

Abstract
A series of hydroxyanthraquinones having an alkylating N-mustard pharmacophore at 1′-position were synthesized via a bioisostere approach to evaluate their cytotoxicity against four tumor cell lines (MDA-MB-231, HeLa, MCF-7 and A549). These compounds displayed significant in vitro cytotoxicity against MDA-MB-231 and MCF-7 cells, reflecting the excellent selectivity for the human breast cancer. Among them, compound 5k was the most cytotoxic with IC50 value of 0.263 nM and is more potent than DXR (IC50 = 0.294 nM) in inhibiting the growth of MCF-7 cells. The excellent cytotoxicity and good selectivity of compound 5k suggest that it could be a promising lead for further design and development of anticancer agents, especially for breast cancer.
Keywords: Anthraquinone, Nitrogen mustard, Synthesis, Antitumor activity
1. Introduction
Nitrogen mustards (N-mustards) are frequently used anticancer agents in the clinic, such as cyclophosphamide, chlorambucil and melphalan [1]. However, the severe host toxicity and drug resistance associated with these drugs continue to present serious impediments in cancer chemotherapy. Moreover, the efficiency of these drugs is limited by low affinity for DNA and rapid quenching via hydrolysis prior to DNA alkylation [2–4]. Many N-mustard structural modifications have been made during the last 40 years in order to increase the target affinity. In this regard, medicinal chemists have focused on changing the carrying parts for N-mustards by linking the N-mustard residue to DNA-binding molecules [5–12] (Fig. 1). However, up to date, despite the fact that many carriers have been investigated, the majority of them focused on benzene derivatives and new clinical candidates with substantially enhanced activities remain elusive.
Fig. 1.

Selected examples of N-mustards with a benzene derivative carrier.
The anthracyclines (doxorubicin, daunorubicin, idarubicin, epirubicin, and the anthraquinone mitoxantrone) are one of the most effective anticancer agents for many types of tumors [13]. Although newer drugs have emerged continually in recent years, the fact that anthracyclines remain a first-line treatment for cancer underscores its sustained relevance as an anticancer drug. Based on the proven efficacy of anthracyclines and related compounds in antitumor application, their molecular motifs may serve as a good platform for structural modification to enhance potency in other types of cytotoxic agents. A unique feature of anthracyclines is the presence of a 1,4-dihydroxyanthraquinone moiety that may play a key role both in the DNA binding and cell or tissue bioavailability [14–16]. Considering the importance of the 1,4-dihydroxyanthraquinone moiety in anticancer drugs and biologically active natural products [17–19], the combination of a 1,4-dihydroxyanthraquinone structure with an N-mustard moiety could lead to novel compounds possessing potent anticancer properties. The anthraquinone portion provides a potential substrate for DNA intercalation [20,21] and bioreductive activation [22,23], and the alkylating group imparts cytotoxic activity. In this regard, some chrysophanol and emodin-nitrogen mustard conjugates have been synthesized and their anticancer activity against Leukemia L1210 and HL-60 cells were evaluated by Watanabe’s group [24]. Thereafter the synthesis of anthraquinonee–chlorambucil conjugates and their antitumor activity have been reported by Ahn’s group [25].
Inspired by these results and based on bioisosterism, we proposed to replace the ether bridge at C-7 of doxorubicin (DXR) with a nitrogen atom in anticipation that the resultant molecules possessing two pharmacophores (hydroxyanthraquinone and N-mustard) with one molecule may exhibit potent antitumor activity while decreasing the drug resistance to certain extent (Fig. 2). The ether bridge of DXR between the anthracyclinone moiety and the aminosugar was thought to be important for its activity [26]. Bioisosteric replacement of functional groups is a frequently employed technique in drug discovery for the generation of more potent and more selective analogues of lead structures [27]. On the other hand, with mitoxatrone and bisantrene in clinical use for the therapy of cancer, the nitrogen atom and bisethylamine moiety seemed to represent an important factor for their high therapeutic indices. In this work, with the goal to develop potent hydroxyanthraquinones with antitumor activity and as a part of our ongoing interest in the synthesis of novel hydroxyanthraquinone compounds and biological evaluation [28–31], we combined these features into one molecule to mimic the structure of DXR and mitoxantrone. Herein, we report our studies on the design and synthesis of a new series of hydroxyanthraquinone N-mustard derivatives and demonstrate their biological activities.
Fig. 2.

The structures of representative anthracyclines and the newly designed compounds.
2. Results and discussion
2.1. Chemistry
The synthetic route for the hydroxyanthraquinone bearing an alkylating N-mustard residue on 1′-position is shown in Scheme 1. The commercially available starting material hydroxyanthraquinone 1 was converted to compound 2 via a modified Marschalk reaction according to the procedure previously developed in our laboratory [29]. Chlorination of 2 with thionyl chloride at 50 °C led to the intermediate 3, which was directly used in the next step without the need for purification. Subsequently, condensation of diethanolamine with intermediate 3 in dichloromethane at room temperature gave intermediate 4. There was no need to purify the crude product after workup in this step and the crude product can be directly used in the next step. A final chlorination of intermediate 4 with thionyl chloride afforded the target compounds 5a-t. This chlorination was successfully performed by using the same condition for the synthesis of intermediate 3. After completion of the reaction, the desired products were easily isolated in pure form by column chromatography. It should be noted that through all the reactions for the synthesis of the target compounds, the protection of hydroxyl groups was not required. All the newly synthesized compounds were characterized by 1H NMR and 13C NMR as well as high resolution mass spectrum (HR-MS).
Scheme 1.

Reagents and conditions: (a) NaOH/Na2S2O4/MeOH/0 °C/N2, then H2O2; (b) SOCl2, 50 °C; (c) HN(CH2CH2OH)2, CH2Cl2, rt; (d) SOCl2, 50 °C.
2.2. Antitumor activity
The in vitro antitumor activities of the anthraquinone N-mustards 5a-t were evaluated against the MDA-MB-231 (triple negative breast cancer), HeLa (cervical cancer), MCF-7 (ER+ breast cancer), and A549 (non-small cell lung cancer) cell lines and were compared with the cytotoxic activity of DXR and chlorambucil, chosen as reference drugs in this assay. The inhibitory activities (IC50) of the tested compounds are shown in Table 1.
Table 1.
In vitro cytotoxicity of compounds 5a-t against four tumor cell lines.

| |||||||
|---|---|---|---|---|---|---|---|
| Compound | R | R1 | R2 | IC50 (nM)a
|
|||
| MDA-MB-231 | Hela | MCF-7 | A549 | ||||
| 5a | H | H | OH | 211 | 139 | 0.714 | 519 |
| 5b | CH3 | H | OH | 1000 | 154 | 426 | 180 |
| 5c | C2H5 | H | OH | 84.5 | 105 | 144 | 155 |
| 5d | n-C3H7 | H | OH | 61.9 | 152 | 69.5 | 302 |
| 5e | i-propyl | H | OH | 77.6 | 217 | 13.5 | 276 |
| 5f | i-butyl | H | OH | 14.6 | 392 | 352 | 686 |
| 5g | PhCH2 | H | OH | 51.1 | 665 | 36.5 | 1360 |
| 5h | Ph | H | OH | 137 | 339 | 15.8 | 217 |
| 5i | 2-FC6H4 | H | OH | 0.59 | 615 | 1.57 | 462 |
| 5j | 3-FC6H4 | H | OH | 59.6 | 166 | 0.295 | 495 |
| 5k | 4-FC6H4 | H | OH | 80.7 | 249 | 0.263 | 421 |
| 5l | 4-ClC6H4 | H | OH | 48.0 | 185 | 10.2 | 95.7 |
| 5m | 4-BrC6H4 | H | OH | 299 | 262 | 78.3 | 299 |
| 5n | 2-MeOC6H4 | H | OH | 2.13 | 210 | 28.5 | 810 |
| 5o | 3-MeOC6H4 | H | OH | 91.2 | 210 | 10.4 | 411 |
| 5p | 4-MeOC6H4 | H | OH | 12.9 | 457 | 5.36 | 1010 |
| 5q | 4-MeC6H4 | H | OH | 22.5 | 93.8 | 23.0 | 720 |
| 5r | 3,4-MeC6H4 | H | OH | 389 | 234 | 0.801 | 1540 |
| 5s | Ph | OH | H | 316 | 308 | 90.8 | 542 |
| 5t | Ph | H | OCH3 | 278 | 480 | 3.69 | 501 |
| Chlorambucil | 520 | 943 | 26.4 | 85.7 | |||
| Doxorubicin | 0.414 | 4.16 | 0.294 | 5.58 | |||
IC50 values were calculated from three independent experiments.
As shown in Table 1, almost all the synthesized target compounds are more potent than chlorambucil against MDA-MB-231, HeLa, and MCF-7 cell lines. For instance, compound 5i showed a ca. 900-fold increase in its inhibitory activity against MDA-MB-231 cells compared to chlorambucil; compound 5q was 10-fold more active against HeLa cells than chlorambucil; compound 5k was found to be over 100 times more potent against MCF-7 cell than chlorambucil. The objective in designing and preparing anthraquinone N-mustard conjugates was to identify new chemical entities with significantly improved cellular antiproliferative activity. To this end, our results suggest that hydroxyanthraquinone derivatives can be combined with N-mustard of low concentration to inhibit tumor cell growth more effectively by lowering the toxic side effects of N-mustard that comes from the use of high concentrations. More importantly, some of the conjugates showed better or comparable activity than DXR, the most active single agent used in advanced breast cancer. For instance, compounds 5j exhibited potent antiproliferative activities against MCF-7 cells with IC50 values of 0.295 nM. The activity is close to those of DXR (IC50 = 0.294 nM). Moreover, compound 5k exhibited higher cellular cytotoxicity than DXR, with an IC50 value of 0.263 nM. This is particularly important for developing DXR analogues because despite the fact that >2000 analogues of DXR have been synthesized and tested, so far no clinical candidates or drugs have emerged with substantially enhanced efficacy [32].
When these compounds were screened against MDA-MB-231 cells, most of the compounds showed excellent activities with IC50 values below 100 nM (chlorambucil IC50 = 520 nM). Among them, compounds 5i and 5n showed the highest growth inhibitory activity, with the IC50 values of 0.59 and 2.13 nM, respectively. In particular, the potency of compound 5i was comparative to the reference drug DXR (IC50 = 0.414 nM). These results confirm that introduction of an ortho-substituted phenyl group at the 1′-position in the side chain of hydroxyanthraquinone could dramatically improve the selectivity over MDA-MB-231 cells. In addition, the length of the alkyl group at the 1′-position could significantly influence the activity. Compound 5f with a four-carbon isobutyl group exhibited IC50 value of 14.6 nM, which was approximately 70-fold stronger than that of compound 5b with a methyl group (IC50 = 1000 nM). Similar trend was observed with compounds 5c, 5d, and 5e. Conversion of the 1,4-dihydroxyanthraquinone core to 1,8-dihydroxyanthraquinone and 1-hydroxy-4-methoxylanthraquinone resulted in a decrease in activity, indicating that the hydroxyl group at the 4-position of anthraquinone was critical for the activity against the triple negative breast cancer cells (5h vs. 5s and 5t).
The conjugates were then screened against HeLa, MCF-7, and A549 cell lines to determine their effect on growth inhibition. In HeLa cells, all the prepared compounds exhibited good cytotoxic activity regardless of the nature of the substituents on the 1′-position. These compounds with the IC50 values ranging from 93.8 to 665 nM were markedly more potent than the positive control chlorambucil (IC50 = 943 nM). For MCF-7 cell line, 12 of the 20 compounds tested showed significant inhibitory activity with IC50 values below 20 nM. Furthermore, the potencies of compounds 5j and 5k (IC50 = 0.295 and 0.263 nM, respectively) were greater than, or comparable to DXR (IC50 = 0.294 nM). Thus, it became clear that MCF-7 cell line was the most sensitive to our hydroxyanthraquinone N-mustard derivatives among the four tested cell lines. On the other hand, for A549 cell line, none of the tested compounds was found more potent than chlorambucil, implying that the newly synthesized compounds, while having no advantage in the treatment of lung cancer, demonstrate a greater extent of selectivity towards differentiated tumor cells than chlormabucil.
Based on the above results, it can be concluded that the novel hydroxyanthraquinone N-mustard derivatives were highly cytotoxic in human breast cancer represented by a metastatic triple negative cell line (MDA-MB-231) and one that expresses the estrogen receptor (MCF-7), but to a much lesser degree in HeLa and A549 cell lines, reflecting the excellent selectivity of the compounds for human breast cancer.
Antitumor agents may be considered potentially more useful when they are more lethal to tumor cell than to non-tumor cell. Hence, two compounds (5i and 5n) exhibiting good activity against MDA-MB-231 and eight compounds (5e, 5h, 5j, 5k, 5o, 5p, 5r, and 5t) exhibiting good activity against MCF-7 were chosen for further evaluation to test their cytotoxicity on normal cell line. As shown in Fig. 3, at a dose of 10 μM, the tested compounds displayed significant toxicities towards MCF-10, the normal mammary epithelial cells. However, they were much less toxic than DXR at the same dose. At 1 μM, the compounds were moderately toxic to MCF-10 cells, but still far less lethal compared to DXR. These results indicate that hydroxyanthraquinone derivatives having an N-mustard pharmacophore at 1′-position improved the selective toxicity against cancer cells over the widely prescribed chemotherapeutic agent DXR.
Fig. 3.
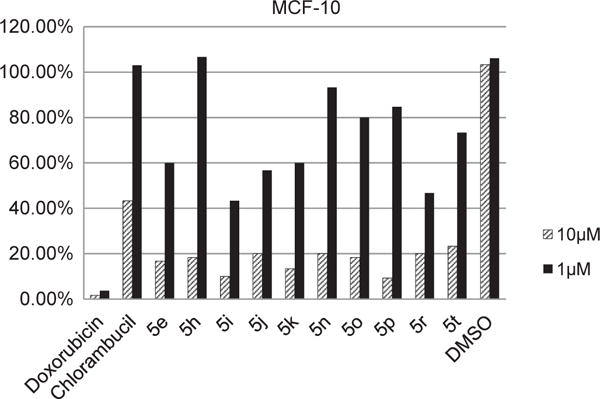
Cytotoxicity of hydroxyanthraquinone N-mustards towards MCF-10 normal mammary epithelial cells.
3. Conclusions
In this study, we designed and synthesized twenty hydroxyanthraquinone N-mustards through efficient synthetic routes. The advantages of the synthesis include easily available raw materials, no protection-deprotection sequence, simple workup, and convenient isolation. Evaluation of the cytotoxic activity of the designed compounds demonstrated significantly greater potency against MDA-MB-231 and MCF-7, two selected breast cancer cell lines than in HeLa and A549 cells. In particular, compound 5k possessed superior activity with an IC50 value of 0.263 nM than DXR against MCF-7, which could be a good candidate for further pharmacological studies. The data presented in this work constitute an important starting point for the design and synthesis of new active compounds based on N-mustards and anthracyclines and may provide valuable information to medicinal chemists working on this field, which will facilitate the development of novel antitumor drugs.
4. Experimental
4.1. Chemistry
Regents and solvents were purchased commercially and used without further purification, unless otherwise stated. Reactions were monitored using thin-layer chromatography (TLC) on silica gel plates. Flash column chromatography was performed on silica gel (200–300 mesh) for purification of the compounds. Melting points were measured on an X-4 melting-point apparatus and were uncorrected. The 1H NMR and 13C NMR spectra were recorded on Bruker Avance spectrometer at 400 MHz and 100 MHz, respectively. Chemical shift values were reported as δ ppm relative to TMS as internal standard. HRMS were recorded on a Bruker TOF-QIIspectrometer with electrospray ionization (ESI).
4.1.1. General procedure for the synthesis of 3
Compound 2 (1.0 mmol) was treated with thionyl chloride (1.0 mL) and stirred for 2 h at 50 °C. The solution was evaporated in vacuo and the residue was used in the next step without further purification.
4.1.2. General procedure for the synthesis of 4
A solution of 3 (1.0 mmol) in dry CH2Cl2 (10 mL) was treated with diethanolamine (1.5 mmol) and stirred at room temperature. After the completion of the reaction (monitored by TLC), the mixture was poured into water, and extracted three times with CH2Cl2. The organic layer was washed five times with water, dried, and concentrated. The residue was used in the next step without further purification.
4.1.3. General procedure for the synthesis of 5
The crude product 4 was treated with thionyl chloride (1.0 mL) and stirred for 2 h at 50 °C. The reaction mixture was concentrated and the residue was partitioned between water and CH2Cl2. The organic layer was extracted three times with CH2Cl2 and washed with saturated NaHCO3 aq, bine, dried, and concentrated. The residue was purified by flash column chromatography (CH2Cl2/petroleum, 1/1, v/v) to afford 5 as an orange solid.
4.1.3.1. 2-((Bis (2-chloroethyl) amino) methyl)-1,4-dihydroxyanthracene-9,10-dione (5a)
Yield 46%; mp 140–141 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.21 (s, 1H), 12.73 (s, 1H), 8.23–8.26 (m, 2H), 7.96–7.98 (m, 2H), 7.64 (s, 1H), 3.90 (s, 2H), 3.72 (t, J = 6.4 MHz, 4H), 2.97 (t, J = 6.4 MHz, 4H). 13C NMR (100 MHz, DMSO-d6) δ 187.0, 186.2, 156.7, 155.5, 135.1, 135.0, 133.0, 132.8, 126.7, 126.6, 112.1, 55.5, 51.5, 42.4. HRMS (ESI): m/z caclcd for C19H17Cl2NO4Na [M + Na]+: 416.0432; Found: 416.0428.
4.1.3.2. 2-(1-(Bis(2-chloroethyl)amino)ethyl)-1,4-dihydroxyanthracene-9,10-dione (5b)
Yield 49%; mp 111–112 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.40 (s, 1H), 12.68 (s, 1H), 8.16–8.21 (m, 2H), 7.90–7.95 (m, 2H), 7.48 (s, 1H), 4.41 (q, J = 6.8 MHz, 1H), 3.52–3.64 (m, 4H), 2.84–2.98 (m, 4H), 1.36 (d, J = 6.8 MHz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 187.0, 186.1, 156.5, 155.7, 135.1, 135.0, 132.9, 132.8, 126.7, 126.6, 126.4, 112.3, 111.5, 52.6, 52.4, 42.9, 16.7. HRMS (ESI): m/z caclcd for C20H19Cl2NO4Na [M + Na]+: 430.0589; Found: 430.0579.
4.1.3.3. 2-(1-(Bis(2-chloroethyl)amino)propyl)-1,4-dihydroxyanthracene-9,10-dione (5c)
Yield 33%; mp 92–93 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.42 (s, 1H), 12.70 (s, 1H), 8.23−8.18 (m, 2H), 7.97−7.92 (m, 2H), 7.50 (s, 1H), 4.43 (t, J = 6.8 Hz, 1H), 3.66−3.55 (m, 4H), 3.00−2.87 (m, 4H), 1.38 (d, J = 6.8 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 187.1, 186.1, 156.4, 156.2, 135.1, 135.0, 132.9, 132.8, 127.0, 126.7, 126.6, 112.4, 111.7, 58.0, 52.4, 42.9, 23.4, 11.2. HRMS (ESI): m/z caclcd for C21H21Cl2NO4Na [M + Na]+: 444.0745; Found: 444.0718.
4.1.3.4. 2-(1-(Bis(2-chloroethyl)amino)butyl)-1,4-dihydroxyanthracene-9,10-dione (5d)
Yield 43%; mp 73–74 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.55 (s, 1H), 12.74 (s, 1H), 8.30−8.26 (m, 2H), 8.01−7.97 (m, 2H), 7.52 (s, 1H), 4.36 (t, J = 7.6 Hz, 1H), 3.63−3.56 (m, 4H), 3.03−2.96 (m, 2H), 2.83−2.76 (m, 2H), 1.94−1.85 (m, 1H), 1.81−1.73 (m, 1H), 1.40−1.35 (m, 1H), 1.32−1.20 (m, 1H), 0.90 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 186.9, 186.0, 156.3, 156.1, 135.0, 134.9, 132.7, 132.6, 127.0, 126.6, 126.5, 112.2, 111.5, 55.8, 52.3, 42.9, 32.5, 19.4, 13.8. HRMS (ESI): m/z caclcd for C22H23Cl2NO4Na [M + Na]+: 458.0902; Found: 458.0902.
4.1.3.5. 2-(1-(Bis(2-chloroethyl)amino)-2-methylpropyl)-1,4-dihydroxyanthracene-9,10-dione (5e)
Yield 54%; mp 114–115 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.57 (s, 1H), 12.73 (s, 1H), 8.26−8.22 (m, 2H), 7.99−7.95 (m, 2H), 7.50 (s, 1H), 3.95 (d, J = 10.8 Hz, 1H), 3.66−3.62 (m, 4H), 3.03−2.96 (m, 2H), 2.69−2.62 (m, 2H), 2.44−2.35 (m, 2H), 1.14 (d, J = 6.0 Hz, 3H), 0.69 (d, J = 6.4 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 187.2, 186.1, 156.3, 156.2, 140.8, 135.1, 135.0, 132.9, 132.8, 127.4, 126.7, 126.5, 112.4, 111.7, 52.0, 42.8, 27.7, 20.6, 20.2. HRMS (ESI): m/z caclcd for C22H23Cl2NO4Na [M + Na]+: 458.0902; Found: 458.0863.
4.1.3.6. 2-(1-(Bis(2-chloroethyl)amino)-3-methylbutyl)-1,4-dihydroxyanthracene-9,10-dione (5f)
Yield 28%; mp 79–80 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.51 (s, 1H), 12.68 (s, 1H), 8.23−8.19 (m, 2H), 7.96−7.92 (m, 2H), 7.49 (s, 1H), 4.43 (t, J = 7.2 Hz, 1H), 3.61−3.55 (m, 4H), 3.03−2.96 (m, 2H), 1.80−1.65 (m, 2H), 1.60−1.52 (m, 1H), 0.90 (t, J = 6.8 Hz, 6H). 13C NMR (100 MHz, DMSO-d6) δ 187.1, 186.1, 156.3, 156.0, 135.1, 135.0, 132.9, 132.8, 127.1, 126.7, 126.5, 112.4, 111.7, 53.9, 52.4, 43.0, 24.6, 22.7, 22.3. HRMS (ESI): m/z caclcd for C23H25Cl2NO4Na [M + Na]+: 472.1058; Found: 472.1058.
4.1.3.7. 2-(1-(Bis(2-chloroethyl)amino)-2-phenylethyl)-1,4-dihydroxyanthracene-9,10-dione (5g)
Yield 38%; mp 121–123 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.49 (s, 1H),12.72 (s,1H), 8.23−8.20 (m, 2H), 7.95−7.91 (m, 2H), 7.82−7.73 (m, 1H), 7.30 (d, J = 7.2 Hz, 2H), 7.23 (t, J = 7.6 Hz, 2H), 7.12 (t, J = 7.2 Hz, 1H), 4.78−4.70 (m, 1H), 3.57−3.43 (m, 4H), 3.33−3.20 (m, 2H), 3.11−2.83 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 187.0, 186.1, 156.2, 156.1, 138.8, 135.0, 134.9, 132.9, 132.8, 129.2, 128.2, 127.4, 126.7, 126.5, 112.3, 111.6, 58.2, 52.7, 42.9, 35.3. HRMS (ESI): m/z caclcd for C26H23Cl2NO4Na [M + Na]+: 506.0902; Found: 506.0903.
4.1.3.8. 2-((Bis(2-chloroethyl)amino) (phenyl)methyl)-1,4-dihydroxyanthracene-9,10-dione (5h)
Yield 36%; mp 86–87 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.26 (s, 1H), 12.68 (s, 1H), 8.14–8.17 (m, 2H), 7.88–7.90 (m, 2H), 7.70 (s, 1H), 7.42 (d, J = 7.6 Hz, 2H), 7.37 (t, J = 7.6 Hz, 2H), 7.27=7.31 (m, 1H), 5.51 (s, 1H), 3.60–3.65 (m, 4H), 2.95–3.00 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 187.0, 186.0, 156.4, 155.1, 143.5, 139.3, 135.0, 134.9, 132.7, 132.6, 128.8, 128.6, 127.8, 126.6, 126.5, 126.4, 112.6, 111.8, 63.0, 52.6, 42.1. HRMS (ESI): m/z caclcd for C25H21Cl2NO4Na [M + Na]+: 492.0745; Found: 492.0736.
4.1.3.9. 2-((Bis(2-chloroethyl)amino) (2-fluorophenyl)methyl)-1,4-dihydroxyanthracene-9,10-dione (5i)
Yield 37%; mp 137–140 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.17 (s, 1H), 12.68 (s, 1H), 8.17 (s, 2H), 7.90 (s, 2H), 7.62 (s, 1H), 7.38−7.29 (m, 2H), 7.25−7.15 (m, 2H), 5.73 (s, 1H), 3.60−3.48 (m, 4H), 3.07−2.95 (m, 4H). 13C NMR (100 MHz, DMSO) δ 187.0, 186.1, 160.3 (d, J = 244.5 Hz), 156.3, 155.0, 142.4, 135.1, 134.9, 132.8, 132.7, 130.2 (d, J = 2.7 Hz), 130.0 (d, J = 8.4 Hz), 127.0, 126.6, 126.5, 126.0 (d, J = 13.2 Hz), 124.6 (d, J = 2.8 Hz), 115.7 (d, J = 22.2 Hz), 112.5, 111.9, 57.0, 53.2, 42.4. HRMS (ESI): m/z caclcd for C25H20Cl2FNO4Na [M + Na]+: 510.0651; Found: 510.0651.
4.1.3.10. 2-((Bis(2-chloroethyl)amino)(3-fluorophenyl)methyl)-1,4-dihydroxyanthracene-9,10-dione (5j)
Yield 35%; mp 124–126 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.26 (s, 1H), 12.68 (s, 1H), 8.14–8.17 (m, 2H), 7.88–7.90 (m, 2H), 7.70 (s, 1H), 7.42 (d, J = 7.6 Hz, 1H), 7.37 (t, J = 7.6 Hz, 2H), 7.27–7.31 (m, 1H), 5.51 (s, 1H), 3.60–3.65 (m, 4H), 2.95–3.00 (m, 4H). 13C NMR (100 MHz, DMSO) δ 187.0, 186.1, 162.1 (d, J = 243.1 Hz), 156.3, 155.1, 142.4 (d, J = 6.9 Hz), 135.0, 134.9, 132.7, 132.6, 130.5, 130.4, 126.8, 126.6, 126.5, 124.7 (d, J = 2.0 Hz), 115.4 (d, J = 21.5 Hz), 114.6 (d, J = 52.5, 20.8 Hz), 112.7, 112.0, 62.3, 42.1. HRMS (ESI): m/z caclcd for C25H20Cl2FNO4Na [M + Na]+: 510.0651; Found: 510.0651.
4.1.3.11. 2-((Bis(2-chloroethyl)amino)(4-fluorophenyl)methyl)-1,4-dihydroxyanthracene-9,10-dione (5k)
Yield 27%; mp 82–84 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.32 (s, 1H), 12.74 (s, 1H), 8.26−8.24 (m, 2H), 8.01−7.97 (m, 2H), 7.74 (s, 1H), 7.54−7.51 (m, 2H), 7.25 (t, J = 8.8 Hz, 2H), 5.58 (s, 1H), 3.73−3.63 (m, 4H), 3.08−2.96 (m, 4H). 13C NMR (100 MHz, DMSO) δ 186.9, 186.0, 161.5 (d, J = 243.1 Hz), 156.4, 155.0, 143.2, 135.5 (d, J = 2.8 Hz), 135.0, 134.9, 132.7, 132.6, 130.8, 130.7126.6, 126.5, 126.4, 115.5, 115.2, 112.6, 111.8, 62.1, 52.5, 42.1. HRMS (ESI): m/z caclcd for C25H20Cl2FNO4Na [M + Na]+: 510.0651; Found: 510.0652.
4.1.3.12. 2-((Bis(2-chloroethyl)amino)(4-chlorophenyl)methyl)-1,4-dihydroxyanthracene-9,10-dione (5l)
Yield 49%; mp 96–97 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.26 (s, 1H), 12.67 (s, 1H), 8.20−8.16 (m, 2H), 7.95−7.90 (m, 2H), 7.66 (s, 1H), 7.47−7.42 (m, 4H), 5.53 (s, 1H), 3.69−3.59 (m, 4H), 3.04−2.92 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 187.0, 186.1, 156.4, 155.0, 142.8, 138.4, 135.1, 134.9, 132.8, 132.3, 130.6, 128.5, 126.6, 126.5, 112.7, 111.9, 62.2, 52.5, 42.1. HRMS (ESI): m/z caclcd for C25H20Cl3NO4Na [M + Na]+: 526.0356; Found: 526.0334.
4.1.3.13. 2-((Bis(2-chloroethyl)amino)(4-bromophenyl)methyl)-1,4-dihydroxyanthracene-9,10-dione (5m)
Yield 51%; mp 93–94 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.26 (s, 1H), 12.67 (s, 1H), 8.18−8.16 (m, 2H), 7.94−7.90 (m, 2H), 7.67 (s, 1H), 7.57 (d, J = 8.0 Hz, 2H), 7.40 (d, J = 8.0 Hz, 1H), 5.52 (s, 1H), 3.70−3.59 (m, 4H), 3.03−2.92 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 187.0, 186.1, 156.4, 155.0, 142.7, 138.8, 135.1, 134.9, 132.7, 132.6, 131.5, 130.9, 126.6, 126.5, 126.4, 120.9, 112.7, 111.9, 62.2, 52.5, 42.1. HRMS (ESI): m/z caclcd for C25H20Cl2BrNO4Na [M + Na]+: 569.9850; Found: 569.9842.
4.1.3.14. 2-((Bis(2-chloroethyl)amino)(2-methoxyphenyl)methyl)-1,4-dihydroxyanthracene-9,10-dione (5n)
Yield 33%; mp 61–63 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.16 (s, 1H), 12.75 (s, 1H), 8.19−8.14 (m, 2H), 7.93−7.87 (m, 2H), 7.64 (s, 1H), 7.31 (t, J = 7.6 Hz, 1H), 7.08 (d, J = 8.0 Hz, 1H), 7.02 (d, J = 7.6 Hz, 1H), 6.89 (t, J = 7.6 Hz, 1H), 5.79 (s, 1H), 3.89 (s, 3H), 3.59−3.45 (m, 4H), 3.04−2.97 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 186.9, 186.0, 157.2, 156.5, 155.5, 144.4, 135.0, 134.9, 132.9, 132.8, 129.2, 128.8, 127.3, 126.9, 126.6, 126.5, 120.4, 112.2, 111.5, 111.4, 57.4, 55.7, 53.6, 42.7. HRMS (ESI): m/z caclcd for C26H23Cl2NO5Na [M + Na]+: 522.0851; Found: 522.0818.
4.1.3.15. 2-((Bis(2-chloroethyl)amino)(3-methoxyphenyl)methyl)-1,4-dihydroxyanthracene-9,10-dione (5o)
Yield 37%; mp 121–123 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.29 (s, 1H), 12.68 (s, 1H), 8.23−8.20 (m, 2H), 7.96−7.91 (m, 2H), 7.65 (s, 1H), 7.27 (t, J = 7.6 Hz, 1H), 6.99−6.96 (m, 2H), 6.87−6.84 (m, 1H), 5.49 (s, 1H), 3.73 (s, 3H), 3.67−3.59 (m, 4H), 3.01−2.94 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 187.1, 186.2, 159.3, 156.3, 155.1, 143.2, 141.0, 135.1, 134.9, 132.9, 132.8, 129.6, 126.7, 126.6, 126.5, 120.8, 114.7, 112.8, 112.7, 112.0, 62.8, 55.0, 52.7, 42.2. HRMS (ESI): m/z caclcd for C26H23Cl2NO5Na [M + Na]+: 522.0851; Found: 522.0811.
4.1.3.16. 2-((Bis(2-chloroethyl)amino)(4-methoxyphenyl)methyl)-1,4-dihydroxyanthracene-9,10-dione (5p)
Yield 27%; mp 61–63 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.17 (s, 1H), 12.76 (s, 1H), 8.21−8.17 (m, 2H), 7.93−7.92 (m, 2H), 7.65 (s, 1H), 7.33−7.29 (m, 1H), 7.09−7.02 (m, 2H), 6.91−6.88 (m, 1H), 5.80 (s, 1H), 3.86 (s, 3H), 3.56−3.46 (m, 4H), 3.04−2.97 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 187.0, 186.1, 159.3, 156.4, 155.1, 143.2, 141.0, 135.0, 134.9, 132.8, 132.7, 129.6, 126.6, 126.5, 120.8, 114.7, 112.8, 112.7, 111.9, 62.8, 55.0, 52.7, 42.2. HRMS (ESI): m/z caclcd for C26H23Cl2NO5Na [M + Na]+: 522.0851; Found: 522.0833.
4.1.3.17. 2-((Bis(2-chloroethyl)amino)(4-methylphenyl)methyl)-1,4-dihydroxyanthracene-9,10-dione (5q)
Yield 19%; mp 63–65 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.26 (s, 1H), 12.7 (s, 1H), 8.20−8.18 (m, 2H), 7.92−7.90 (m, 2H), 7.71 (s, 1H), 7.31 (d, J = 8.0 Hz, 2H), 7.16 (d, J = 7.6 Hz, 1H), 5.45 (s, 1H), 3.67−3.57 (m, 4H), 3.02−2.90 (m, 4H), 2.27 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 187.1, 186.2, 156.4, 155.1, 143.8, 137.1, 136.2, 135.1, 134.9, 132.9, 132.8, 129.1, 128.7, 126.7, 126.5, 126.3, 112.7, 111.8, 62.7, 52.6, 42.1, 20.6. HRMS (ESI): m/z caclcd for C26H23Cl2NO4Na [M + Na]+: 506.0902; Found: 506.0898.
4.1.3.18. 2-((Bis(2-chloroethyl)amino)(3,4-dimethylphenyl)methyl)-1,4-dihydroxyanthracene-9,10-dione (5r)
Yield 31%; mp 118–121 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.26 (s, 1H), 12.70 (s, 1H), 8.20−8.17 (m, 2H), 7.93−7.88 (m, 2H), 7.70 (s, 1H), 7.19 (s, 1H), 7.14−7.09 (m, 2H), 5.42 (s, 1H), 3.67−3.57 (m, 4H), 3.02−2.90 (m, 4H), 2.20 (s, 3H), 2.17 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 187.0, 186.1, 156.5, 155.1, 143.9, 136.5, 136.3, 135.8, 135.0, 134.9, 132.8, 132.7, 129.8, 129.6, 126.6, 126.5, 126.3, 126.1, 112.6, 111.7, 62.7, 52.6, 42.1, 19.5, 19.0. HRMS (ESI): m/z caclcd for C27H25Cl2NO4Na [M + Na]+: 520.1058; Found: 520.1059.
4.1.3.19. 2-((Bis(2-chloroethyl)amino)(phenyl)methyl)-1,8-dihydroxyanthracene-9,10-dione (5s)
Yield 46%; mp 123–124 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.47 (s, 1H), 11.76 (s, 1H), 8.08 (d, J = 7.6 Hz, 1H) 7.78−7.74 (m, 2H), 7.66 (d, J = 7.2 Hz, 1H), 7.42 (d, J = 7.6 Hz, 2H), 7.36−7.32 (m, 3H), 7.28−7.24 (m, 1H), 5.53 (s, 1H), 3.62−3.59 (m, 4H), 2.97−2.93 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 192.4, 181.1, 161.3, 159.1, 140.1, 137.5, 135.6, 133.3, 131.9, 128.5, 127.6, 124.4, 119.3, 119.1, 116.0, 115.8, 62.5, 52.9, 42.0. HRMS (ESI): m/z caclcd for C25H21Cl2NO4 [M]: 470.0926; Found: 470.0926.
4.1.3.20. 2-((Bis(2-chloroethyl)amino)(phenyl)methyl)-1-hydroxy-4-methoxyanthracene-9,10-dione (5t)
Yield 39%; mp 114–115 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.34 (s, 1H), 8.15−8.07 (m, 2H), 7.97 (s, 1H), 7.91−7.81 (m, 2H), 7.47 (d, J = 7.2 Hz, 2H), 7.38−7.34 (m, 2H), 7.30−7.26 (m, 1H), 5.51 (s, 1H), 3.99 (s, 3H), 3.68−3.62 (m, 4H), 3.03−2.90 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 189.0, 180.1, 154.3, 153.7, 140.1, 139.4, 135.2, 134.5, 133.5, 131.7, 129.0, 128.5, 127.7, 126.5, 126.0, 122.1, 117.4, 115.5, 63.3, 56.7, 52.5, 42.1. HRMS (ESI): m/z caclcd for C26H23Cl2NO4Na [M + Na]+: 506.0902; Found: 506.0903.
4.2. Cell culture and cytotoxicity assays
The two breast cancer cell lines, MDA-MB-231 and MCF-7, the cervical cancer cell line, HeLa, and the non-small cell lung carcinoma cell line, A549 were routinely cultured in DMEM medium supplemented with 10% FBS, 4 mM glutamine, 1 mM sodium pyruvate, 100 IU/mL penicillin, 100 ug/mL streptomycin and 0.25 ug/mL amphotericin. Cultures were maintained in 5% carbon dioxide at a temperature of 37 °C. The cells were plated in 24-well plates at a density of 20,000 per well in 10% FBS DMED medium. The cells were then treated with Doxorubicin, Chlorambucil, or synthesized hydroxyanthraquinone N-mustards derivatives separately at 5 different doses ranging from 0.01 μM to 10 μM for 5 days, while equal treatment volumes of DMSO were used as vehicle control. Cell numbers were counted with a cell viability analyzer (Beckmane–Coulter). The ratio of drug treated viable cell numbers to vehicle treated viable cell numbers was defined as percentage viability. IC50 values were obtained from dose response curves for each tested compound.
Supplementary Material
Acknowledgments
We are thankful for the financial support from Major Program of Natural Science Foundation of Jiangsu Higher Education Institutions (12KJA150005) (L.-M. Zhao), NIGMS grant number 1U54GM104940 (G. Wang), the Natural Science Foundation of Jiangsu Province (BK2012576) (L.-M. Zhao), and NIMHD grant number 2G12MD007595 (G. Wang).
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.ejmech.2015.08.006.
References
- 1.Chabner BA, Bertino J, Cleary J, Ortiz T, Lane A, Supko JG, Ryan D. Cytotoxic agents. In: Brunton LL, C B, Knollmann BC, editors. Goodman & Gilman’s Pharmacologic Basis of Therapeutics. twelfth. McGraw-Hill; New York: 2011. Chapter 61. [Google Scholar]
- 2.Nabarro JDN. J Pharm Pharmacol. 1950;2:865–879. doi: 10.1111/j.2042-7158.1950.tb13010.x. [DOI] [PubMed] [Google Scholar]
- 3.Povirk LF, Shuker DE. Mutat Res. 1994;318:205–226. doi: 10.1016/0165-1110(94)90015-9. [DOI] [PubMed] [Google Scholar]
- 4.Rajski SR, Williams RM. Chem Rev. 1998;98:2723–2795. doi: 10.1021/cr9800199. [DOI] [PubMed] [Google Scholar]
- 5.Carroll FI, Philip A, Blackwell JT, Taylor DJ, Wall ME. J Med Chem. 1972;15:1158–1161. doi: 10.1021/jm00281a016. [DOI] [PubMed] [Google Scholar]
- 6.Cullis PM, Merson-Davies L, Weaver R. J Am Chem Soc. 1995;17:8033–8034. [Google Scholar]
- 7.Cozzi P, Beria I, Biasoli G, Caldarelli M, Capolongo L, Geroni C, Mongelli N. Bioorg Med Chem Lett. 1997;7:2979–2984. doi: 10.1016/s0960-894x(00)00204-3. [DOI] [PubMed] [Google Scholar]
- 8.Baraldi PG, Beria I, Cozzi P, Geroni C, Espinosa A, Gallo MA, Entrena A, Bingham JP, Hartley JA, Romagnoli R. Bioorg Med Chem. 2004;12:3911–3921. doi: 10.1016/j.bmc.2004.04.045. [DOI] [PubMed] [Google Scholar]
- 9.Reux B, Weber V, Galmier MJ, Borel M, Madesclaire M, Madelmont JC, Debiton E, Coudert P. Bioorg Med Chem. 2008;16:5004–5020. doi: 10.1016/j.bmc.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 10.Li S, Wang X, He Y, Zhao M, Chen Y, Xu J, Feng M, Chang J, Ning H, Qi C. Eur J Med Chem. 2013;67:293–301. doi: 10.1016/j.ejmech.2013.06.055. [DOI] [PubMed] [Google Scholar]
- 11.Marvania B, Kakadiya R, Christian W, Chen T-J, Wu M-H, Suman S, Tala K, Lee T-C, Shah A, Su T-L. Eur J Med Chem. 2014;83:695–708. doi: 10.1016/j.ejmech.2014.06.066. [DOI] [PubMed] [Google Scholar]
- 12.Xu Y-Z, Gu X-Y, Peng S-J, Fang J-G, Zhang Y-M, Huang D-J, Chen J-J, Gao K. Eur J Med Chem. 2015;94:284–297. doi: 10.1016/j.ejmech.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 13.Arcamone F. Doxorubicin, Anticancer Antibiotics. Vol. 17. New York: 1981. (Medicinal Chemistry Series). [Google Scholar]
- 14.Traganos F, Evenson DP, Staiano-Coico L, Darzynkiewicz Z, Melamed MR. Cancer Res. 1980;40:671–681. [PubMed] [Google Scholar]
- 15.Patterson LH. Cancer Metastasis Rev. 1993;12:119–134. doi: 10.1007/BF00689805. [DOI] [PubMed] [Google Scholar]
- 16.Gholivand MB, Kashanian S, Peyman H, Roshanfekr H. Eur J Med Chem. 2011;46:2630–2638. doi: 10.1016/j.ejmech.2011.03.034. [DOI] [PubMed] [Google Scholar]
- 17.Konishi M, Ohkuma H, Matsumoto K, Tsuno T, Kamei H, Miyaki T, Oki T, Kawaguchi HJ. J Antibiot. 1989;42:1449–1452. doi: 10.7164/antibiotics.42.1449. [DOI] [PubMed] [Google Scholar]
- 18.Konishi M, Ohkuma H, Tsuno T, Oki T, VanDuyne GD, Clardy J. J Am Chem Soc. 1990;112:3715–3716. [Google Scholar]
- 19.Allevi P, Anastasia M, Bingham S, Ciuffreda P, Fiecchi A, Cighetti G, Muir M, Scala A, Tyman J. J Chem Soc Perkin Trans. 1998;1:575–582. [Google Scholar]
- 20.Abramson HN, Banning JW, Nachtman JP, Roginski ET, Sardessai M, Wormser HC, Wu J, Nagia Z, Schroeder RR, Bernardo MM. J Med Chem. 1986;29:1709–1714. doi: 10.1021/jm00159a024. [DOI] [PubMed] [Google Scholar]
- 21.Frederick CA, Williams LD, Ughetto G, Van der Marel GA, Van Boom JH, Rich A, Wang AH. J Biochem. 1990;29:2538–2549. [PubMed] [Google Scholar]
- 22.Lin TS, Tiecher BA, Sartorelli AC. J Med Chem. 1980;23:1237–1242. doi: 10.1021/jm00185a019. [DOI] [PubMed] [Google Scholar]
- 23.Fisher GR, Gutierrez PL, Oldcorne MA, Patterson LH. Biochem Pharmacol. 1992;43:575–585. doi: 10.1016/0006-2952(92)90581-3. [DOI] [PubMed] [Google Scholar]
- 24.Koyama M, Takahashi K, Chou TC, Darzynkiewicz Z, Kapuscinski J, Kelly TR, Watanabe KA. J Med Chem. 1989;32:1594–1599. doi: 10.1021/jm00127a032. [DOI] [PubMed] [Google Scholar]
- 25.Jin G-Z, You Y-J, Yong K, Nam N-H, Ahn B-Z. Eur J Med Chem. 2001;36:361–366. doi: 10.1016/s0223-5234(01)01229-6. [DOI] [PubMed] [Google Scholar]
- 26.Jin GZ, Song GY, Zheng XG, Kim Y, Sok DE, Ahn BZ. Arch Pharm Res. 1998;21:198–206. doi: 10.1007/BF02974028. [DOI] [PubMed] [Google Scholar]
- 27.Langdon SR, Ertl P, Brown N. Mol Inf. 2010;29:366–385. doi: 10.1002/minf.201000019. [DOI] [PubMed] [Google Scholar]
- 28.Zhao L-M, Zhang L-M, Liu J-J, Wan L-J, Cheng Y-Q, Zhang S-Q, Yan Z-W, Jiang J-H. Eur J Med Chem. 2012;47:255–260. doi: 10.1016/j.ejmech.2011.10.050. [DOI] [PubMed] [Google Scholar]
- 29.Zhao LM, Ma FY, Jin HS, Ma J, Wang H, Fu CZ. Eur J Org Chem. :7193–7199. [Google Scholar]
- 30.Zhao LM, Zhang LM, Ma FY, Wang XS, Jin HS. Tetrahedron Lett. 2013;54:2802–2805. [Google Scholar]
- 31.Cao FX, Zhao LM. Tetrahedron Lett. 2015;56:2724–2727. [Google Scholar]
- 32.Weiss RB. Semin Oncol. 1992;19:670–686. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.