

Abstract
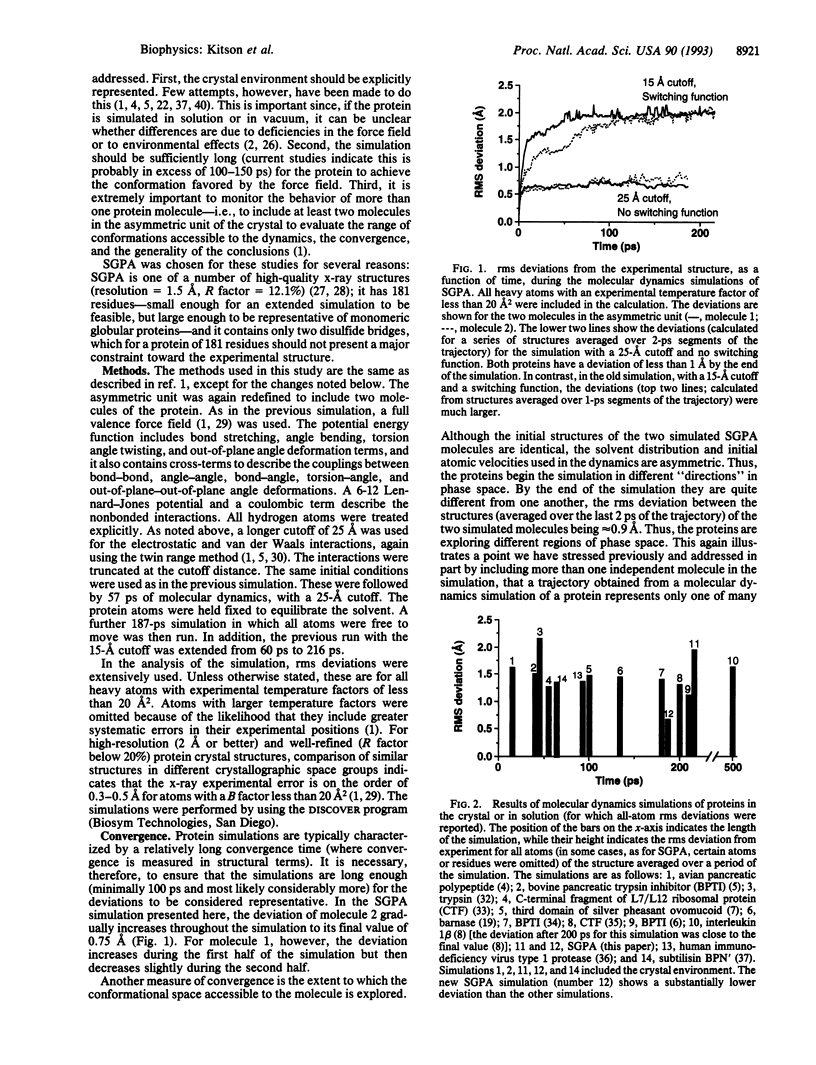
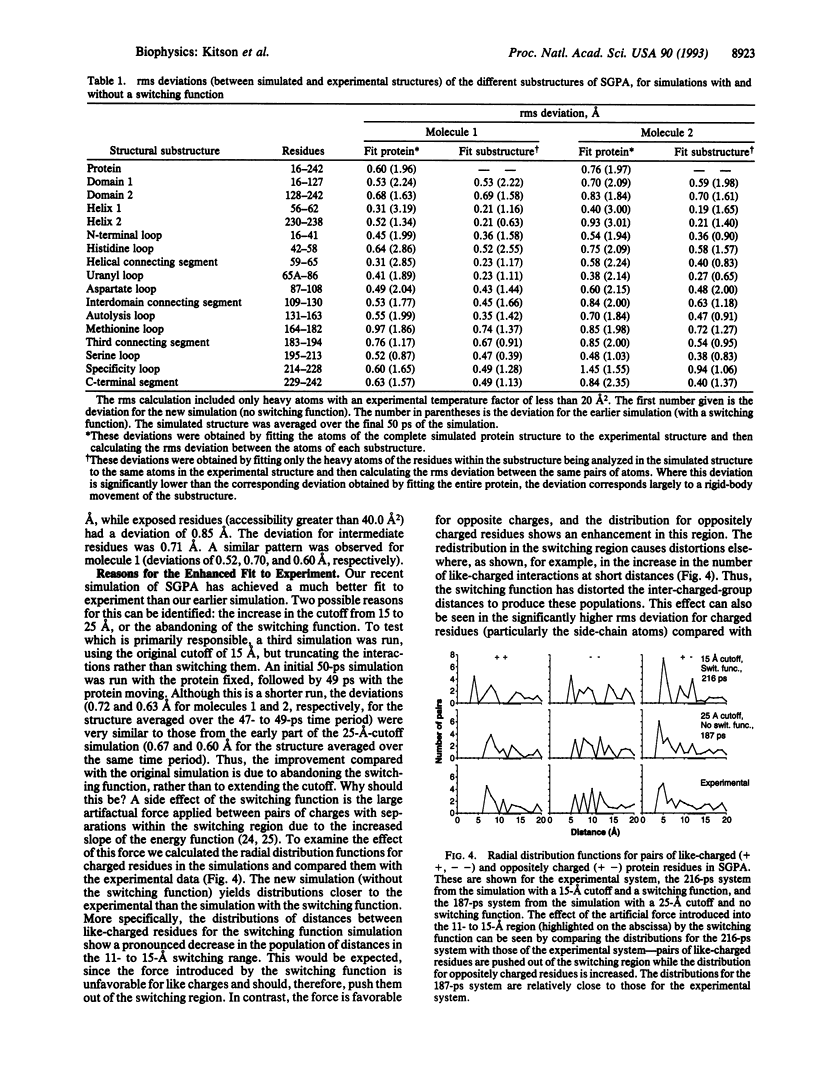
Computational methods are frequently used to simulate the properties of proteins. In these studies accuracy is clearly important, and the improvement of accuracy of protein simulation methodology is one of the major challenges in the application of theoretical methods, such as molecular dynamics, to structural studies of biological molecules. Much effort is being devoted to such improvements. Here, we present an analysis of a 187-ps molecular dynamics simulation of the serine protease Streptomyces griseus protease A in its crystal environment. The reproduction of the experimental structure is considerably better than has been achieved in earlier simulations--the root mean square deviation of the simulated structure from the x-ray structure being less than 1 A, a significant step toward the goal of simulating proteins to within experimental error. The use of a longer cutoff with truncation rather than a switching function, inclusion of all crystalline water and the counterions in the crystallization medium, and use of the consistent valence force field characterize the differences in this calculation.
Full text
PDF



Images in this article
Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Avbelj F., Moult J., Kitson D. H., James M. N., Hagler A. T. Molecular dynamics study of the structure and dynamics of a protein molecule in a crystalline ionic environment, Streptomyces griseus protease A. Biochemistry. 1990 Sep 18;29(37):8658–8676. doi: 10.1021/bi00489a023. [DOI] [PubMed] [Google Scholar]
- Berendsen H. J., Van Gunsteren W. F., Zwinderman H. R., Geurtsen R. G. Simulations of proteins in water. Ann N Y Acad Sci. 1986;482:269–286. doi: 10.1111/j.1749-6632.1986.tb20961.x. [DOI] [PubMed] [Google Scholar]
- Brünger A. T., Kuriyan J., Karplus M. Crystallographic R factor refinement by molecular dynamics. Science. 1987 Jan 23;235(4787):458–460. doi: 10.1126/science.235.4787.458. [DOI] [PubMed] [Google Scholar]
- Chandrasekhar I., Clore G. M., Szabo A., Gronenborn A. M., Brooks B. R. A 500 ps molecular dynamics simulation study of interleukin-1 beta in water. Correlation with nuclear magnetic resonance spectroscopy and crystallography. J Mol Biol. 1992 Jul 5;226(1):239–250. doi: 10.1016/0022-2836(92)90136-8. [DOI] [PubMed] [Google Scholar]
- Clore G. M., Gronenborn A. M., Brünger A. T., Karplus M. Solution conformation of a heptadecapeptide comprising the DNA binding helix F of the cyclic AMP receptor protein of Escherichia coli. Combined use of 1H nuclear magnetic resonance and restrained molecular dynamics. J Mol Biol. 1985 Nov 20;186(2):435–455. doi: 10.1016/0022-2836(85)90116-0. [DOI] [PubMed] [Google Scholar]
- Dauber-Osguthorpe P., Roberts V. A., Osguthorpe D. J., Wolff J., Genest M., Hagler A. T. Structure and energetics of ligand binding to proteins: Escherichia coli dihydrofolate reductase-trimethoprim, a drug-receptor system. Proteins. 1988;4(1):31–47. doi: 10.1002/prot.340040106. [DOI] [PubMed] [Google Scholar]
- Harte W. E., Jr, Swaminathan S., Beveridge D. L. Molecular dynamics of HIV-1 protease. Proteins. 1992 Jul;13(3):175–194. doi: 10.1002/prot.340130302. [DOI] [PubMed] [Google Scholar]
- Harte W. E., Jr, Swaminathan S., Mansuri M. M., Martin J. C., Rosenberg I. E., Beveridge D. L. Domain communication in the dynamical structure of human immunodeficiency virus 1 protease. Proc Natl Acad Sci U S A. 1990 Nov;87(22):8864–8868. doi: 10.1073/pnas.87.22.8864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiner A. P., Berendsen H. J., van Gunsteren W. F. MD simulation of subtilisin BPN' in a crystal environment. Proteins. 1992 Dec;14(4):451–464. doi: 10.1002/prot.340140406. [DOI] [PubMed] [Google Scholar]
- Joseph D., Petsko G. A., Karplus M. Anatomy of a conformational change: hinged "lid" motion of the triosephosphate isomerase loop. Science. 1990 Sep 21;249(4975):1425–1428. doi: 10.1126/science.2402636. [DOI] [PubMed] [Google Scholar]
- Kitson D. H., Hagler A. T. Catalysis of a rotational transition in a peptide by crystal forces. Biochemistry. 1988 Sep 20;27(19):7176–7180. doi: 10.1021/bi00419a002. [DOI] [PubMed] [Google Scholar]
- Kitson D. H., Hagler A. T. Theoretical studies of the structure and molecular dynamics of a peptide crystal. Biochemistry. 1988 Jul 12;27(14):5246–5257. doi: 10.1021/bi00414a045. [DOI] [PubMed] [Google Scholar]
- Krüger P., Strassburger W., Wollmer A., van Gunsteren W. F. A comparison of the structure and dynamics of avian pancreatic polypeptide hormone in solution and in the crystal. Eur Biophys J. 1985;13(2):77–88. doi: 10.1007/BF00256528. [DOI] [PubMed] [Google Scholar]
- Lee B., Richards F. M. The interpretation of protein structures: estimation of static accessibility. J Mol Biol. 1971 Feb 14;55(3):379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- Levitt M., Sharon R. Accurate simulation of protein dynamics in solution. Proc Natl Acad Sci U S A. 1988 Oct;85(20):7557–7561. doi: 10.1073/pnas.85.20.7557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loncharich R. J., Brooks B. R. The effects of truncating long-range forces on protein dynamics. Proteins. 1989;6(1):32–45. doi: 10.1002/prot.340060104. [DOI] [PubMed] [Google Scholar]
- McCammon J. A., Gelin B. R., Karplus M. Dynamics of folded proteins. Nature. 1977 Jun 16;267(5612):585–590. doi: 10.1038/267585a0. [DOI] [PubMed] [Google Scholar]
- Moult J., Sussman F., James M. N. Electron density calculations as an extension of protein structure refinement. Streptomyces griseus protease A at 1.5 A resolution. J Mol Biol. 1985 Apr 20;182(4):555–566. doi: 10.1016/0022-2836(85)90241-4. [DOI] [PubMed] [Google Scholar]
- Sielecki A. R., Hendrickson W. A., Broughton C. G., Delbaere L. T., Brayer G. D., James M. N. Protein structure refinement: Streptomyces griseus serine protease A at 1.8 A resolution. J Mol Biol. 1979 Nov 15;134(4):781–804. doi: 10.1016/0022-2836(79)90486-8. [DOI] [PubMed] [Google Scholar]
- Singh U. C., Benkovic S. J. A free-energy perturbation study of the binding of methotrexate to mutants of dihydrofolate reductase. Proc Natl Acad Sci U S A. 1988 Dec;85(24):9519–9523. doi: 10.1073/pnas.85.24.9519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struthers R. S., Tanaka G., Koerber S. C., Solmajer T., Baniak E. L., Gierasch L. M., Vale W., Rivier J., Hagler A. T. Design of biologically active, conformationally constrained GnRH antagonists. Proteins. 1990;8(4):295–304. doi: 10.1002/prot.340080403. [DOI] [PubMed] [Google Scholar]
- Warshel A., Sussman F., Hwang J. K. Evaluation of catalytic free energies in genetically modified proteins. J Mol Biol. 1988 May 5;201(1):139–159. doi: 10.1016/0022-2836(88)90445-7. [DOI] [PubMed] [Google Scholar]
- Weiner S. J., Seibel G. L., Kollman P. A. The nature of enzyme catalysis in trypsin. Proc Natl Acad Sci U S A. 1986 Feb;83(3):649–653. doi: 10.1073/pnas.83.3.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York D. M., Darden T. A., Pedersen L. G., Anderson M. W. Molecular dynamics simulation of HIV-1 protease in a crystalline environment and in solution. Biochemistry. 1993 Feb 16;32(6):1443–1453. doi: 10.1021/bi00057a007. [DOI] [PubMed] [Google Scholar]
- van Gunsteren W. F., Berendsen H. J., Hermans J., Hol W. G., Postma J. P. Computer simulation of the dynamics of hydrated protein crystals and its comparison with x-ray data. Proc Natl Acad Sci U S A. 1983 Jul;80(14):4315–4319. doi: 10.1073/pnas.80.14.4315. [DOI] [PMC free article] [PubMed] [Google Scholar]