were inverse normal transformed and used in genetic association analysis, performed separately for EAs and AAs within each study. In CARDIA, the residuals based on measurements of each exam year were averaged across years and the averaged values were used in the inverse normal transformation. The genetic analysis used an allele dosage for each SNP, assuming an additive genetic effect, and adjusted for the first 10 principal components derived from EIGENSTRAT [35] to account for potential population stratification. For cohorts of unrelated individuals, a linear regression model was used with PLINK V 1.0.7 [36]; for the FHS family data, a linear mixed effects model was used to account for correlation between individuals due to family structure [37]. Ethnic-specific results across studies were combined using a fixed effects, inverse variance-weighted meta-analysis as implemented in METAL [38]. Genomic control correction was applied during the meta-analysis. SNPs with MAF-weighted sample size (MAF × N) < 10 or imputed SNPs with MAF < 1% were excluded from individual cohorts. The significance threshold was P < 2.0 × 10−6, to account for the effective number of independent tests [39]. Heterogeneity was assessed using the I2 inconsistency metric. To obtain clinically meaningful effect size estimates, association analyses were repeated for the most significantly associated SNPs with untransformed FVIII:C and VWF:Ag measurements. The proportion of variance explained by SNPs was based on the ARIC data, the largest study in the consortium, and calculated by subtraction of variance explained by non-SNP covariates from the total variance explained by all covariates in linear regression models.
Analysis of X chromosome SNPs
The analytical approach for X chromosome SNPs was similar to that described above for autosomal SNPs except as follows: for males, genotypes for SNPs on the X chromosome were coded as 0 or 2 [40]. In PLINK linear regression, sex was automatically adjusted for as a covariate for the analysis of X chromosome SNPs. The results from the PLINK analysis were highly consistent with those from an alternative approach in which males and females were analyzed separately in linear regression in SAS and meta-analyzed across cohort and gender groups. The percentage of variance explained by X chromosome SNPs (i.e., F8 SNPs) was calculated by GCTA (genome-wide complex trait analysis) with full dosage compensation [41].
When multiple statistically significant SNPs clustered at a region, sequential conditional analyses were performed to adjust for the most significant SNP from each adjustment step until no other SNP attained significance. Since the association of ABO blood group with VWF:Ag and FVIII:C was known and the ABO locus was significant in this study, we used the following SNPs to tag the O, B, A2, and O2 groups in conditional analyses: rs529565 (tag for O in EA, r2=0.67), rs8176693 (for O in AA, r2=0.55), rs8176749 (one of the functional variants for B group), rs8176704 (for A2, r2=1 in both EA and AA), and rs512770 (the functional variant for O2) [42]. Linkage disequilibrium (LD) between SNPs, represented by r2, was used to evaluate the independence of associations from a region. The r2 statistics from the CEU sample of the HapMap phase 2 were used as a reference for the CARe EA data, while those from the combined African and African American samples of the HapMap phases 2 and 3 were used for the CARe AA data.
Results
The distributions of demographic variables, FVIII:C, and VWF:Ag in 18,556 EA and 5,047 AA participants are shown in Table I. The race-specific quantile-quantile (Q-Q) plots for observed vs. expected –log10 P values for both traits are shown in Supporting Information Figs. S1-S4. There was little influence of population stratification on the data, as evidenced by inflation factors for FVIII:C and VWF:Ag of 1.06 and 1.01, respectively, in EAs, and 0.97 and 1.00, respectively, in AAs. Table II presents the most significantly associated SNPs with FVIII:C and VWF:Ag in EAs and AAs.
Table II. Top Significant SNP Associations for FVIII:C and VWF:Ag at P value <2.0 × 10−6 in EAs and AAs of CARe (Ordered by Chromosome).
| Trait | Region | Top SNP | Position | A1/A2 | AFA1 | Gene (var) | Beta/SE, % | P value | Var% | Imput | Note |
|---|---|---|---|---|---|---|---|---|---|---|---|
| EA | |||||||||||
| FVIII | 3q27 | rs698078a | 187941921 | A/G | 0.59 | KNG1 (intr) | 1.99/0.38 | 4.26 × 10−7 | 0.33 | 0.95–1.00 | New |
| FVIII | 3q27 | rs710446a | 187942621 | T/C | 0.59 | KNG1 (cns) | 1.97/0.39 | 5.10 × 10−7 | 0.33 | – | New |
| FVIII | 9q34.1-.2 | rs529565 | 135139321 | T/C | 0.65 | ABO (intr) | 17.03/0.37 | <1.0 × 10−199 | 11.57 | 0.98–1.05 | Repl |
| FVIII | 12p13.3 | rs1063856 | 6023795 | T/C | 0.64 | VWF (cns) | 2.68/0.40 | 5.84 × 10−12 | 0.32 | – | Repl |
| FVIII | 12p13.3 | rs7962217 | 5931820 | C/T | 0.94 | VWF (cns) | 4.84/0.83 | 6.30 × 10−9 | 0.21 | – | New |
| FVIII | 12p13.3 | rs216321 | 6014245 | C/T | 0.91 | VWF (cns) | −3.99/0.67 | 4.70 × 10−10 | 0.19 | – | New |
| FVIII | Xq28 | rs12557310b | 154388892 | C/T | 0.72 | TMLHE (intr) | −2.94/0.48 | 8.02 × 10−10 | 1.86 | – | New |
| FVIII | Xq28 | rs2096362b | 153885468 | G/A | 0.74 | F8 (intr) | −2.82/0.49 | 1.88 × 10−9 | 0.13 | – | Repl |
| VWF | 9q34.1-.2 | rs529565 | 135139321 | T/C | 0.66 | ABO (intr) | 22.18/0.50 | <1.0 × 10−199 | 13.93 | 0.98–1.00 | Repl |
| VWF | 12p13.3 | rs1063856 | 6023795 | T/C | 0.64 | VWF (cns) | 4.83/0.54 | 1.06 × 10−19 | 0.88 | – | Repl |
| VWF | 12p13.3 | rs216321 | 6014245 | C/T | 0.91 | VWF (cns) | −7.24/0.92 | 1.71 × 10−17 | 0.48 | – | Repl |
| AA | |||||||||||
| FVIII | 9q34.1-.2 | rs8176693 | 135127478 | C/T | 0.90 | ABO (intr) | 37.24/1.65 | 2.51 × 10−114 | 8.62 | 0.66–0.78 | Repl |
| FVIII | 10q22 | rs2236568 | 82025903 | A/C | 0.76 | MAT1A (intr) | 5.28/1.06 | 1.69 × 10−6 | 0.69 | – | New |
| FVIII | 12p13.3 | rs2229446 | 5973333 | C/T | 0.81 | VWF (cns) | −9.47/1.13 | 1.95 × 10−20 | 1.16 | – | Repl |
| FVIII | 12p13.3 | rs1800380 | 6008856 | C/T | 0.70 | VWF (cs) | 5.72/0.95 | 5.62 × 10−11 | 0.35 | 0.92–1.00 | New |
| FVIII | 12p13.3 | rs4764482c | 6039994 | T/C | 0.80 | VWF (intr) | −5.74/1.10 | 8.12 × 10−8 | 0.74 | – | Repl |
| VWF | 9q34.1-.2 | rs8176693 | 135127478 | C/T | 0.90 | ABO (intr) | 48.62/2.37 | 1.66 × 10−89 | 10.18 | 0.65–0.74 | Repl |
| VWF | 12p13.3 | rs2229446 | 5973333 | C/T | 0.81 | VWF (cns) | −12.67/1.68 | 1.13 × 10−16 | 1.75 | – | Repl |
| VWF | 12p13.3 | rs1063856c | 6023795 | C/T | 0.59 | VWF (cns) | −8.69/1.32 | 1.72 × 10−10 | 1.21 | – | Repl |
All data presented are independent associations except those labeled with a, b, and c, A1 = allele 1 (major allele), A2 = allele 2 (minor allele), AFA1 = average allele frequency for A1 allele in the meta-analysis, var = variant class (intr = intron, cns = coding-nonsynonymous or missense, cs = coding-synonymous), Beta = change in trait level per 1 allele increase in the minor allele based on untransformed measurements, SE = standard error, Var% = variance % explained by the SNP based on the ARIC data; imput = ratio of observed to expected variance as a measure of imputation quality (range presented, “- ” for genotyped SNPs), Repl = replication.
r2 = 1.0 in the HapMap CEU sample.
r2 = 0.08 in the HapMap CEU sample.
r2 = 0.16 in the HapMap African and African American samples.
Genetic associations in EAs
In EAs, 119 SNPs for FVIII:C and 140 SNPs for VWF:Ag exceeded the pre-specified significance threshold of P < 2 × 10−6. The significant SNP associations for FVIII:C clustered at 4 loci: KNG1 (kininogen 1) on chromosome 3q27; ABO on chromosome 9q34.1-q34.2; VWF on chromosome 12p13.3; and F8/TMLHE (trimethyllysine hydroxylase, epsilon) on chromosome Xq28. The significant SNP associations for VWF:Ag clustered at 2 loci, ABO and VWF.
At the KNG1 locus, 3 SNPs that are in high LD (r2=0.84–1.00) were associated with FVIII:C – rs698078 (P=4.26 × 10−7, Table II), rs710446 (coding, nonsynonymous, P=5.10 × 10−7, Table II), and rs5030062 (P=1.81 × 10−6) (Fig. 1). The P values for the other two SNPs did not attain statistical significance after adjustment for rs710446 (P > 0.05). At the ABO locus, the strongest association for both FVIII:C and VWF:Ag was with rs529565 (P < 1.0 × 10−199 for both FVIII:C and VWF:Ag, Table II). rs529565 is intronic to the ABO gene and tags the O blood group (r2=0.67 with the O group variant). Simultaneous adjustment for the SNPs that tag the ABO O, B, A2, and O1v/O2 groups abolished all the other ABO SNP associations for both traits.
Figure 1.
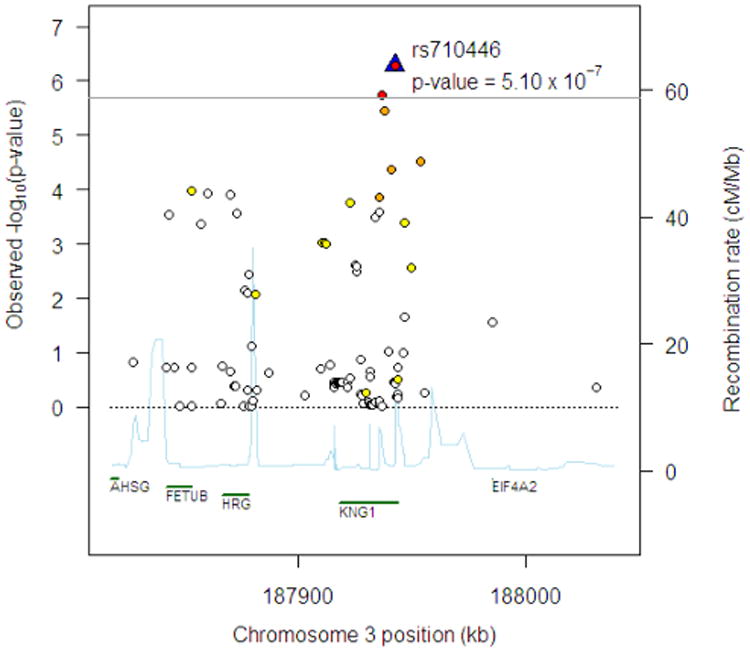
Regional association plot at chromosome 3q27 region for FVIII:C in EAs. The horizontal line indicates the pre-specified significance threshold of P = 2.0 × 10−6. The top SNP is shown by the blue triangle and labeled. The colors of the remaining SNPs reflect the r2 with the top SNP based on the HapMap CEU data with the following color scheme: r2≥0.8—red, 0.5≤r2 < 0.8—orange, 0.2≤r2 < 0.5—yellow, r2 < 0.2—white. The light blue line represents the recombination rate (the y axis at right side) based on the data from the HapMap CEU, YRI and JPT1CHB populations. Gene annotations are shown along the x axis.
At the VWF locus, three sets of independent SNP associations, spanning 24 SNPs, were identified for FVIII:C (Table II, Supporting Information Table SII). The three sets were led by rs1063856 (coding-nonsynonymous, Thr789Ala, P=5.84 × 10−12), rs7962217 (coding-nonsynonymous, Gly2705Arg, P=6.30 × 10−9; P=4.03 × 10−8 after adjustment for rs1063856), and rs216321 (coding-nonsynonymous, Ala852Gln, P=6.30 × 10−9; P=4.03 × 10−8 after adjustment for rs1063856 and rs7962217), respectively. The top variants rs1063856, rs7962217, and rs216321 are in low LD (r2=0.008–0.035) and are consistent with three independent sites of genetic contribution within a single gene to variation in FVIII:C. For VWF:Ag at the VWF locus, two sets of independent associations of 42 SNPs emerged (Table II, Supporting Information Table SIII), led by rs1063856 (P=1.06 × 10−19) and rs216321 (P=1.71 × 10−17; P=6.36 × 10−12 after adjusting for rs1063856), respectively. Association plots are presented for FVIII:C (Fig. 2) and VWF:Ag (Supporting Information Fig. S5) in the VWF locus.
Figure 2.
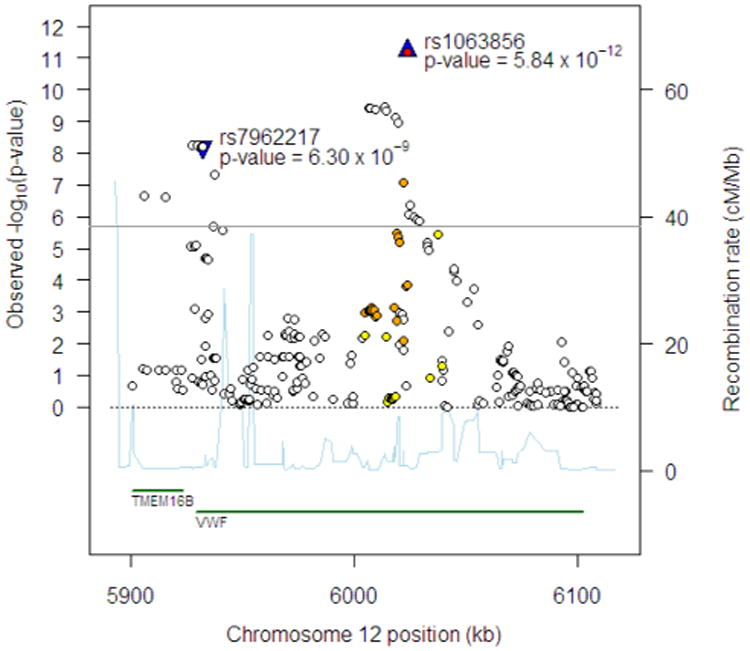
Regional association plot at chromosome 12p13.3 region for FVIII:C in EAs. The 1st and 2nd top independent SNPs are shown by the blue triangle and labeled.
Interestingly, the second set of independent SNPs in VWF for FVIII:C, led by rs7962217, was not associated with VWF:Ag in either the meta-analysis of all cohorts (P=0.38 for rs7962217) or individual cohorts (e.g., in ARIC, P=0.75 for rs7962217).
On the X chromosome, one TMLHE SNP (rs12557310, P=8.02 × 10−10) and 4 SNPs from F8 (most significant SNP, rs2096362, P=1.88 × 10−9) were significantly associated with FVIII:C (Supporting Information Tables SIV, Supporting Information Fig. S6). rs12557310 is the only SNP from the TMLHE gene that was genotyped. Of the 4 F8 SNPs, one is coding-nonsynonymous (rs1800291, P=9.74 × 10−7) and the remaining 3 SNPs (including the most significant, rs2096362) are intronic. There was weak to moderate LD between the TMLHE SNP rs12557310 and the four F8 SNPs (r2=0.08–0.31), with stronger LD among the four F8 SNPs (r2=0.20–1.00), as expected. Adjustment for rs1800291 only modestly attenuated the association at TMLHE (P=3.12 × 10−5 for rs12557310, Supporting Information Fig. S6). In contrast, adjusting for rs12557310 in TMLHE removed the majority of the association for the F8 SNPs (P=0.08–0.0028).
Of the associations identified above in EAs, those with the KNG1 locus, VWF (rs7962217 and rs216321), and TMLHE (rs12557310) for FVIII:C have not been previously reported. The total variance explained by the significant and independent associated SNPs was 14.5% for FVIII:C (12.6% for autosomal SNPs and 1.9% for X chromosome SNPs) and 15.6% for VWF:Ag.
Genetic associations in AAs
In African Americans, 85 SNPs were significantly associated with FVIII:C and 76 SNPs were associated with VWF:Ag. The SNPs were located in three loci for FVIII:C – ABO, MAT1A (methionine adenosyltransferase I, alpha, chromosome 10q22), and VWF. In contrast to FVIII:C, the SNP associations for VWF:Ag were located in ABO and VWF.
In the ABO locus, 68 SNPs were significantly associated with FVIII:C and/or VWF:Ag. The most significantly associated SNP for both traits was rs8176693 (P=2.51 × 10−114 for FVIII:C and P=1.66 × 10−89 for VWF:Ag; Table II). rs8176693 is intronic in ABO and a tag for the O blood group (r2=0.55). Adjustment for SNPs that tag the O, B, A2, and O1v/O2 groups did not abolish all the other ABO associations for either trait, with a few intergenic and intronic SNPs remaining significant (data not shown). These remaining SNP associations could be attributable to incomplete adjustment, as a result of relatively poor imputation quality for the O proxy SNP rs8176693 (0.65–0.78), as well as modest LD between rs8176693 and the O functional variant in African Americans (r2=0.55). The remaining SNP associations at the ABO locus were not further investigated.
At the MAT1A locus, one intronic variant (rs2236568) was significantly associated with FVIII:C (P=1.69 × 10−6) while other SNPs showed similar, but weaker, association with FVIII:C (Supporting Information Figures).
At the VWF locus, 16 and 8 SNPs exceeded the P < 2 × 10−6 threshold in their associations with FVIII:C and VWF:Ag (Supporting Information Tables SV and SVI), respectively. The significant SNPs for FVIII:C included three independent sets, led by rs2229446 (coding-nonsynonymous, Arg2185Gln, P=1.95 × 10−20), rs1800380 (coding-synonymous, P=5.62 × 10−11; P=7.60 × 10−10 after conditioning on rs2229446), and rs4764482 (intronic, P=8.12 × 10−8; P=9.57 × 10−8 after 2nd conditional analysis). The significant SNPs for VWF:Ag at VWF included two independent sets and were led by rs2229446 (P=1.13 × 10−16) and rs1063856 (P=1.72 × 10−10; P=5.14 × 10−12 after conditioning on rs2229446). There is low LD among the leading variants rs2229446, rs1800380, and rs1063856 (r2=0.004–0.02). rs1063856, the second top independent SNP for VWF:Ag in AAs, is tagged by the 3rd independent set for FVIII:C (r2=0.54–0.16, Supporting Information Results). Association plots for FVIII:C and VWF:Ag at the VWF region in AAs are presented in Supporting Information Figures.
Of the associations in AAs, those with the MAT1A locus and VWF rs1800380 for FVIII:C have not been previously reported. The total variance explained by the significant and independent SNPs was 11.0% for FVIII:C and 13.2% for VWF:Ag.
Details on the conditional analysis results can be found in Supporting Information Results, and association plots for other loci in Supporting Information Figures. There was no evidence of significant heterogeneity across studies for most of the associated SNPs, with the exception of one between ABO rs529565 with VWF:Ag in EAs (P=1.92 × 10−8); however, the effect sizes were not substantially different across studies (data not shown).
Cross-ethnic comparison
The most associated SNPs for FVIII:C and VWF:Ag among EAs and AAs were compared in Table III. Most of the significantly associated SNPs identified in either EAs or AAs were replicated in the other ethnic group, with similar direction of association and nominal p < 0.05. An exception was noted for FVIII:C with the TMLHE/F8 SNPs in AAs and the MAT1A SNP association in EAs. The strongest SNP association for FVIII:C at F8 in AAs was different—rs5945270 (intronic, MAF=0.006, P=0.03). In both African/African American and European samples of the HapMap Project, there is low LD between the non-overlapping VWF SNPs identified in our EA (rs7962217 and rs216321) and AA (rs2229446 and rs1800380) populations (Supporting Information Tables SVIIa and SVIIb).
Table III. Top Significant SNP Associations in EAs and AAs (in Bold) and the Corresponding Results in the Other Ethnic Population.
| Trait | SNP | Gene | A1/A2 | EA | AA | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||||
| AFA1 | N | Beta/SE, % | P value | AFA1 | N | Beta/SE, % | P value | ||||
| FVIII | rs698078 | KNG1 | A/G | 0.59 | 15,921 | 1.99/0.38 | 4.26 × 10−07 | 0.49 | 5,020 | 2.08/0.88 | 3.38 × 10−3 |
| rs710446 | KNG1 | T/C | 0.59 | 15,915 | 1.97/0.39 | 5.10 × 10−07 | 0.49 | 5,005 | 1.99/0.88 | 3.88 × 10−3 | |
| rs529565 | ABO | T/C | 0.65 | 15,921 | 17.03/0.37 | <1.0 × 10−199 | 0.62 | 5,020 | 17.40/0.88 | 1.81 × 10−92 | |
| rs8176693 | ABO | C/T | 0.93 | 15,921 | 14.90/0.70 | 5.65 × 10−105 | 0.90 | 5,020 | 37.24/1.65 | 2.51 × 10−114 | |
| rs2236568 | MAT1A | A/C | 0.45 | 15,917 | −0.15/0.38 | 0.75 | 0.76 | 5,004 | 5.28/1.06 | 1.69 × 10−6 | |
| rs1063856 | VWF | T/C | 0.64 | 15,913 | 2.68/0.40 | 5.84 × 10−12 | 0.42 | 5,005 | 4.61/0.89 | 3.92 × 10−06 | |
| rs7962217 | VWF | C/T | 0.94 | 15,914 | 4.84/0.83 | 6.30 × 10−9 | 0.98 | 4,372 | 6.96/3.40 | 2.98 × 10−2 | |
| rs216321 | VWF | C/T | 0.91 | 15,917 | −3.99/0.67 | 4.7 × 10−10 | 0.95 | 5,005 | −6.64/1.97 | 1.13 × 10−3 | |
| rs2229446 | VWF | C/T | 0.99 | 9,039 | 2.85/4.78 | 0.89 | 0.81 | 4,992 | −9.47/1.13 | 1.95 × 10−20 | |
| rs1800380 | VWF | C/T | 0.75 | 15,921 | 1.47/0.43 | 8.70 × 10−4 | 0.70 | 5,020 | 5.72/0.95 | 5.62 × 10−11 | |
| rs4764482 | VWF | T/C | 0.48 | 15,916 | −0.75/0.38 | 0.07 | 0.80 | 5,005 | −5.74/1.10 | 8.12 × 10−8 | |
| rs12557310 | TMLHE | C/T | 0.72 | 15,909 | −2.94/0.48 | 8.02 × 10−10 | 0.15 | 5,005 | −1.19/1.39 | 0.34 | |
| rs2096362 | F8 | G/A | 0.74 | 15,814 | −2.82/0.49 | 1.88 × 10−9 | 0.47 | 4,981 | 0.13/0.97 | 0.84 | |
| VWF | rs529565 | ABO | T/C | 0.66 | 12,863 | 22.18/0.50 | <1.0 × 10−199 | 0.62 | 3,460 | 23.90/1.30 | 1.23 × 10−76 |
| rs8176693 | ABO | C/T | 0.93 | 12,863 | 20.50/0.96 | 1.16 × 10−101 | 0.90 | 3,460 | 48.62/2.37 | 1.66 × 10−89 | |
| rs1063856 | VWF | T/C | 0.64 | 12,855 | 4.83/0.54 | 1.06 × 10−19 | 0.41 | 3,446 | 8.69/1.32 | 1.72 × 10−10 | |
| rs216321 | VWF | C/T | 0.91 | 12,858 | −7.24/0.92 | 1.71 × 10−17 | 0.95 | 3,267 | −9.86/2.96 | 2.50 × 10−3 | |
| rs2229446 | VWF | C/T | 0.99 | 9,043 | −16.72/6.14 | 0.05 | 0.81 | 3,435 | −12.67/1.68 | 1.13 × 10−16 | |
| rs1063856 | VWF | C/T | 0.36 | 12,855 | −4.83/0.54 | 1.06 × 10−19 | 0.59 | 3,446 | −8.69/1.32 | 1.72 × 10−10 | |
A1 = allele 1 (major allele in the ethnic population in which the associations were the middle significant ones, i.e. in bold), A2 = allele 2 (minor allele in the ethnic population in which the associations were the middle significant ones), AFA1 = average allele frequency for A1 allele in the corresponding ethnic population, Beta = change in trait level per 1 allele increase in the A2 allele based on untransformed measurements, SE = standard error.
Discussion
We investigated genetic determinants of FVIII:C and VWF:Ag levels based on 50,000 SNPs from a cardiovascular gene-centric chip (supplemented by imputation data) in 18,556 EAs and 5,047 AAs from five population-based cohort studies in the USA. We identified novel loci, KNG1 and TMLHE in EAs, and novel associations in VWF (rs7962217 in EAs and rs1800380 in AAs) that have not been reported previously for FVIII:C and VWF:Ag. The newly identified associations with KNG1 and VWF SNPs were replicated across our ethnic populations. Of note, the locus represented by VWF rs7962217 was specific to FVIII:C, with no significant association for VWF:Ag. We also extended the association of VWF rs216321 with VWF:Ag previously reported in EAs [43,44] to FVIII:C in both EA and AA populations. Finally we confirmed previously reported associations of FVIII:C and VWF:Ag levels with VWF rs1063856 from EA and AA populations [13–15,43–46], VWF rs2229446 from an AA population [14], and the ABO locus [10] from a European population.
At the KNG1 locus, the most associated SNP, rs710446, is in exon 10 of kininogen isoform 1 (high molecular weight kininogen, HMWK) and codes for an Ile to Thr substitution at amino acid 581, predicted to be involved in splicing regulation [47]. KNG1 has not been previously associated with FVIII:C. HMWK is a cofactor for activation of kallikrein and factors XI and XII. Defects in KNG1 are the cause of HMWK deficiency (MIM #228960), an autosomal recessive disorder. rs710446 has been associated with the activated partial thromboplastin time (aPTT), FXI, and VTE risk in EA populations [48–52]. The aPTT is highly correlated with FVIII:C and is a global coagulation test that reflects the interacting effects of factors in the classical intrinsic (FXII, FXI, FIX and FVIII) and common coagulation cascades as well as contact activation [53]. There are two possible interpretations of the identified association of KNG1 with FVIII:C. The first is that the association was observed by virtue of the aPTT reflecting HMWK levels (in part) and that FVIII:C is measured using an aPTT-based assay; the second is that this association reflects an unknown functional link between HWMK and FVIII. Further investigations, including functional assays for HMWK levels, are needed to understand the underlying mechanisms for this association.
Because VWF serves as a carrier for FVIII, we expected overlapping genetic associations for these two phenotypes at ABO and VWF. However, we observed discrepant results for VWF rs7962217; this SNP was only associated with FVIII:C, indicating that the locus tagged by VWF rs7962217 influences FVIII:C through other pathways than VWF:Ag levels. VWF rs7962217 is located in exon 50 of the VWF gene and codes for Gly to Arg substitution at amino acid 2705 (G2705R), which is predicted to be deleterious, affecting a splicing site [47,54]. Exon 50 is within the carboxy-terminus (CK) domain of the VWF protein. The CK domain is crucial for the dimerization of the VWF subunit and mutations in this domain have been identified in type 2A von Willebrand Disease (VWD2A) [55], thought to be associated with synthesis or proteolysis of VWF multimers. Future studies are needed to investigate the mechanisms by which this VWF variant influences FVIII:C level, including the possibility of mediation through VWF dimerization on the interaction of VWF with FVIII.
We detected significant associations at the F8/TMLHE region for FVIII:C in EAs, in contrast to the published GWAS study [15] that observed no significant associations in the F8 locus. In that GWAS, imputation data for the X chromosome were not available in two of the four studies. In our study, the F8 SNPs on the IBC chips were selected as tag SNPs to provide a greater LD coverage than genome-wide arrays, enabling a more comprehensive investigation of F8 effects on phenotypes. Of the four F8 SNPs that were significantly associated with FVIII:C in our study, rs1800291 (D1241E) has been previously associated with FVIII:C in two studies of European populations [9,11]. In our study, the effect size of rs1800291 was weaker compared to the other three F8 SNPs and TMLHE rs12557310. Adjustment for rs1800291 only modestly reduced the significance of association for the other SNPs, in contrast to more substantial reduction in significance after adjusting for rs12557310. A previously published report of the ARIC data identified eight F8 intronic SNPs associated with FVIII:C or FVIII:C/VWF:Ag in EAs [56]. There is weak to modest LD between rs12557310 and the eight reported SNPs (r2=0.11–0.36), in contrast to stronger LD between rs1800291 and the eight SNPs (r2=0.14–0.67). Therefore, it is possible that the associations we observed at the TMLHE/F8 locus are attributable to a different underlying F8 variant than those reflected by rs1800291 or the eight F8 SNPs. TMLHE encodes trimethyllysine dioxygenase, whose relevance to coagulation pathways is currently unknown. It was noted that the associations at the TMLHE/F8 region were not observed in AAs of our study. This discrepancy could be due to different LD between the two populations as well as interactions by other ethnicity-related factors.
The association between FVIII:C and MAT1A SNPs has not been previously reported in any ethnic populations. MAT1A encodes hepatic methionine adenosyltransferase I/III, which is responsible for the synthesis of S-adenosylmethionine (SAM). SAM donates methyl groups for most biological methylations including the generation of homocysteine [57], and hyperhomocysteinemia is a modest risk factor for VTE [57]. The MAT1A SNP associations detected with FVIII:C should be confirmed in independent populations, given the lack of replication for this association in the EAs in this study.
Recently, Johnsen et al. [14] reported associations of FVIII:C and VWF:Ag with nonsynonymous variants in VWF in four AA populations, discovered by exome sequencing in the NHLBI Exome Sequencing Project; three of the cohorts overlapped with those in this study (ARIC, CARDIA and MESA). In that study [14], six VWF SNPs were independently associated with VWF:Ag and/or FVIII:C levels: rs1063856, rs57950734, rs11063988, rs149424724, rs76342212 (i.e., rs2229446), and rs61750625. Our study confirmed the findings for rs1063856 and extended the association for rs2229446 to EA populations. Of the other three VWF SNPs identified in our study (rs7962217, rs216321, and rs1800380), Johnsen et al. [14] analyzed only rs7962217 but did not find significant association (P=0.25–0.55). rs7962217 was primarily identified in EAs of our study (P=6.30 × 10−9) with nominal significance in AAs (P=0.03).
While AA populations in the two studies largely overlapped, all participants in our study were genotyped for rs7962217, in contrast to 595 participants in the Johnsen et al. [14] study being sequenced and the rest imputed for all VWF variants. The difference in genotyping coverage might explain the discrepant significance for this SNP in AAs. Between our VWF SNPs (rs216321 and rs1800380) and the other four SNPs of that study, LD information in HapMap is only available for rs61750625, which was not in LD with our variants (r2=0). In the pooled ARIC IBC data with the VWF sequencing and imputation data from Johnsen et al. [14], LD between our SNPs (rs216321 and rs1800380) and the other three of that study [14] (rs57950734, rs11063988, rs149424724) approached zero for most pairs (r2 < 0.05), with the highest between rs1800380 and rs11063988 (r2=0.11).
Strengths and limitations
The CARe IBC custom genotyping array provides a great advantage for this study because a majority of the genes relevant to the coagulation pathway were included and the array has better LD coverage, including more non-synonymous variants, than in the previously published GWAS [15]. While the EA populations in this study partially overlapped with those in the published GWAS [15], we included new populations (CARDIA and MESA) which, in combination with the advantages of the CARe IBC array and sequential conditional analysis, provided additional information beyond the published GWAS study. The increased percentage of variance explained by the most associated, independent SNPs for both traits compared to the previously published GWAS [15] demonstrates the value of the gene-centric genotyping array and contribution of this study to the understanding of genetic basis of both traits. Also, the analysis of AA data in our study represents the largest of this kind and provided an opportunity to compare associations across the two ethnic groups. However, there are limitations to our study. The assays for FVIII:C and VWF:Ag were conducted in each individual study, which may have added variation in the levels of these two measurements across studies. This variation should have a minimal influence on the genetic contribution to phenotypes, as the genetic association analysis was conducted within each study to correlate the rank of the phenotype measurements with the rank of genotype allele dosage, and the results were meta-analyzed across studies. Also, our study investigated common and low-frequency SNPs included on the pre-defined genotyping arrays and might miss rare and new SNPs that were unknown at the time of array design, or novel loci that could be detected with a denser genome-wide array and imputation.
In conclusion, using a gene-centric approach, we identified and replicated new associations for FVIII:C at KNG1 as well as with SNPs in VWF that have not been reported previously. With the availability of both EA and AA samples, we were able to delineate the similarities and differences in the genetic associations between the two ethnic groups. Findings from this study expand our understanding of genetic determinants for FVIII:C and VWF:Ag and will guide future efforts to examine the contributions of the newly identified variants to the risk of thrombosis in EA and AA populations.
Supplementary Material
Acknowledgments
The authors acknowledge the support of the National Heart, Lung, and Blood Institute and the contributions of the research institutions, study investigators, field staff and study participants in creating the CARe resource for biomedical research. The following five parent studies have contributed parent study data, ancillary study data, and DNA samples through the Broad Institute of Harvard University and the Massachusetts Institute of Technology (N01-HC-5226) to create the CARe data base for this project:
ARIC: N01-HC-5015, N01-HC-5016, N01-HC-5021, N01-HC-5019, N01-HC-5020, N01-HC-5017, and N01-HC-5018;
CARDIA: HHSN268201300025C, HHSN268201300026C, HHSN268201300027C, HHSN268201300028C, HHSN268201300029C, HHSN268200900041C, and AG032136;
CHS: N01-HC-5239, N01-HC-5079 through N01-HC-5086, N01-HC-5129, N01 HC-15103, N01 HC-55222, N01-HC-5150, N01-HC-5133, HL080295, AG-023269, AG-15928, AG-20098, AG-027058, HL-075366, and P30-AG-4827.
FHS: N01-HC-5195;
MESA: N01-HC-5159, N01-HC-5160, N01-HC-5161, N01-HC-5162, N01-HC-5163, N01-HC-5164, N01-HC-5165, N01-HC-5166, N01-HC-5167, N01-HC-5168, and N01-HC-5169;
The authors thank the University of Minnesota Supercomputing Institute for use of the Blade and Calhoun supercomputers.
Contract grant sponsor: NIH; Contract grant number: R01-HL095603, R01-HL59367.
Footnotes
Additional Supporting Information may be found in the online version of this article.
Conflicts of interest: The authors declare no competing financial interests.
References
- 1.Tuddenham EGD, Cooper DN. The Molecular Genetics of Haemostasis and its Inherited Disorders. Oxford: Oxford University Press; 1994. p. 585. [Google Scholar]
- 2.Thompson AR. Structure and function of the factor VIII gene and protein. Semin Thromb Hemost. 2003;29:11–22. doi: 10.1055/s-2003-37935. [DOI] [PubMed] [Google Scholar]
- 3.Spiel AO, Gilbert JC, Jilma B. von willebrand factor in cardiovascular disease: Focus on acute coronary syndromes. Circulation. 2008;117:1449–1459. doi: 10.1161/CIRCULATIONAHA.107.722827. [DOI] [PubMed] [Google Scholar]
- 4.Kamphuisen PW, Eikenboom JC, Bertina RM. Elevated factor VIII levels and the risk of thrombosis. Arterioscler Thromb Vasc Biol. 2001;21:731–738. doi: 10.1161/01.atv.21.5.731. [DOI] [PubMed] [Google Scholar]
- 5.Warren DM, Soria JM, Souto JC, et al. Heritability of hemostasis phenotypes and their correlation with type 2 diabetes status in Mexican Americans. Hum Biol. 2005;77:1–15. doi: 10.1353/hub.2005.0034. [DOI] [PubMed] [Google Scholar]
- 6.Souto JC, Almasy L, Borrell M, et al. Genetic determinants of hemostasis phenotypes in Spanish families. Circulation. 2000;101:1546–1551. doi: 10.1161/01.cir.101.13.1546. [DOI] [PubMed] [Google Scholar]
- 7.de Lange M, Snieder H, Ariens RA, et al. The genetics of haemostasis: A twin study. Lancet. 2001;357:101–105. doi: 10.1016/S0140-6736(00)03541-8. [PMC][10.1016/S0140-6736(00)03541-8] [11197396] [DOI] [PubMed] [Google Scholar]
- 8.Souto JC, Almasy L, Borrell M, et al. Genetic susceptibility to thrombosis and its relationship to physiological risk factors: The GAIT study. Genetic analysis of idiopathic thrombophilia. Am J Hum Genet. 2000;67:1452–1459. doi: 10.1086/316903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scanavini D, Legnani C, Lunghi B, et al. The factor VIII D1241E polymorphism is associated with decreased factor VIII activity and not with activated protein C resistance levels. Thromb Haemost. 2005;93:453–456. doi: 10.1160/TH04-09-0629. [DOI] [PubMed] [Google Scholar]
- 10.Souto JC, Almasy L, Muniz-Diaz E, et al. Functional effects of the ABO locus polymorphism on plasma levels of von willebrand factor, factor VIII, and activated partial thromboplastin time. Arterioscler Thromb Vasc Biol. 2000;20:2024–2028. doi: 10.1161/01.atv.20.8.2024. [DOI] [PubMed] [Google Scholar]
- 11.Viel KR, Machiah DK, Warren DM, et al. A sequence variation scan of the coagulation factor VIII (FVIII) structural gene and associations with plasma FVIII activity levels. Blood. 2007;109:3713–3724. doi: 10.1182/blood-2006-06-026104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harvey PJ, Keightley AM, Lam YM, et al. A single nucleotide polymorphism at nucleotide -1793 in the von willebrand factor (VWF) regulatory region is associated with plasma VWF:ag levels. Br J Haematol. 2000;109:349–353. doi: 10.1046/j.1365-2141.2000.02000.x. [DOI] [PubMed] [Google Scholar]
- 13.Lacquemant C, Gaucher C, Delorme C, et al. Association between high von willebrand factor levels and the Thr789Ala vWF gene polymorphism but not with nephropathy in type I diabetes. The GENEDIAB study group and the DESIR study group. Kidney Int. 2000;57:1437–1443. doi: 10.1046/j.1523-1755.2000.00988.x. [DOI] [PubMed] [Google Scholar]
- 14.Johnsen JM, Auer PL, Morrison AC, et al. Common and rare von willebrand factor (VWF) coding variants, VWF levels, and factor VIII levels in African Americans: The NHLBI exome sequencing project. Blood. 2013;122:590–597. doi: 10.1182/blood-2013-02-485094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith NL, Chen MH, Dehghan A, et al. Novel associations of multiple genetic loci with plasma levels of factor VII, factor VIII, and von willebrand factor: the CHARGE (cohorts for heart and aging research in genome epidemiology) consortium. Circulation. 2010;121:1382–1392. doi: 10.1161/CIRCULATIONAHA.109.869156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Desch KC, Ozel AB, Siemieniak D, et al. Linkage analysis identifies a locus for plasma von willebrand factor undetected by genome-wide association. Proc Natl Acad Sci USA. 2013;110:588–593. doi: 10.1073/pnas.1219885110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tregouet DA, Heath S, Saut N, et al. Common susceptibility alleles are unlikely to contribute as strongly as the FV and ABO loci to VTE risk: Results from a GWAS approach. Blood. 2009;113:5298–5303. doi: 10.1182/blood-2008-11-190389. [PMC] [10.1182/blood-2008-11-190389] [19278955] [DOI] [PubMed] [Google Scholar]
- 18.Heit JA, Armasu SM, Asmann YW, et al. A genome-wide association study of venous thromboembolism identifies risk variants in chromosomes 1q24.2 and 9q. J Thromb Haemost. 2012;10:1521–1531. doi: 10.1111/j.1538-7836.2012.04810.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang W, Teichert M, Chasman DI, et al. A genome-wide association study for venous thromboembolism: The extended cohorts for heart and aging research in genomic epidemiology (CHARGE) consortium. Genet Epidemiol. 2013;37:512–521. doi: 10.1002/gepi.21731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keating BJ, Tischfield S, Murray SS, et al. Concept, design and implementation of a cardiovascular gene-centric 50 k SNP array for large-scale genomic association studies. PLoS One. 2008;3:e3583. doi: 10.1371/journal.pone.0003583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Musunuru K, Lettre G, Young T, et al. Candidate gene association resource (CARe): Design, methods, and proof of concept. Circ Cardiovasc Genet. 2010;3:267–275. doi: 10.1161/CIRCGENETICS.109.882696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.The ARIC investigators. The atherosclerosis risk in communities (ARIC) study: Design and objectives. Am J Epidemiol. 1989;129:687–702. [PubMed] [Google Scholar]
- 23.Friedman GD, Cutter GR, Donahue RP, et al. CARDIA: Study design, recruitment, and some characteristics of the examined subjects. J Clin Epidemiol. 1988;41:1105–1116. doi: 10.1016/0895-4356(88)90080-7. [DOI] [PubMed] [Google Scholar]
- 24.Fried LP, Borhani NO, Enright P, et al. The cardiovascular health study: Design and rationale. Ann Epidemiol. 1991;1:263–276. doi: 10.1016/1047-2797(91)90005-w. [DOI] [PubMed] [Google Scholar]
- 25.Tell GS, Fried LP, Hermanson B, et al. Recruitment of adults 65 years and older as participants in the cardiovascular health study. Ann Epidemiol. 1993;3:358–366. doi: 10.1016/1047-2797(93)90062-9. [DOI] [PubMed] [Google Scholar]
- 26.Feinleib M, Kannel WB, Garrison RJ, et al. The framingham offspring study. Design and preliminary data. Prev Med. 1975;4:518–525. doi: 10.1016/0091-7435(75)90037-7. [DOI] [PubMed] [Google Scholar]
- 27.Bild DE, Bluemke DA, Burke GL, et al. Multiethnic study of atherosclerosis: Objectives and design. Am J Epidemiol. 2002;156:871–881. doi: 10.1093/aje/kwf113. [DOI] [PubMed] [Google Scholar]
- 28.Chambless LE, McMahon R, Wu K, et al. Short-term intraindividual variability in hemostasis factors. The ARIC study. Atherosclerosis risk in communities intraindividual variability study. Ann Epidemiol. 1992;2:723–733. doi: 10.1016/1047-2797(92)90017-k. [DOI] [PubMed] [Google Scholar]
- 29.Folsom AR, Wu KK, Rosamond WD, et al. Prospective study of hemostatic factors and incidence of coronary heart disease: The atherosclerosis risk in communities (ARIC) study. Circulation. 1997;96:1102–1108. doi: 10.1161/01.cir.96.4.1102. [DOI] [PubMed] [Google Scholar]
- 30.Cushman M, Yanez D, Psaty BM, et al. Association of fibrinogen and coagulation factors VII and VIII with cardiovascular risk factors in the elderly: The cardiovascular health study. Cardiovascular health study investigators. Am J Epidemiol. 1996;143:665–676. [Google Scholar]
- 31.Lutsey PL, Cushman M, Steffen LM, et al. Plasma hemostatic factors and endothelial markers in four racial/ethnic groups: The MESA study. J Thromb Haemost. 2006;4:2629–2635. doi: 10.1111/j.1538-7836.2006.02237.x. [DOI] [PubMed] [Google Scholar]
- 32.Green D, Ruth KJ, Folsom AR, et al. Hemostatic factors in the coronary artery risk development in young adults (CARDIA) study. Arterioscler Thromb. 1994;14:686–693. doi: 10.1161/01.atv.14.5.686. [DOI] [PubMed] [Google Scholar]
- 33.Frankel DS, Meigs JB, Massaro JM, et al. Von willebrand factor, type 2 diabetes mellitus, and risk of cardiovascular disease: The Framingham offspring study. Circulation. 2008;118:2533–2539. doi: 10.1161/CIRCULATIONAHA.108.792986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Y, Abecasis GR. Mach 1.0: Rapid haplotype reconstruction and missing genotype inference. Am J Hum Genet. 2006;S79:2290. [Google Scholar]
- 35.Price AL, Patterson NJ, Plenge RM, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 36.Purcell S, Neale B, Todd-Brown K, et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen MH, Yang Q. GWAF: An R package for genome-wide association analyses with family data. Bioinformatics. 2010;26:580–581. doi: 10.1093/bioinformatics/btp710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Available at: http://www.sph.umich.edu/csg/abe-casis/Metal/index.html.
- 39.Wassel CL, Lange LA, Keating BJ, et al. Association of genomic loci from a cardiovascular gene SNP array with fibrinogen levels in European Americans and African-Americans from six cohort studies: The candidate gene association resource (CARe) Blood. 2011;117:268–275. doi: 10.1182/blood-2010-06-289546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clayton D. Testing for association on the X chromosome. Biostatistics. 2008;9:593–600. doi: 10.1093/biostatistics/kxn007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang J, Lee SH, Goddard ME, et al. GCTA: A tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olsson ML, Chester MA. Polymorphism and recombination events at the ABO locus: A major challenge for genomic ABO blood grouping strategies. Transfus Med. 2001;11:295–313. doi: 10.1046/j.1365-3148.2001.00320.x. [DOI] [PubMed] [Google Scholar]
- 43.van Loon JE, Kavousi M, Leebeek FW, et al. von willebrand factor plasma levels, genetic variations and coronary heart disease in an older population. J Thromb Haemost. 2012;10:1262–1269. doi: 10.1111/j.1538-7836.2012.04771.x. [DOI] [PubMed] [Google Scholar]
- 44.Zabaneh D, Gaunt TR, Kumari M, et al. Genetic variants associated with von willebrand factor levels in healthy men and women identified using the HumanCVD BeadChip. Ann Hum Genet. 2011;75:456–467. doi: 10.1111/j.1469-1809.2011.00654.x. [DOI] [PubMed] [Google Scholar]
- 45.Campos M, Sun W, Yu F, et al. Genetic determinants of plasma von willebrand factor antigen levels: A target gene SNP and haplotype analysis of ARIC cohort. Blood. 2011;117:5224–5230. doi: 10.1182/blood-2010-08-300152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klemm T, Mehnert AK, Siegemund A, et al. Impact of the Thr789Ala variant of the von willebrand factor levels, on ristocetin co-factor and collagen binding capacity and its association with coronary heart disease in patients with diabetes mellitus type 2. Exp Clin Endocrinol Diabetes. 2005;113:568–572. doi: 10.1055/s-2005-872896. [DOI] [PubMed] [Google Scholar]
- 47.Yuan HY, Chiou JJ, Tseng WH, et al. FASTSNP: An always up-to-date and extendable service for SNP function analysis and prioritization. Nucleic Acids Res. 2006;34:w635–w641. doi: 10.1093/nar/gkl236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Houlihan LM, Davies G, Tenesa A, et al. Common variants of large effect in f12, kng1, and HRG are associated with activated partial thromboplastin time. Am J Hum Genet. 2010;86:626–631. doi: 10.1016/j.ajhg.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morange PE, Oudot-Mellakh T, Cohen W, et al. kng1 Ile581Thr and susceptibility to venous thrombosis. Blood. 2011;117:3692–3694. doi: 10.1182/blood-2010-11-319053. [DOI] [PubMed] [Google Scholar]
- 50.Tang W, Schwienbacher C, Lopez LM, et al. Genetic associations for activated partial thromboplastin time and prothrombin time, their gene expression profiles, and risk of coronary artery disease. Am J Hum Genet. 2012;91:152–162. doi: 10.1016/j.ajhg.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sabater-Lleal M, Martinez-Perez A, Buil A, et al. A genome-wide association study identifies kng1 as a genetic determinant of plasma factor XI level and activated partial thromboplastin time. Arterioscler Thromb Vasc Biol. 2012;32:2008–2016. doi: 10.1161/ATVBAHA.112.248492. [DOI] [PubMed] [Google Scholar]
- 52.Gaunt TR, Lowe GD, Lawlor DA, et al. A genecentric analysis of activated partial thromboplastin time and activated protein C resistance using the HumanCVD focused genotyping array. Eur J Hum Genet. 2013;21:779–783. doi: 10.1038/ejhg.2012.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zakai NA, Ohira T, White R, et al. Activated partial thromboplastin time and risk of future venous thromboembolism. Am J Med. 2008;121:231–238. doi: 10.1016/j.amjmed.2007.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang QY, Song J, Gibbs RA, et al. Characterizing polymorphisms and allelic diversity of von willebrand factor gene in the 1000 genomes. J Thromb Haemost. 2013;11:261–269. doi: 10.1111/jth.12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Enayat MS, Guilliatt AM, Surdhar GK, et al. Aberrant dimerization of von willebrand factor as the result of mutations in the carboxy-terminal region: Identification of 3 mutations in members of 3 different families with type 2A (phenotype IID) von willebrand disease. Blood. 2001;98:674–680. doi: 10.1182/blood.v98.3.674. [DOI] [PubMed] [Google Scholar]
- 56.Campos M, Buchanan A, Yu F, et al. Influence of single nucleotide polymorphisms in factor VIII and von willebrand factor genes on plasma factor VIII activity: The ARIC study. Blood. 2012;119:1929–1934. doi: 10.1182/blood-2011-10-383661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Undas A, Brozek J, Szczeklik A. Homocysteine and thrombosis: From basic science to clinical evidence. Thromb Haemost. 2005;94:907–915. doi: 10.1160/TH05-05-0313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.