

Abstract
Changes in the cardiac metabolic milieu during the perinatal period redirect mitochondrial substrate preference from carbohydrates to fatty acids. Mechanisms responsible for this mitochondrial plasticity are unknown. Here we found that PINK1-Mfn2-Parkin mediated mitophagy directs this metabolic transformation in mice. A mitofusin (Mfn) 2 mutant lacking PINK1 phosphorylation sites necessary for Parkin binding (Mfn2 AA) inhibited mitochondrial Parkin translocation, suppressing mitophagy without impairing mitochondrial fusion. Cardiac Parkin deletion or expression of Mfn2 AA from birth, but not after weaning, prevented postnatal mitochondrial maturation essential to survival. Five week old Mfn2 AA hearts retained a fetal mitochondrial transcriptional signature without normal increases in fatty acid metabolism and mitochondrial biogenesis genes. Myocardial fatty acylcarnitine levels and cardiomyocyte respiration induced by palmitoylcarnitine were concordantly depressed. Thus, instead of transcriptional reprogramming, fetal cardiomyocyte mitochondria undergo perinatal Parkin-mediated mitophagy and replacement by mature adult mitochondria. Mitophagic mitochondrial removal underlies developmental cardiomyocyte mitochondrial plasticity and metabolic transitioning of perinatal hearts.
Mammalian hearts depend upon mitochondrial oxidative phosphorylation to fuel myocardial contraction and pump function; catabolism of carbohydrates or fats generates ATP that powers excitation-contraction coupling. Under conditions of optimal intrinsic mitochondrial functioning, i.e. when mitochondria are “fit”, energy demands and access to metabolic substrates and oxygen are central determinants of mitochondrial respiration. During organism development both substrate availability and tissue oxygen content change. Accordingly, the increase in trans-placental oxygen exchange in early embryos provokes a shift from anaerobic glycolysis to aerobic mitochondrial respiration (1). After birth, loss of trans-placental carbohydrate substrates promotes a further transition to fatty acid metabolism (2) as small, fetal cardiomyocyte mitochondria are supplanted by adult organelles (3). The normal perinatal developmental conversion from glucose to fatty acid cardiac metabolism, and its maladaptive reversal back toward glucose in diseased adult hearts, have been linked to changes in metabolic gene expression, so-called “metabolic reprogramming” (4). Cellular mechanisms underlying these cardiac metabolic transitions are poorly described, and conventional wisdom has been that mitochondria are “flexible fuel” organelles capable of switching back and forth between carbohydrate and fatty acid metabolism (5).
Stochastic damage to cardiomyocyte mitochondria places hearts at risk from metabolic insufficiency or reactive oxygen species (ROS)-mediated cytotoxicity (6). It is believed that maintaining mitochondrial functional integrity requires continuous surveillance and culling of dysfunctional organelles. In cultured fibroblasts depolarized mitochondria are identified, sequestered, and eliminated through directed autophagy followed by lysosomal destruction (mitophagy). This form of mitochondrial quality control is mediated in part by two Parkinson’s disease factors, the E3 ubiquitin ligase Parkin and its upstream activating kinase, PTEN-induced putative kinase 1 (PINK1) (7, 8). By removing impaired organelles the overall fitness of the mitochondrial collective is preserved.
The observation that glycolytic metabolism is preferred in fetal hearts, but is maladaptive in diseased adult hearts (4), indicates that mitochondrial fitness is neither a specific nor a unique condition. Rather, mitochondria are “fit” when they demonstrate optimal functional compatibility for a given developmental or pathophysiological milieu. In this context, the idea that mitochondrial quality must be actively controlled applies not just to selective culling of individual damaged mitochondria, but also to generalized cell- and organ-wide promotion of mitochondrial turnover during developmental or disease-related transitions of cellular fuel and energy metabolism. A possible role for mitophagic mitochondrial replacement during metabolic transitions has not been addressed, in part because developmental phenotypes are not observed in otherwise normal mice systemically lacking Parkin (9, 10). However, absence of phenotypes in Parkin-deficient mice may simply reflect opportunistic compensation by Parkin-independent mitophagy pathways (11).
Here, we set out to clarify the role of Parkin-mediated mitochondrial turnover in the normal developmental switch from carbohydrate to fatty-acid based metabolism in perinatal mouse hearts. To avoid confounding effects of germ-line and cardiac-specific Parkin ablation we developed and deployed in vitro and in vivo systems in which expression of a mitofusin 2 (Mfn2) mutant lacking PINK1 phosphorylation sites essential for its binding of Parkin suppressed Parkin translocation to mitochondria, thus specifically interrupting Parkin-dependent mitophagy.
Parkin is essential for perinatal mitochondrial maturation in cardiomyocytes
Cardiomyocyte mitochondria of 1 day old mice exhibit the typical elongated, curvilinear morphology of human fetal heart mitochondria (3), but mature over three weeks into the larger ovoid mitochondrial structure characteristic of adult mammalian hearts (Fig. 1A). Cardiomyocyte-specific ablation of Park2, encoding Parkin, on perinatal day 1 (P1) was rapidly lethal in most mice. However, a small number of “escapers” survived until P21. PCR analysis revealed incomplete Cre-mediated Park2 gene recombination in hearts of these mice (Fig. 1B), suggesting that partial Parkin insufficiency induced a forme fruste of the early lethal cardiac Parkin deletion phenotype. Compared to normal littermates, P21 Parkin-insufficient hearts appeared slightly smaller (Figs. 1C, 1D). Myocardial histology of Parkin-deficient hearts was unremarkable (Figs. 1C, 1D), but ultrastructural imaging of myocardium from surviving 3 week old perinatal cardiac Parkin-insufficient mice revealed mitochondria having a morphology typical of fetal hearts (Fig. 1C), whereas control (Park2 fl/fl) littermate mitochondria had the morphology of normal adult hearts (Fig. 1D). Furthermore, perinatal Parkin-deficient hearts had abundant inclusions of homogenous appearance with smooth borders, characteristic of lipid droplets (Fig. 1C). Lethality of perinatal cardiac-specific Park2 ablation, together with findings of surviving escaper mice, suggested that Parkin may be essential for maturational development of cardiomyocyte mitochondria. However, the aggressive phenotype confounded attempts to determine underlying mechanisms. Accordingly, we built upon previous observations that Parkin fails to translocate to depolarized mitochondria of Mfn2-deficient mice (12, 13), and the discovery that PINK1-phosphorylation of Mfn2 promotes Mfn2-Parkin binding (12), to develop a system in which Parkin-mediated mitophagy could be conditionally interrupted in vitro or in vivo without primarily targeting either Parkin or PINK1.
Fig. 1. Early lethality of perinatal cardiomyocyte-specific Parkin-deficient mice.
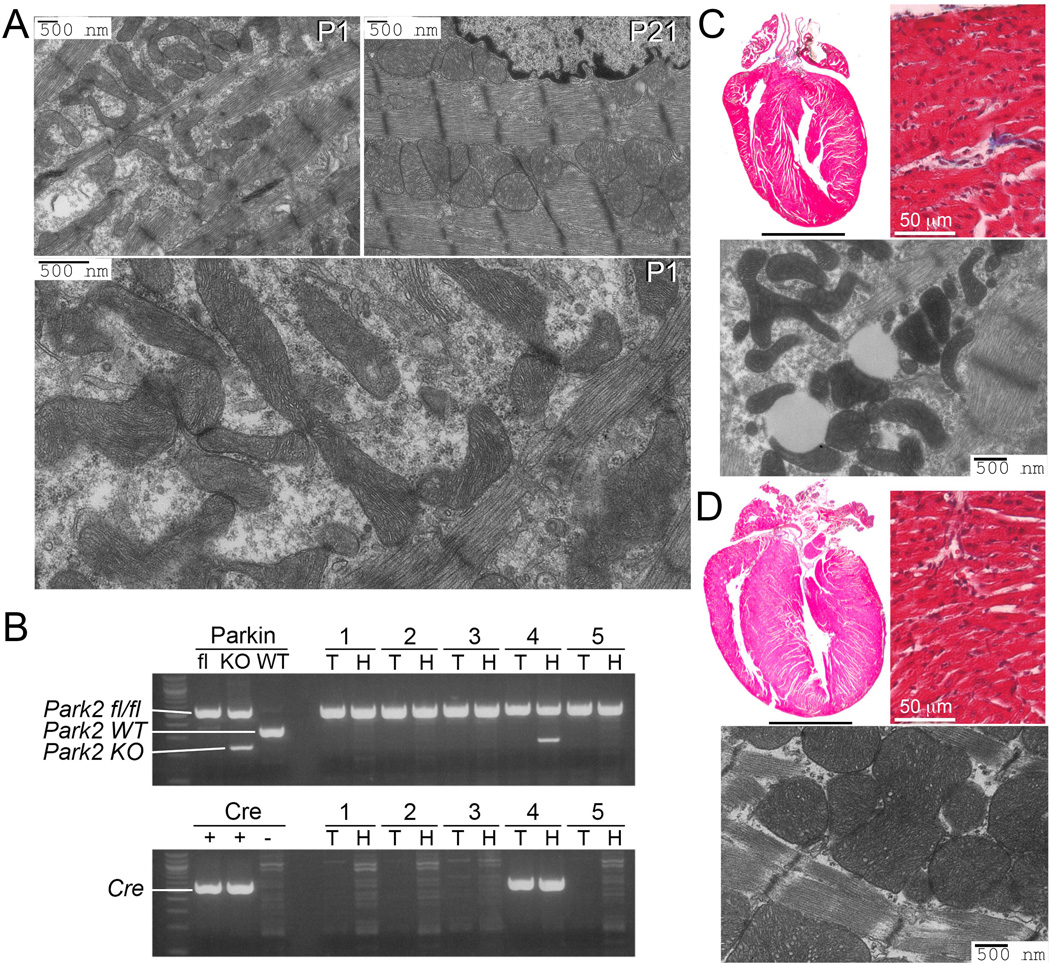
(A) Transmission electron microscopy (TEM) showing normal cardiomyocyte mitochondria on the first (P1) and 21st (P21) day of life. Enlargement shows structural details at P1. (B) PCR genotyping of floxed Park2 gene (top) and tamoxifen-inducible cardiac Cre (bottom) of surviving mice from a representative litter at P21; 3 mice died before weaning. KO indicates Cre recombined Park2 fl/fl allele. T is tail DNA; H is heart DNA. (C and D) Representative (of 3) hearts, histological sections, and TEMs from P21 cardiac Parkin deficient (top) and control (bottom) mice. Scale bars for hearts is 2 mm.
Mutational interdiction of Mfn2 phosphorylation by PINK1 inhibits Parkin-mediated mitophagy
PINK1 is stabilized and accumulates in depolarized mitochondria (14) and phosphorylates Mfn2 (Fig. 2A), enabling its binding to Parkin (12). Mimicking Mfn2 phosphorylation by substituting glutamic acid (E) for the critical threonine and serine (Mfn2 T111E/S442E; Mfn2 EE) induces PINK1-independent Mfn2-Parkin association (12) and promoted spontaneous Parkin recruitment to mitochondria of cultured fibroblasts (Fig. 2B; fig. S1). Non-phosphorylatable alanine (A) substitution at the same sites (Mfn2 AA) had reciprocal effects, preventing PINK1-mediated Mfn2-Parkin binding (12) and suppressing Parkin translocation provoked by the mitochondrial uncoupling agent carbonylcyanide-p-trifluoromethoxyphenylhydrazone (FCCP) (Fig. 2B; fig. S1). Effects of Mfn2 EE and Mfn2 AA on Parkin translocation were concordant with stimulation or inhibition, respectively, of mitochondrial-lysosomal interactions (Fig. 2C; fig. S1). In agreement with Mfn2 EE functioning as a constitutive mitochondrial Parkin receptor, it failed to evoke mitophagy in Parkin-deficient cells (fig. S2). Although the mitophagy response to FCCP was reduced in PINK1-deficient cells, Mfn2 EE nevertheless promoted spontaneous Parkin translocation (fig. S3). Because non-phosphorylatable Mfn2 AA inhibited Parkin localization to mitochondria without primarily affecting PINK1 or Parkin, we used it to interrupt PINK1-Parkin mediated mitophagy without adversely impacting non-mitophagic Parkin functionality (15).
Fig. 2. Mitochondrial Parkin mobilization directed by pseudo-PINK1 phosphorylated Mfn2.
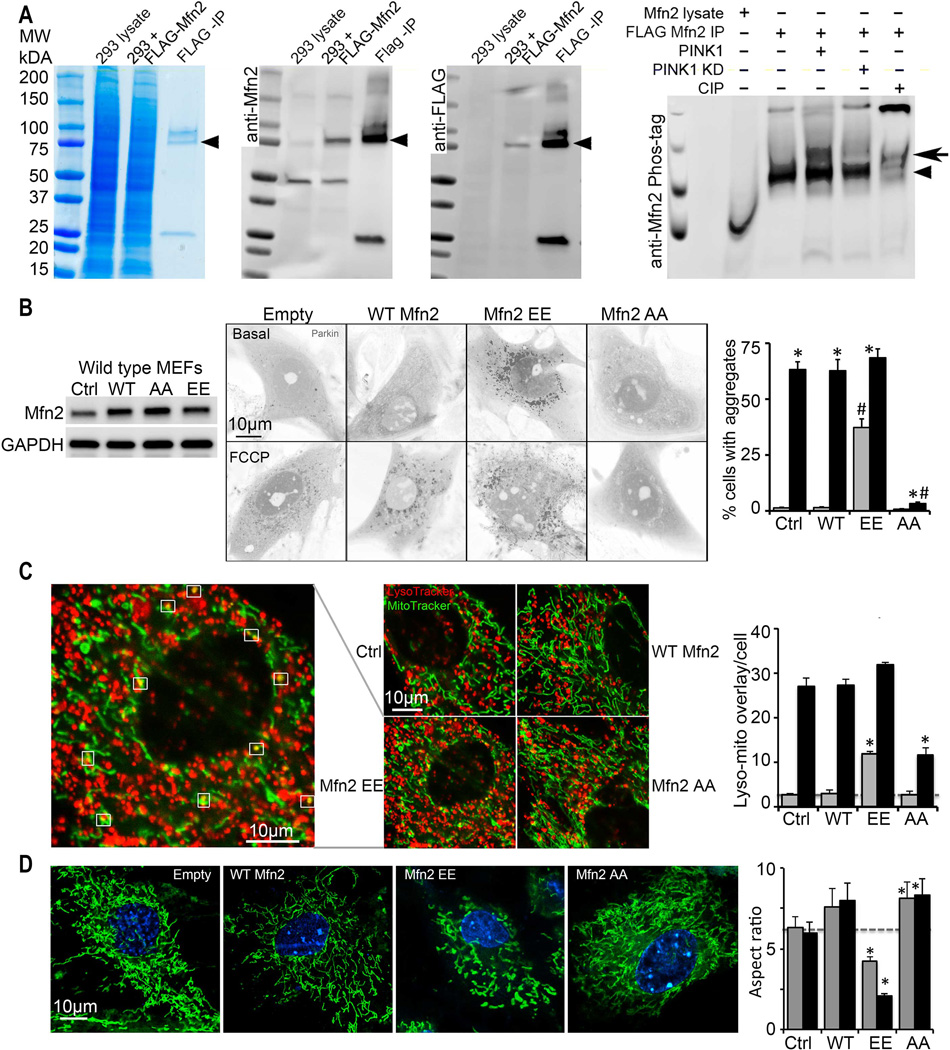
(A) Phosphorylation of Mfn2 by recombinant PINK1 in a cell-free system. First three panels show enrichment of FLAG-Mfn2 by anti-FLAG immunoprecipitation (IP); left is Coomassie blue stained gel, middle is anti-Mfn2 immunoblot, right is anti-FLAG immunoblot. Fourth panel shows anti-Mfn2 Phos-Tag immunoblot of in vitro PINK1 phosphorylation reactants; KD is kinase dead PINK1, CIP is calf intestinal phosphatase. Arrowheads show FLAG-Mfn2; bold arrow indicates phospho-Mfn2. (B) Spontaneous mcParkin translocation in MEFs provoked by adeno-Mfn2 EE, and FCCP-mediated Parkin translocation suppressed by adeno-Mfn2 AA. To the left is immunoblot of Mfn2. (C) Lysosomal-mitochondrial interactions (white squares) provoked by adeno-Mfn2 EE and suppressed by adeno-Mfn2 AA. (D) Mitochondrial elongation (aspect ratio) inhibited by adeno-Mfn2 EE and stimulated by adeno-Mfn2 AA. WT is wild type adeno-Mfn2. In B and C grey bars are basal; black bars are after FCCP or antimycin A. In D grey bars are 24h and black bars are 48h after adeno-Mfn virus infection. * is p<0.05 vs adeno β-gal control (Ctrl); # is p<0.05 vs same condition WT adeno-Mfn2.
We asked if Mfn2 phosphorylation of T111 and S442 modified other cellular actions of Mfn2. Mfn2 promotes outer mitochondrial membrane tethering and fusion (16). Non-phosphorylatable Mfn2 AA was as effective as WT Mfn2 for inducing mitochondrial fusion in either wild-type or Mfn2-deficient fibroblasts; pseudo-phosphorylated Mfn2 EE did not promote fusion, instead evoking mitochondrial shortening (Fig. 2D and figs. S1, S2, S3). Thus, Parkin binding and mitochondrial outer membrane fusion are mutually exclusive functions of Mfn2 regulated by PINK1-mediated phosphorylation.
Mitochondrial fragmentation is functionally linked to mitophagy (17). We asked if absence of fusion-promoting activity for the phospho-mimic Mfn2 EE was sufficient to provoke mitophagy. FCCP-stimulated Parkin translocation and lysosomal-mitochondrial colocalization were examined in cells expressing a naturally occurring fusion-defective human Mfn2 mutant, R400Q, in which PINK1 phosphorylation sites are intact (18). Mfn2 R400Q induced mitochondrial shortening similar to Mfn2 EE, but did not promote Parkin translocation or mitophagy (fig. S4). Thus, inhibition of mitochondrial fusion and mitochondrial recruitment of Parkin by Mfn2 are not functionally coupled, except through PINK1-mediated Mfn2 phosphorylation. Finally, we determined if phosphorylation of Mfn2 on T111 and S442 affected its binding of Miro mitochondrial transport proteins (19). WT Mfn2, Mfn2 AA, and Mfn2 EE each bound Miro1 and Miro2, exhibiting the previously reported preference for Miro2 (fig. S5). Mitochondrial fusion and Parkin translocation are therefore reciprocally and uniquely regulated by PINK1-mediated phosphorylation of Mfn2 T111 and S442.
Perinatal inhibition of Parkin-mediated mitophagy induces lethal cardiomyopathy
Because Mfn2 AA promoted mitochondrial fusion similar to wild-type Mfn2, but inhibited mitochondrial Parkin localization, we used it to dissect the role of Parkin-mediated mitophagy in mouse hearts. Both separation-of-function Mfn2 mutants (EE and AA) were expressed using a bi-transgenic doxycycline-suppressible cardiomyocyte-specific Myh6 promoter system (Fig. 3A); cardiac transgene expression can be suppressed with doxycycline, but in its absence it begins shortly after birth (20). WT Mfn2 was expressed separately as a control for increased myocardial Mfn2 content (Fig. 3A, top). In vivo regulation of cardiac mitophagy by Mfn2 EE and Mfn2 AA was assessed by crossing Mfn2 EE and Mfn2 AA mice to mice conditionally expressing Parkin using the same doxycycline-suppressible Myh6-driven expression system (21), and concomitantly inducing the Mfn2 and Parkin transgenes in adult mice. Mitochondrial localization of Parkin, increased ubiquitination of mitochondrial proteins, and mitochondrial localization of the autophagosomal docking protein, p62/Sequestosome 1 (SQSTM1) were enhanced in Mfn2 EE/Parkin hearts, but reduced in Mfn2 AA/Parkin hearts, compared to Parkin-overexpressing controls (Fig. 3A, bottom). These in vivo results recapitulate the in vitro effects of the Mfn2 phosphorylation mutants on mitophagy.
Fig. 3. Perinatal cardiomyopathy evoked by non-phosphorylated Mfn2 AA.
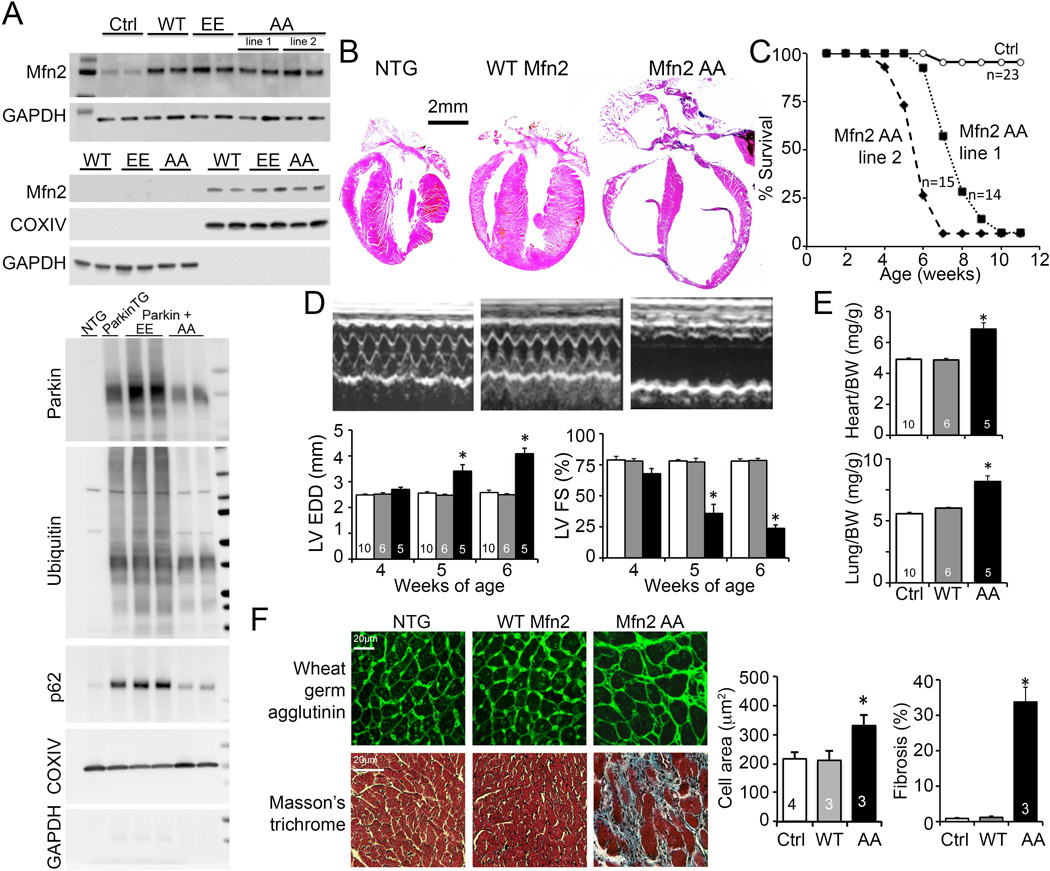
(A) Immunoblot analysis of Mfn2 expression (top) and mitochondrial Parkin localization (bottom) in transgenic mouse hearts. Upper panel - top pair is cardiac homogenate; bottom pair is mitochondrial-enriched 10,000g pellet (cytochrome oxidase IV; COX IV) and post-mitochondrial supernatant (GAPDH). Lower panel - Immunoblot analysis of mitochondrial-associated Parkin and downstream mitophagy events and their modulation by cardiac-expressed Mfn2 EE and Mfn2 AA. (B) Representative hearts of 6 week old mice. (C) Survival. (D) Serial echocardiographic data of 4-6 week old mice; white bars are Ctrl, grey is WT Mfn2, and black is Mfn2 AA. (E) Heart (top) and lung (bottom) weights of 6 week old mice indexed to body weight (BW). (F) Histological studies of cardiomyocyte cross sectional area (top) and myocardial fibrosis (bottom); quantitative data are on the right. *is p<0.05 vs WT Mfn2 and NTG control.
Mice expressing WT Mfn2 or Mfn2 EE from birth (i.e. that never received doxycycline) appeared normal (Fig. 3B-E; fig S6). In contrast, cardiac expression of Mfn2 AA at similar levels from birth (Fig 3A) was uniformly lethal by 7-8 weeks of age in two independent transgenic lines (Fig. 3B, 3C). Cardiac dilatation (Fig. 3B, 3D), worsening pump function (Fig. 3D) and pulmonary congestion (Fig. 3E) identified progressive cardiomyopathy as the underlying process and heart failure as the terminal event. Myocardium from surviving 6 week old Mfn2 AA mice exhibited cardiomyocyte enlargement and replacement fibrosis (Fig. 3F). TUNEL staining was increased proportionally to in vivo Evans blue labeling without caspase 3 processing, revealing necrosis to be the likely mechanism for Mfn2 AA-induced cardiomyocyte dropout (fig. S7A-C). However, there were no differences in the size of Mfn2-dependent mitochondrial-sarcoplasmic reticulum interfaces (22) in WT-Mfn2 and Mfn2 AA expressing mice (fig. S7D), showing that inter-organelle tethering by Mfn2 (23) was not perturbed by preventing T111 and S442 phosphorylation. Remarkably, cardiac Mfn2 AA induction at the time of weaning (~3 weeks) or in young adult mice (8 weeks) evoked no cardiac pathology (fig. S8). Thus, Parkin localization to Mfn2 on cardiomyocyte mitochondria is essential only between birth and weaning.
Parkin-mediated mitophagy is essential for perinatal cardiomyocyte mitochondrial maturation
The only molecular differences between cardiac WT Mfn2 and Mfn2 AA mice were two non-phosphorylatable amino acids (fig. S9). Yet, the cardiac phenotype of the former was benign whereas the latter developed lethal juvenile cardiomyopathy. Because Mfn2 AA promoted mitochondrial fusion as well as WT Mfn2 (see Fig. 2D and fig. S1D), and the Mfn2 AA mutation did not affect other Mfn2 functions (see figs. S5 and S7), we reasoned that the Mfn2 AA cardiomyopathy was caused by suppressing Parkin-mediated mitophagy. In agreement with this idea, mitochondria-associated Parkin and p62/SQSTM1 were both reduced in the hearts of 2-3 week old Mfn2 AA mice, compared to age-matched WT Mfn2 mice (Fig. 4A, top). Mitochondrial Parkin association was likewise depressed by Mfn2 AA in hearts of 2-3 week old food-deprived mice, but mitochondrial-associated p62/SQSTMI was increased (Fig. 4A, bottom). Thus, Mfn2 AA inhibits developmentally programmed Parkin-mediated mitophagy, but not Parkin-independent mitochondrial autophagy during starvation-induced macroautophagy.
Fig. 4. Abnormalities in mitophagy and mitochondria induced by perinatal cardiac Mfn2 AA.
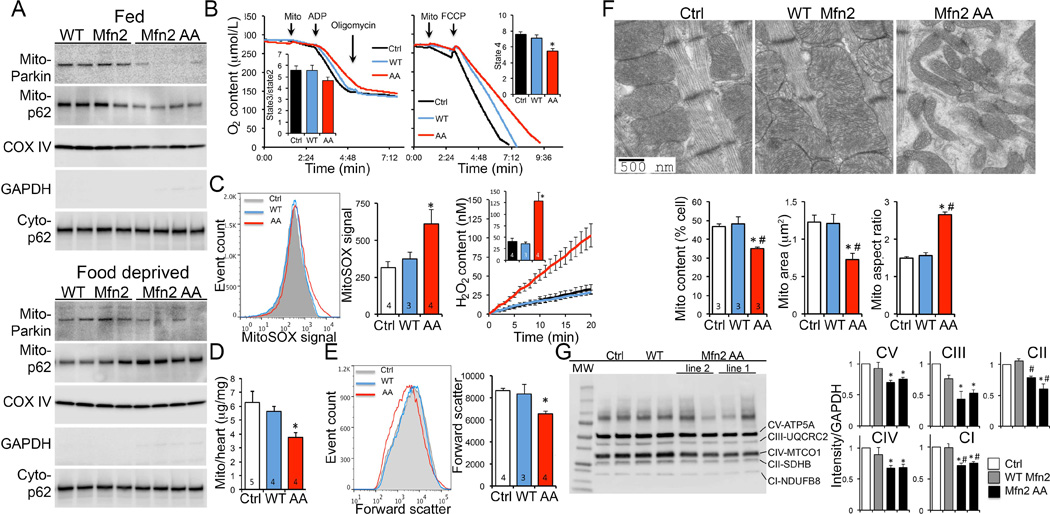
(A) Immunoreactive Parkin and p62/SQSTM1 in 2-3 week old mitochondrial-enriched mouse heart fractions (mito-) of fed mice (top) and food-deprived mice (bottom). Cyto-p62 is p62/SQSTM1 in the cytosolic fraction. (B) Substrate-stimulated (left) and maximum uncoupled (right) respiration of isolated cardiac mitochondria. (C) Isolated mitochondrial O2− (MitoSOX; left) and H2O2 (Amplex red; right) production studies. (D) Cardiac mitochondrial protein content. (E) Flow cytometric mitochondrial forward scatter. (F) Ultrastructural studies of cardiomyocyte mitochondria; mitochondrial content is % area occupied by mitochondria, mitochondrial area is mean area of individual organelles, mitochondrial aspect ratio is long axis/short axis. (G) Immunoblot analysis of respiratory complex proteins. Quantitative data to right are n=4. * is p<0.05 vs Ctrl; # is p<0.05 vs WT Mfn2.
Multiple lines of evidence revealed a deterioration in mitochondrial quality specific to Mfn2 AA hearts, including impaired maximal mitochondrial respiration (Fig 4B), increased mitochondrial production of O2− and H2O2 (Fig. 4C), and modest increases in levels of the mitochondrial stress proteins Fibroblast growth factor 21 (FGF21), Heat shock protein 60 (Hsp60), Lon peptidase 1 (LONP1), and ATPase family gene 3-like 2 (AFG3L2) (fig. S10). By comparison, liver and skeletal muscle mitochondria from cardiac-expressing Mfn2 AA mice were normal (fig. S11).
Reduced myocardial content of mitochondrial proteins (Fig. 4D; fig S12) and decreased flow cytometric forward scatter (Fig. 4E) pointed to abnormalities in the abundance and morphology of cardiac mitochondria of 6 week old Mfn2 AA mice. Ultrastructural examination revealed small, unusually shaped cardiomyocyte mitochondria (Fig 4F; fig. S13). Decreased respiratory complex protein abundance also reflected lower mitochondrial content in Mfn2 AA hearts (Fig. 4G). In agreement with absence of cardiac phenotypes when Mfn2 AA expression was induced at or after weaning (see fig. S8A), mitochondrial respiration, morphometry, and ROS production were normal in those mice (fig. S14).
Mitochondrial morphology of young adult Mfn2 AA mouse hearts resembled that of both normal postnatal (P1) mouse hearts and P21 cardiac Parkin deficient mouse hearts (compare Fig 4F to Figs. 1A and 1D). The transition of mouse cardiac mitochondria from fetal to adult morphology normally occurs within the first three weeks of life (see Fig. 1), concurrent with a functional shift favoring fatty acid metabolism (5, 24, 25). We posited that suppression of Parkin-mediated mitophagy by Mfn2 AA caused fetal mitochondria to be retained. Consistent with this idea, Mfn2 AA cardiac mitochondria did not undergo the time-dependent transformation in size and shape observed by P21 in WT Mfn2 and control hearts (Fig. 5A). Interruption of normal mitochondrial morphological maturation at P21 was associated with dilated cardiomyopathy that developed within 2 weeks thereafter (Figs. 3D, 5B and 5C; fig. S15).
Fig. 5. Fetal mitochondria persist in young adult Mfn2 AA mouse hearts.
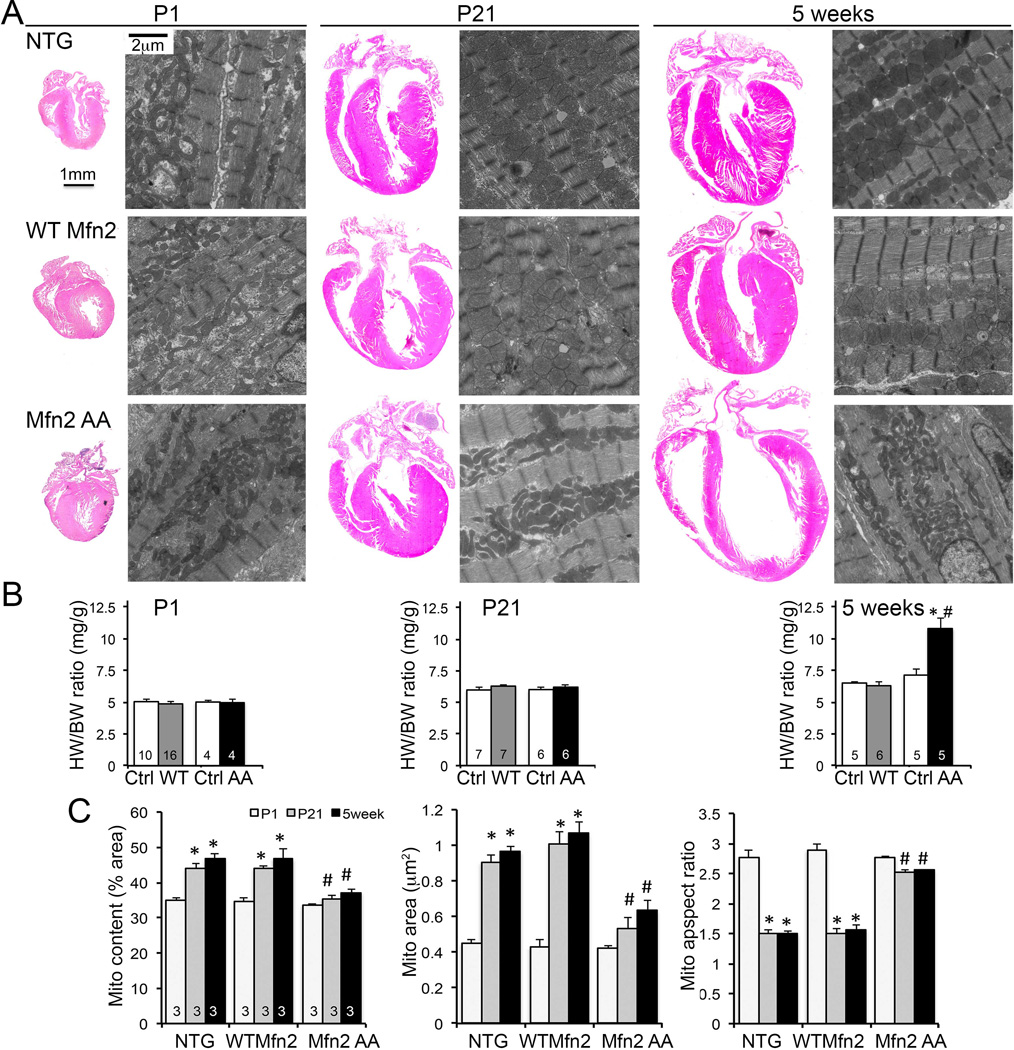
(A) Representative 4-chamber heart sections and transmission electron micrographs of cardiomyocyte mitochondria from P1, P21, and 5 week old mouse hearts. NTG controls are top row, WT Mfn2 middle row, Mfn2 AA bottom row. Quantitative data for heart weights are in (B) and for mitochondrial ultrastructure in (C); * = p<0.05 vs P1, # = p<0.05 vs WT Mfn2 at same stage.
Mitophagy is essential for the perinatal transformation of cardiac metabolism
Cardiac metabolic transitioning after birth is linked to increased expression of mitochondrial replication and transcription factors, i.e. mitochondrial biogenesis (26). We measured the changes in transcript levels for over 400 cardiac-expressed mitochondrial proteins from late embryo through adulthood, thereby defining normal transcriptional reprogramming of cardiac metabolism (fig. S16). RNA-sequencing of perinatal day 1 (P1), day 21 (P21) and 5 week old hearts showed that WT Mfn2 did not perturb normal mitochondrial gene reprogramming. By contrast, the mitochondrial transcript profile of 5 week old Mfn2 AA hearts co-segregated with P1 hearts (Fig. 6A), driven largely by failure of electron transport, fatty acid catabolism, and ketone body metabolism gene abundance to increase normally during the perinatal-to-adult transition (Fig. 6A; Supplemental datasets 1 and 2). Hemodynamic stress in adult hearts did not fully recapitulate fetal metabolic gene expression (fig. S17), demonstrating that Mfn2 AA caused true retention of the embryonic mitochondrial transcriptome and not cardiomyopathy-related re-expression of embryonic genes (25). Suppression of metabolic gene reprogramming in 5 week old Mfn2 AA hearts affected genes encoding fatty acid (Fig. 6B) and branched chain amino acid (fig. S18) tricarboxylic acid (TCA) cycle entry factors and some TCA cycle enzymes themselves (Fig. 6B), while largely sparing glycolysis genes (Fig. 6B); effects on oxidative phosphorylation (OXPHOS) genes were variable (Fig. 6B). Characteristic perinatal increases in abundance of transcriptional activators of metabolism and mitochondrial replication factors, i.e. mitochondrial biogenesis genes, were also suppressed in Mfn2 AA hearts (Fig. 6C).
Fig. 6. Failure of metabolic gene reprogramming after perinatal Mfn2 AA expression.
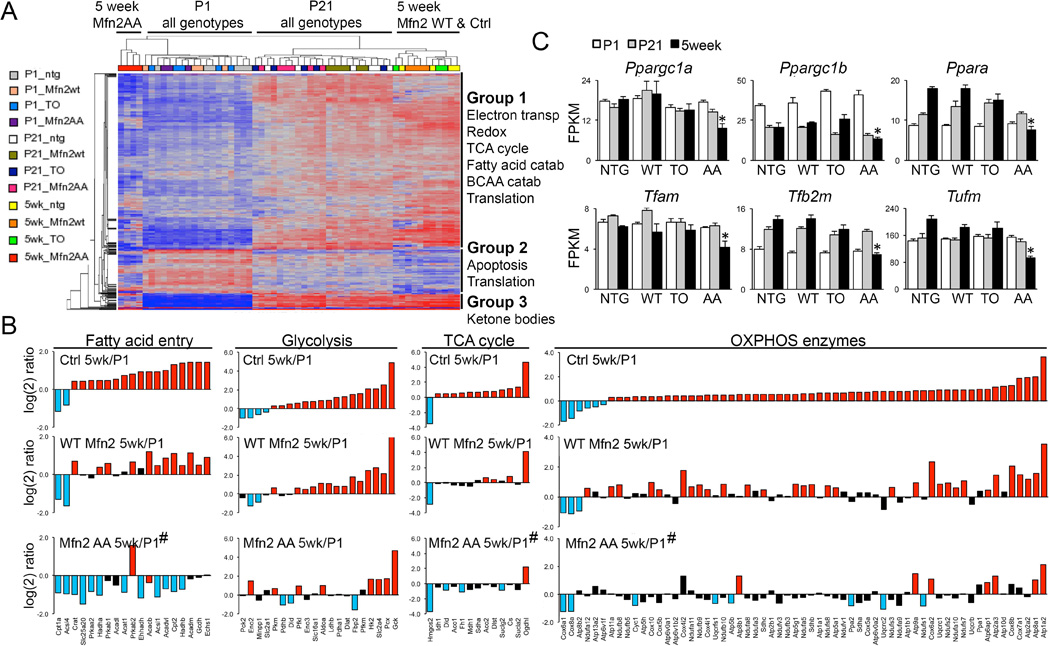
(A) Heat map of mitochondrial gene expression in P1, P21, and 5 week mouse hearts; functional annotation of Mfn2 AA gene clusters is to the right. (B) Postnatal reprogramming of mitochondrial genes by metabolic function. Bars are mean values from results in Fig 4B; log(2) gene expression at 5 weeks vs P1 for Ctrl (top), WT Mfn2 (middle) and Mfn2 AA (lower) hearts. Blue and red bars are significantly down and upregulated, respectively (1.25 fold, FDR <0.02); black bars are not significantly regulated. (C) Regulated expression of mitochondrial biogenesis and replication genes during the perinatal-adult transition.
* = FDR<0.02 vs littermate control mice (TO). # is p<0.0001 vs WT Mfn2 (2-way ANOVA).
To define the metabolic consequences of interrupting Parkin-mediated mitophagy in Mfn2 AA mouse hearts we compared mitochondrial respiration stimulated by the fetal-preferred glycolytic substrate pyruvate to respiration stimulated by the adult-preferred fatty acid substrate palmitoylcarnitine (2, 4, 24). Oxygen consumption by permeabilized cardiomyocytes was similar in control, WT- Mfn2, and Mfn2 AA cells given pyruvate. However, Mfn2 AA cardiomyocytes exhibited impaired respiration when provided with palmitoylcarnitine (Fig. 7A). Moreover, myocardial metabolite profiling revealed abnormally low levels of multiple fatty acid acylcarnitines, which are products of mitochondrial fatty acid oxidative flux, in comparison with age-matched control and WT-Mfn2 hearts. Instead, acylcarnitine levels in 5 week Mfn2 AA hearts were comparable to those of normal P1 hearts (Figs. 7B-D). By contrast, myocardial abundance of organic acids in young adult Mfn2 AA and control hearts was similar. The concordant abnormalities of mitochondrial gene expression, substrate preference for mitochondrial respiration, and metabolic profile exhibited by cardiac Mfn2 AA mice (fig. S19) illustrate the global impact of Parkin-mediated mitochondrial removal on normal developmental metabolic transitioning of the perinatal heart. Taken together, the data point to a mismatch between mitochondrial programming and metabolic substrate availability as the underlying cause of progressive cardiomyopathy in juvenile Mfn2 AA mice with defective cardiomyocyte mitophagy.
Fig. 7. Adult Mfn2 AA hearts retain a fetal-like glycolytic metabolism.
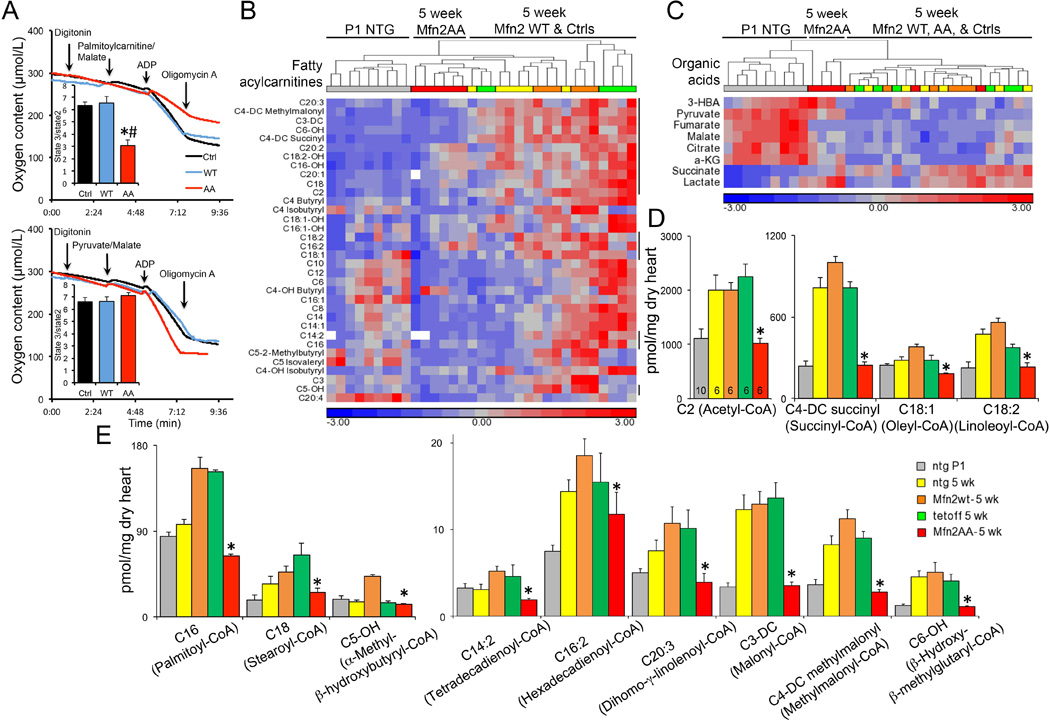
(A) Palmitoylcarnitine-stimulated (top) and pyruvate-stimulated (bottom) respiration of isolated permeabilized cardiomyocytes. (B and C) Standardized heat map showing unsupervised clustering of myocardial acylcarnitine (B) and organic acid (C) metabolite content in P1 NTG and 5 week old NTG, WT Mfn2, and Mfn2 AA mouse hearts. Vertical lines to the right of (B) indicate significantly dysregulated metabolites in 5 week Mfn2 AA hearts vs contemporaneous controls. (D) Quantitative data for absolute myocardial content of dysregulated metabolites in (B). –DC and -OH designate monohydroxylated and dicarboxylic acid acylcarnitine species, respectively. Common names for parent species are in parentheses. *=p<0.05 vs WT Mfn2 (ANOVA).
Discussion
Here we have shown that Parkin-mediated mitophagy is essential for normal perinatal cardiac mitochondrial and metabolic maturation. By expressing from birth an engineered Mfn2 AA mutant that cannot be PINK1-phosphorylated on T111 and S442 as required for Mfn2-Parkin binding (12), the normal developmental perinatal transformation of cardiac metabolism was disrupted. Persistence of fetal carbohydrate-metabolizing mitochondria in adult Mfn2 AA hearts revealed the requirement for organelle removal through the PINK1-Mfn2-Parkin mitophagy mechanism pior to mitochondrial transitioning to normal adult fatty acid metabolism. Even the genetic program encoding critical fatty acid metabolism pathways was repressed when fetal mitochondria were retained. Parkin thus promotes mitochondrial removal and suppresses biogenesis, consistent with previously described Parkin-dependent regulation of the mitochondrial biogenesis factors PARIS and PGC-1α (11). Organelle replacement rather than simple reprogramming may be necessary because mitochondrial respiratory supercomplexes are organized as paracrystalline arrays (27, 28) whose disassembly, reorganization, and reassembly in pre-existing embryonic cardiac mitochondria could be disruptive (7). An alternate means of mitochondrial transitioning might be through cycles of fission and fusion. However, the normal half time for turning over adult cardiomyocyte mitochondria through mitochondrial dynamism is ~ 3 weeks (29), within which time our studies reveal the perinatal transformation from fetal to adult mitochondria to be complete. Thus, we propose that the Parkin-Mfn2 interaction drives general mitophagic turnover of fetal mitochondria in the perinatal heart, enabling their replacement with mitochondria incorporating biogenically-derived metabolic systems optimized for the high energetic demands of contracting adult hearts.
The metabolic preference for glycolysis in growing fetal hearts is reminiscent of the Warburg effect observed in cancer, wherein the transformation from restrained to malignant growth is associated with a transition to increased glycolysis and less dependence upon aerobic mitochondrial ATP production (30). Although the specific molecular determinants of glycolytic metabolism in fetal hearts undoubtedly differ from those of tumors, in both instances increased glycolysis is adaptive for a hypoxic environment and is optimized for increasing biomass, i.e. for promoting cell growth (31). Thus, the growing fetal heart derives two benefits from a predominantly glycolytic metabolism: 1) The ability to generate ATP sufficient for the comparatively modest needs of fetal heart contraction despite a relatively hypoxic environment; and 2) A metabolism that facilitates uptake and incorporation of amino acids and fatty acids into new cellular structures for cell and organ growth, rather than metabolizing them for energy production. On the other hand, fatty acid metabolism in fully grown adult hearts provides more efficient ATP production to fuel increased contractile demand. Our findings reveal this cardiac metabolic transition to be an essential adaptation for extra-uterine life.
The developmental role for Parkin-dependent mitophagy described herein was unsuspected because germ-line Parkin gene ablation in mice has not produced robust phenotypes (9, 10). As deployed here, Mfn2 AA perturbed endogenous Parkin strictly by interdicting its localization to mitochondria, i.e. by acting as a non-functioning cardiomyocyte Parkin receptor. The lethal cardiomyopathy evoked by Mfn2 AA, but not WT Mfn2, exposed novel Parkin functionality specific to the postnatal period during which myocardial carbohydrate-dependent metabolism transitions to fatty acid-dependence. Early lethality after perinatal cardiomyocyte-specific Parkin gene deletion likewise demonstrated an essential role for Parkin in postnatal hearts. However, perinatal cardiomyocyte Parkin deficiency was generally fatal within the first 3 weeks of life, whereas perinatal cardiac Mfn2 AA expression provoked a slower progressing (but ultimately equally deadly) cardiomyopathy. The aggressive phenotype induced by cardiomyocyte-directed Parkin deletion likely results from complete disruption of Parkin activity that, in addition to promoting mitophagy, includes regulation of mitochondrial dynamism, modulation of subcellular mitochondrial motility, mediation of mitochondrial protein degradation, and stimulation of mitochondrial biogenesis (15). By comparison, expressing Mfn2 AA is a more precise intevention that only inhibits Parkin activity dependent upon its binding to PINK1-phosphorylated Mfn2, i.e. mitophagy. Indeed, because endogenous Mfn2, PINK1, and Parkin are still present, mitophagy was not entirely abrogated by Mfn2 AA. Accordingly, cardiomyocyte-specific perinatal Parkin gene deletion and Mfn2 AA expression had similar effects on mitochondrial maturation in the heart, but the Mfn2 AA phenotype was an attenuated form of the full-blown phenotype that can be evoked by perinatal mitophagic dysfunction.
Our findings add to evidence that in vivo Parkin functionality is underestimated from conventional systemic Parkin deletion. Whereas germ-line Parkin knockout mice exhibit few manifestations of Parkinson’s disease or cardiac involvement at baseline (9, 10, 32), conditional Parkin ablation in the substantia nigra recapitulates a central pathological feature of Parkinson’s disease, degeneration of dopaminergic neurons (11). Likewise, shRNA-mediated Parkin suppression reduces mitophagy in mouse livers, whereas mitophagy is intact in germ-line Parkin knockout mouse livers (33). Finally, cardiomyocyte-specific Parkin ablation alleviates the cardiomyopathy induced by dynamin-related protein 1 (Drp1) deficiency, whereas global Parkin deficiency fails to rescue a similar cardiac Drp1-deficient model (21, 34).
These results also clarify the specific function of PINK1-Mfn2-Parkin signaling in mitophagy. The need for PINK1-phophorylated Mfn2 to function as a Parkin binding protein in mitophagic mitochondrial clearance is not absolute, because Parkin will translocate to mitochondria of FCCP-treated cells lacking Mfn2 (fig. S1). Thus, just as there are Parkin-independent pathways for mitochondrial elimination (35), we would infer one or more Mfn2-independent means of recruiting Parkin to depolarized mitochondria. Nonetheless, in normal and Mfn2-deficient fibroblasts non-phosphorylatable Mfn2 AA abolished Parkin translocation to, and lysosomal engulfment of, depolarized mitochondria without affecting other Mfn2 activities such as mitochondrial fusion, mitochondrial-SR tethering, and Miro binding. These results define a central role for the PINK1-Mfn2 phosphorylation interaction in conventional mitochondrial quality control, and suggest a mechanism whereby PINK1-phosphorylated Mfn2 induces primary mitochondrial Parkin binding, with PINK1-phosphorylated ubiquitin as the preferred substrate for mitochondrial localized Parkin (36).
The current findings suggest why Mfn2 and Parkin are present in hearts that seemingly have little use for the canonical functions assigned to both proteins. In adult hearts cardiomyocyte mitochondria are static, distinct, rounded organelles. Lacking extensive interconnected mitochondrial networks, adult cardiomyocytes have little requirement for the mitochondrial remodeling that would be promoted by a redundant mitochondrial fusion protein. Indeed, in our hands Mfn1 (but not Mfn2) is completely dispensable to adult mouse hearts (12). Likewise, the PINK1-Parkin pathway clearly mediates autophagic elimination of damaged mitochondria in cultured mammalian cells (14), but evidence from germ-line Park2 knockout mice (9), adult cardiomyocyte-specific Parkin-deficient mice (21), and from interrupting Parkin signaling in adult mice with Mfn2 AA (current study) does not support an important role for Parkin-mediated mitophagy as a homeostatic mitochondrial quality control mechanism in hearts; normal surveillance and culling of individual damaged mitochondria seems to be accomplished through Parkin-independent mechanisms (35). Rather, Parkin-directed mitophagy is invoked as needed to accelerate generalized mitochondrial turnover during developmental transitions of myocardial metabolism (current study), after myocardial injury (9), and when mitochondrial homeostasis is disrupted (21).
Our findings demonstrate that mitochondria are not simply flexible fuel organelles readily switching between carbohydrate and fatty acid substrates. Rather, they have intrinsic metabolic and respiratory systems optimized for different substrate-specific metabolic pathways, and metabolic transitioning requires existing organelles to be mitophagically removed, enabling their replacement with mitochondria appropriate for a given biological context. Parkin and PINK1 gene mutations cause hereditary Parkinson’s disease, and Mfn2 mutations cause Charcot Marie Tooth syndrome type IIa. These are chronic neuromuscular disorders in which cardiac involvement is uncommon (37). It remains to be determined if PINK1-Parkin-Mfn2 mediated mitophagy regulates metabolic function in neurons or skeletal muscle as it does in perinatal hearts. Moreover, the discovery that PINK1-Mfn2-Parkin directed mitophagy is essential to cardiomyocyte metabolic remodeling only during the brief period following birth supports intensive genetic evaluation of the Parkin signaling pathway in neonatal, in addition to adult, cardiomyopathy.
Supplementary Material
Acknowledgments
We gratefully acknowledge the invaluable contributions of S J Gardell of the Sanford-Burnham-Prebys Medical Discovery Institute for LC/MS/MS studies, and M Levy of the Laboratory of Electron Microscopy Sciences, Department of Cell Biology, Washington University School of Medicine, S de la Fuenta, J Mishra, and D Weaver of the Thomas Jefferson University Mito Care Center for TEM analyses. Park2 floxed allele mice were obtained from and used under an MTA with Lexicon Pharmaceuticals. Both unprocessed and transcriptome-aligned RNA-sequencing reads have been deposited in the NCBI Gene Expression Omnibus with accession number GSE68921. Supported by NIH HL59888, HL108943, HL128071 (GWD), HL122124 (GC), HL058493 (DPK), and American Heart Association grants 14PRE18970093 (MS) and 14GRNT20410000 (SJM).
Footnotes
The authors declare no conflicts.
The data presented in this manuscript are tabulated in the supplementary materials.
References and Notes
- 1.Porter GA, Jr, et al. Bioenergetics, mitochondria, and cardiac myocyte differentiation. Prog. Pediatr. Cardiol. 2011;31:75–81. doi: 10.1016/j.ppedcard.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bartelds B, et al. Perinatal changes in myocardial metabolism in lambs. Circulation. 2000;102:926–931. doi: 10.1161/01.cir.102.8.926. [DOI] [PubMed] [Google Scholar]
- 3.Kim HD, et al. Human fetal heart development after mid-term: morphometry and ultrastructural study. J. Mol. Cell. Cardiol. 1992;24:949–965. doi: 10.1016/0022-2828(92)91862-y. [DOI] [PubMed] [Google Scholar]
- 4.Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 2005;85:1093–1129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- 5.Taegtmeyer H, Sen S, Vela D. Return to the fetal gene program: a suggested metabolic link to gene expression in the heart. Ann. N. Y. Acad. Sci. 2010;1188:191–198. doi: 10.1111/j.1749-6632.2009.05100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Song M, Dorn GW., 2nd Mitoconfusion: noncanonical functioning of dynamism factors in static mitochondria of the heart. Cell Metab. 2015;21:195–205. doi: 10.1016/j.cmet.2014.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shirihai OS, Song M, Dorn GW., 2nd How mitochondrial dynamism orchestrates mitophagy. Circ. Res. 2015;116:1835–1849. doi: 10.1161/CIRCRESAHA.116.306374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kubli DA, et al. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J. Biol. Chem. 2013;288:915–926. doi: 10.1074/jbc.M112.411363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee Y, Dawson VL, Dawson TM. Animal models of Parkinson's disease: vertebrate genetics. Cold Spring Harb. Perspect. Med. 2012;2:a009324. doi: 10.1101/cshperspect.a009324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shin JH, et al. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson's disease. Cell. 2011;144:689–702. doi: 10.1016/j.cell.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Y, Dorn GW., 2nd PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340:471–475. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee S, et al. Mitofusin 2 is necessary for striatal axonal projections of midbrain dopamine neurons. Hum. Mol. Genet. 2012;21:4827–4835. doi: 10.1093/hmg/dds352. [DOI] [PubMed] [Google Scholar]
- 14.Narendra DP, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scarffe LA, Stevens DA, Dawson VL, Dawson TM. Parkin and PINK1: much more than mitophagy. Trends Neurosci. 2014;37:315–324. doi: 10.1016/j.tins.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koshiba T, et al. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004;305:858–862. doi: 10.1126/science.1099793. [DOI] [PubMed] [Google Scholar]
- 17.Twig G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eschenbacher WH, et al. Two rare human mitofusin 2 mutations alter mitochondrial dynamics and induce retinal and cardiac pathology in Drosophila. PLoS One. 2012;7:e44296. doi: 10.1371/journal.pone.0044296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Misko A, Jiang S, Wegorzewska I, Milbrandt J, Baloh RH. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J. Neurosci. 2010;30:4232–4240. doi: 10.1523/JNEUROSCI.6248-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Syed F, et al. Physiological growth synergizes with pathological genes in experimental cardiomyopathy. Circ. Res. 2004;95:1200–1206. doi: 10.1161/01.RES.0000150366.08972.7f. [DOI] [PubMed] [Google Scholar]
- 21.Song M, et al. Interdependence of Parkin-Mediated Mitophagy and Mitochondrial Fission in Adult Mouse Hearts. Circ. Res. 2015;117:346–351. doi: 10.1161/CIRCRESAHA.117.306859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y, et al. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca(2+) crosstalk. Circ. Res. 2012;111:863–875. doi: 10.1161/CIRCRESAHA.112.266585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 24.Lopaschuk GD, Spafford MA, Marsh DR. Glycolysis is predominant source of myocardial ATP production immediately after birth. Am. J. Physiol. 1991;261:H1698–H1705. doi: 10.1152/ajpheart.1991.261.6.H1698. [DOI] [PubMed] [Google Scholar]
- 25.Razeghi P, et al. Metabolic gene expression in fetal and failing human heart. Circulation. 2001;104:2923–2931. doi: 10.1161/hc4901.100526. [DOI] [PubMed] [Google Scholar]
- 26.Lehman JJ, et al. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Invest. 2000;106:847–856. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dudkina NV, Kouril R, Peters K, Braun HP, Boekema EJ. Structure and function of mitochondrial supercomplexes. Biochim. Biophys. Acta. 2010;1797:664–670. doi: 10.1016/j.bbabio.2009.12.013. [DOI] [PubMed] [Google Scholar]
- 28.Lapuente-Brun E, et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science. 2013;340:1567–1570. doi: 10.1126/science.1230381. [DOI] [PubMed] [Google Scholar]
- 29.Chen Y, Liu Y, Dorn GW., 2nd Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ. Res. 2011;109:1327–1331. doi: 10.1161/CIRCRESAHA.111.258723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Christofk HR, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 31.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhandari P, Song M, Chen Y, Burelle Y, Dorn GW., 2nd Mitochondrial contagion induced by Parkin deficiency in Drosophila hearts and its containment by suppressing mitofusin. Circ. Res. 2014;114:257–265. doi: 10.1161/CIRCRESAHA.114.302734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams JA, et al. Chronic Deletion and Acute Knockdown of Parkin Have Differential Responses to Acetaminophen-induced Mitophagy and Liver Injury in Mice. J. Biol. Chem. 2015;290:10934–10946. doi: 10.1074/jbc.M114.602284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kageyama Y, et al. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J. 2014;33:2798–2813. doi: 10.15252/embj.201488658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song M, et al. Super-suppression of mitochondrial reactive oxygen species signaling impairs compensatory autophagy in primary mitophagic cardiomyopathy. Circ. Res. 2014;115:348–353. doi: 10.1161/CIRCRESAHA.115.304384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron. 2015;85:257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dorn GW., 2nd Mitochondrial dynamism and heart disease: changing shape and shaping change. EMBO Mol. Med. 2015;7:865–877. doi: 10.15252/emmm.201404575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim KY, et al. Parkin is a lipid-responsive regulator of fat uptake in mice and mutant human cells. J. Clin. Invest. 2011;121:3701–3712. doi: 10.1172/JCI44736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kitada T, et al. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc. Natl. Acad. Sci. U S A. 2007;104:11441–11446. doi: 10.1073/pnas.0702717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sohal DS, et al. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ. Res. 2001;89:20–25. doi: 10.1161/hh1301.092687. [DOI] [PubMed] [Google Scholar]
- 41.Porrello ER, et al. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331:1078–1080. doi: 10.1126/science.1200708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kabaeva Z, Zhao M, Michele DE. Blebbistatin extends culture life of adult mouse cardiac myocytes and allows efficient and stable transgene expression. Am. J. Physiol. Heart Circ. Physiol. 2008;294:H1667–H1674. doi: 10.1152/ajpheart.01144.2007. [DOI] [PubMed] [Google Scholar]
- 43.Matkovich SJ, Dorn GW., 2nd Deep sequencing of cardiac microRNA-mRNA interactomes in clinical and experimental cardiomyopathy. Methods Mol. Biol. 2015;1299:27–49. doi: 10.1007/978-1-4939-2572-8_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matkovich SJ, Hu Y, Dorn GW., 2nd Regulation of cardiac microRNAs by cardiac microRNAs. Circ. Res. 2013;113:62–71. doi: 10.1161/CIRCRESAHA.113.300975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trapnell C, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pagliarini DJ, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dorn GW, 2nd, Matkovich SJ. Menage a Trois: intimate relationship among a microRNA, long noncoding RNA, and mRNA. Circ. Res. 2014;114:1362–1365. doi: 10.1161/CIRCRESAHA.114.303786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matkovich SJ, Edwards JR, Grossenheider TC, de Guzman Strong C, Dorn GW., 2nd Epigenetic coordination of embryonic heart transcription by dynamically regulated long noncoding RNAs. Proc. Natl. Acad. Sci. U S A. 2014;111:12264–12269. doi: 10.1073/pnas.1410622111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kanehisa M, et al. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–D205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.