

Abstract
Objective:
To evaluate serum cytokine profiles for their utility to determine the heterogeneous responses to interferon (IFN)–β treatment in patients with multiple sclerosis (MS).
Methods:
Patients with relapsing-remitting MS (RRMS) or clinically isolated syndrome receiving de novo IFN-β treatment were included in this prospective, observational study. Number of relapses and changes in disability were assessed 2 years prior to and 2 years after initiation of treatment. Sera were collected at baseline and after 3 months on therapy. Cytokine levels in sera were assessed by Luminex multiplex assays. Baseline cytokine profiles were grouped by hierarchical clustering analysis. Demographic features, changes in cytokines, and clinical outcome were then assessed in the clustered patient groups.
Results:
A total of 157 patients were included in the study and clustered into 6 distinct subsets by baseline cytokine profiles. These subsets differed significantly in their clinical and biological response to IFN-β therapy. Two subsets were associated with patients who responded poorly to therapy. Two other subsets, associated with a good response to therapy, showed a significant reduction in relapse rates and no worsening of disability. Each subset also had differential changes in cytokine levels after 3 months of IFN-β treatment.
Conclusions:
There is heterogeneity in the immunologic pathways of the RRMS population, which correlates with IFN-β response.
Relapsing-remitting multiple sclerosis (RRMS) is a heterogeneous disease that extends from clinical course to underlying pathology.1,2 There is a wide variation in treatment response to therapies including interferon (IFN)–β. Though neutralizing antibodies (NAb) against IFN-β are associated with treatment failure,3 they only explain a part of nonresponsiveness. Hence, the limited efficacy of IFN-β may be influenced by the heterogeneity of multiple sclerosis (MS).4
Many biomarker studies for IFN-β response assessed molecular differences in relation to a clinical outcome.5–11 A common bifurcated result defines either responders or nonresponders by metrics based on relapses or disability. While this methodology has identified some candidate biomarkers, these studies do not consider disease heterogeneity.
Here, we measured serum cytokines from patients with RRMS or clinically isolated syndrome (CIS) before the initiation of IFN-β therapy (baseline) and at 3 months while on therapy to assess their utility to predict treatment response. For analysis, we used 3 approaches. The first 2 approaches compare cytokines at baseline and 3 months in responders and nonresponders defined by relapses and Expanded Disability Status Scale (EDSS) score, respectively. The third approach uses the variability of baseline cytokine levels to cluster patients into subsets, and subsequently compares clinical outcomes between these subsets.
METHODS
Patients and specimens.
The prospective European multicenter study Neutralizing Antibodies on Interferon-beta in Multiple Sclerosis (NABINMS)12 enrolled patients with CIS and patients with RRMS, as described previously.13 We assessed cytokines from patients who had available data on relapses 2 years prior to and 2 years after initiation of IFN-β therapy, resulting in a total of 157 patients (table 1). Patients received 1 of the 3 available IFN-β preparations: intramuscular IFN-β-1a (Avonex), subcutaneous (SC) IFN-β-1a (Rebif), or SC IFN-β-1b (Betaferon). Clinical assessments (number of relapses and EDSS scoring14) were performed at baseline and 3, 12, and 24 months after initiation of therapy. Within the observation period, a relapse was defined as patient-reported symptoms (backed up by objective findings) or objectively observed signs typical of an acute inflammatory demyelinating event with a duration of at least 24 hours, in the absence of fever or infection.13 Number of relapses in the 2 years prior to study inclusion was assessed at baseline visit, i.e., taken from patients' charts together with a profound anamnesis.
Table 1.
Characteristics of the patient cohort

Serum and whole blood RNA samples were collected at baseline, immediately before the first IFN-β injection, and at 3 months (±2 weeks) within 4–12 hours postinjection.
Grouping of patients.
Grouping of patients was performed as follows: (1) According to relapse rate: Relapse nonresponders were defined by having ≥1 relapse between baseline and 24 months. (2) According to EDSS score: EDSS nonresponders were defined by showing an increase ≥1 EDSS point from baseline to 24 months. (3) Splitting patients into subsets by baseline cytokine profile was achieved by clustering analysis. Subsequently, relapse rate and change in EDSS score were compared between these subsets.
Serum cytokine, NAb, and myxovirus protein A (MxA) measurements.
Luminex assays (eBiosciences/Affymetrix, San Diego, CA) were performed at Stanford University according to the manufacturer's recommendations with modifications as described in the e-Methods at Neurology.org/nn. NAb to IFN-β and MxA mRNA expression were determined at Innsbruck Medical University as described previously.12,15
Statistical and cluster analysis.
For differences between responders and nonresponders cytokine levels were log2-transformed, and p values, odds ratios, and 95% confidence intervals were calculated using logistic regression.
For cluster analysis of baseline cytokines, the coefficient of variation (CV) of each cytokine in all serum samples was determined. Cytokines with CV values >100% were used to perform hierarchical clustering of baseline samples. Using Gene Cluster software,16 the cytokine values were normalized and centered to the mean, then ordered by complete linkage clustering and presented as a heat map using TreeView.16
Differences in individual cytokine concentrations in the resulting groups were assessed using Kruskal-Wallis with Dunn multiple comparison tests. Differences of cytokines between baseline and 3 months were analyzed using paired t tests. Comparisons of baseline demographic and clinical features within the clustered groups were performed using nonparametric analyses of variance with Dunn multiple comparisons or χ2 tests. Changes in EDSS and number of relapses between baseline and 24 months were assessed using paired t tests.
Approvals and patient consents.
The protocol was approved by the ethic committees (approval number AM2538 239/4.8) of all participating centers. Prior to any study-related investigations, written informed consent was obtained from all patients. All patient data were anonymized.
RESULTS
Comparison of cytokines at baseline and month 3 between responders and nonresponders defined by relapses.
Sixty-seven (42.7%) of 157 patients were relapse nonresponders (table 1). Baseline clinical and demographic features of responders and nonresponders showed no significant differences in disease type, age at onset, disease duration, sex, EDSS, or drug type. We observed that nonresponders had significantly greater relapse rates prior to treatment compared to responders (table e-1). Responders and nonresponders had similar percentages of patients previously on a disease-modifying therapy (DMT) and similar duration between last high-dose methylprednisolone (HDMP) use and initiation of IFN-β (tables e-1 and e-2).
We compared baseline serum cytokine levels between relapse responders and nonresponders. We observed that only interleukin (IL)–1β was significantly different between these 2 groups (table e-3, figure 1A).
Figure 1. Comparison of serum cytokines in responders and nonresponders defined by relapse rate or disability progression.
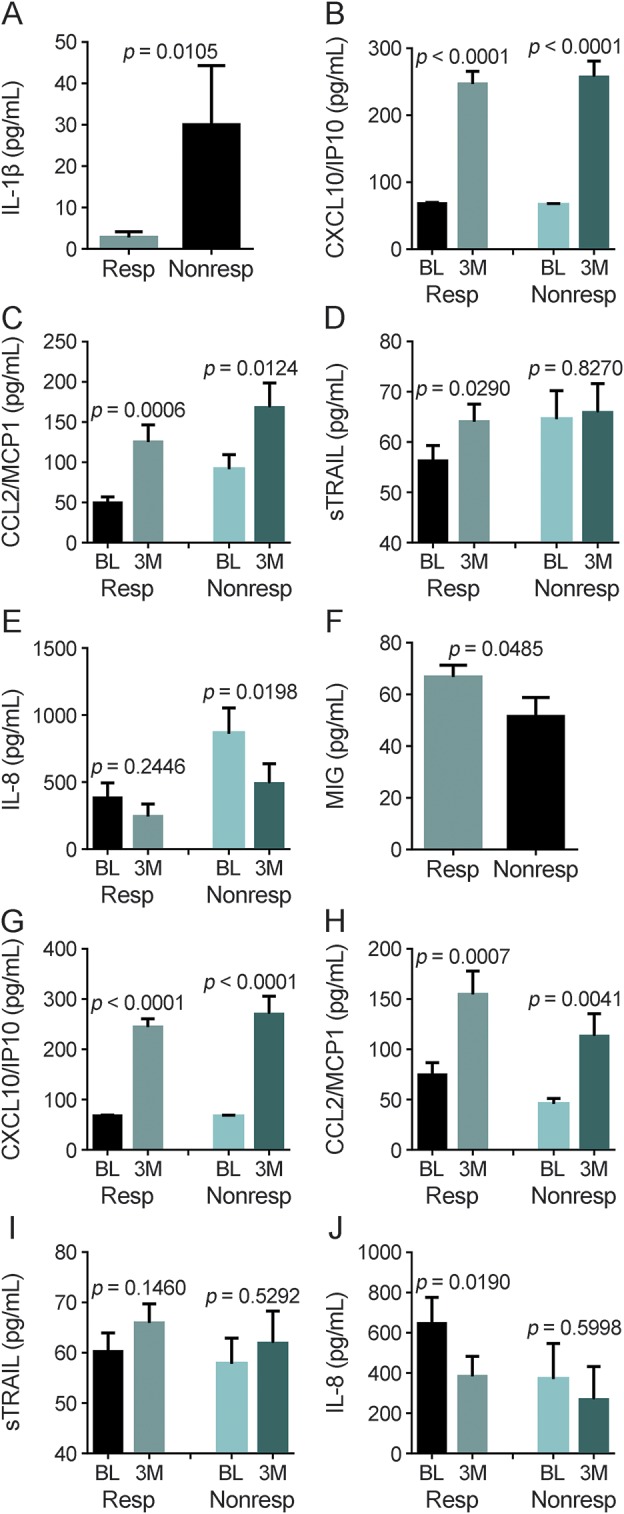
(A) Comparison of baseline serum concentrations of interleukin (IL)–1β in relapse responders (Resp) and nonresponders (Nonresp). p Values were determined using a Student t test. The data represent the mean ± SEM. Changes in serum concentrations of (B) CXCL10/IP10, (C) CCL2/monocyte chemoattractant protein-1 (MCP1), (D) soluble tumor necrosis factor-related apoptosis-inducing ligand (sTRAIL), and (E) IL-8 from baseline (BL) to month 3 (3M) in relapse responders and nonresponders. p Values were determined using a paired t test. The data represent the mean ± SEM. (F) Comparison of baseline serum concentrations of monokine induced by interferon-γ (MIG) in Expanded Disability Status Scale (EDSS) Resp and Nonresp. p Values were determined using a Student t test. The data represent the mean ± SEM. Changes in serum concentrations of (G) CXCL10/IP10, (H) CCL2/MCP1, (I) sTRAIL, and (J) IL-8 from BL to 3M in EDSS Resp and Nonresp. p Values were determined using a paired t test. The data represent the mean ± SEM.
We next compared changes in MxA mRNA (an established surrogate for IFN-β bioavailability17) and cytokine levels at 3 months vs baseline in relapse responders and nonresponders. At 3 months, both groups had significantly elevated levels of CXCL10/IP10, CCL2/monocyte chemoattractant protein-1 (MCP1), and MxA (figures 1, B and C, and e-1), demonstrating that IFN-β is biologically active in both responders and nonresponders.17–19 We also found increases in IL-1α, soluble vascular cell adhesion molecule (sVCAM)-1, and IFN-β in responders and nonresponders (table e-4). Soluble tumor necrosis factor-related apoptosis-inducing ligand (sTRAIL), granulocyte-colony stimulating factor, transforming growth factor β, intercellular adhesion molecule, IL-12p40, macrophage inflammatory protein (MIP) 1β, CD40L, IL-1RA, and MIP1α were significantly elevated by IFN-β in responders but not in nonresponders (figure 1D, table e-4). Finally, IL-8 was significantly decreased, while IFN-α and tumor necrosis factor β were significantly increased at 3 months in the nonresponders but not the responders (figure 1E, table e-4).
Frequency of NAb against IFN-β within 2 years did not differ between relapse responders and nonresponders (9 of 90 and 7 of 67, respectively). However, levels of CXCL10/IP10 at 3 months were significantly lower in patients who developed NAb later on (NAbpos 147.1 ± 29.3 pg/mL vs NAbneg 262.7 ± 16.1 pg/mL, p = 0.0082), thus confirming that NAbs inhibit the biological response to IFN-β.12
Comparison of cytokines at baseline and month 3 between responders and nonresponders defined by EDSS.
Thirty-nine (25.5%) of 153 patients were EDSS nonresponders (table 1). Baseline clinical and demographic features showed no significant differences in disease type, age at onset, disease duration, sex, number of previous relapses, or drug type. However, responders had significantly greater EDSS score at baseline and shorter duration between last HDMP use to initiation of IFN-β compared to nonresponders (table e-5). Responders and nonresponders had similar percentages of patients previously on DMT (tables e-5 and e-6).
We compared baseline serum cytokine levels in the EDSS responders and nonresponders. We found that only monokine induced by interferon-γ (MIG) was significantly higher in the responders compared to nonresponders (figure 1F, table e-7).
We next compared changes in cytokine and MxA transcription levels (3 months vs baseline) in responders and nonresponders. We found that at 3 months CXCL10/IP10, CCL2/MCP1, and MxA were significantly elevated in both groups (figures 1, G and H, and e-2); sTRAIL did not significantly change in the 2 groups (figure 1I); IL-8, plasminogen activator inhibitor–1, CXCL5/ENA78, and hepatocyte growth factor were all significantly decreased in the responders but not the nonresponders (figure 1J, table e-8); CD40L, IL-17A, and nerve growth factor were significantly increased in the responders but not the nonresponders; and sVCAM was significantly increased in the nonresponders but not the responders (table e-8).
Baseline cytokine profiles cluster patients into 6 distinct groups.
Many cytokines including IL-1β do not have a Gaussian distribution in our cohort (figure e-3), suggesting there is heterogeneity in patients. We hypothesized that selecting cytokines with the most variability in the population could be used for clinically meaningful stratification of patients. To select the most variable serum cytokines, CVs of each cytokine from all samples were calculated (table e-9). Cytokines with a CV >100% were used for clustering of the patients' baseline serum concentrations.
Clustering baseline cytokines split the patients into 6 groups (figure 2A). Group 3 (n = 20) had significantly higher concentrations of IL-8, CXCL1/Gro-α, IL-1β, IL-1RA, and CCL2/MCP1 compared to all other groups (figure 2, B–F). Group 6 (n = 8) had significantly higher levels of IFN-β and IL-17F compared to all other groups (figure 2, G–H). Cytokines in group 2 (n = 53) were all low compared to all other groups (figure 2A). Patients in group 1 (n = 23) also had low cytokine levels except for CD40L, which distinguished them from group 2 (figure 2, A and I). The differences that distinguished groups 4 from 5 were more subtle. Group 4 (n = 37) had higher levels of G-CSF and CD40L compared to group 5 (n = 16) and conversely group 5 had higher levels of CXCL10/IP10 and IL-6 compared to group 4 (figure e-4).
Figure 2. Baseline serum cytokine profiles of patients with multiple sclerosis segregated by cluster analysis.

(A) Heatmap depicts relative levels of cytokines and hierarchical clustering of individual patients. Baseline concentration of (B) interleukin (IL)–8, (C) CXCL1/Gro-α, (D) IL-1β, (E) IL-1RA, (F) CCL2/monocyte chemoattractant protein-1 (MCP1), (G) interferon (IFN)–β, (H) IL-17F, and (I) CD40L in the groups of patients. p Values were determined using Kruskal-Wallis test with Dunn multiple comparisons. *p < 0.05, **p < 0.01, ***p < 0.001.
We found no significant differences in most baseline clinical and demographic features of these 6 patient groups (table e-10). There was no statistical difference in in the timing between HDMP and the initiation of IFN-β within the groups. Also, we found that group 2 had statistically greater pretreatment disease duration and EDSS score compared to all other patients (figure e-5). This difference is of note because group 2 had low cytokine concentrations. There were no statistical differences in patients with prior DMT in each group (table e-11).
Differential clinical outcome to IFN-β treatment in the 6 clustered patient groups.
First, we compared the change in relapse rates 2 years prior to and 2 years post initiation of IFN-β therapy. Groups 1, 2, 4, and 5 all had significantly decreased relapse rates while on IFN-β therapy, whereas there were no significant changes in relapse rates in groups 3 and 6 (figure 3A). Furthermore, groups 1 and 5 had a low percentage of patients with relapses while on therapy compared to other groups; conversely, groups 3 and 6 had high percentages of patients who had relapses (table e-12).
Figure 3. Clinical response to interferon (IFN)–β treatment in the different patient groups defined by baseline cytokine profile.

(A) Comparison of the number of relapses 2 years prior to (pre) and 2 years after initiation (post) of IFN-β therapy. (B) Comparison of the Expanded Disability Status Scale (EDSS) score at baseline (BL) and 2 years after initiation of IFN-β therapy. Statistical significance was determined with a paired t test with p < 0.05 considered significant. The data represent the mean ± SEM.
We next assessed worsening in disability from baseline to 24 months. Groups 2 and 6 had significant increases in EDSS; groups 3 and 4 had trends suggesting increased EDSS; and groups 1 and 5 showed no worsening of EDSS after initiation of therapy (figure 3B). Taking both relapse rates and EDSS scores into account, these data suggest that groups 1 and 5 are populations that are likely to respond to IFN-β therapy, whereas groups 3 and 6 represent patients who are likely to be nonresponders. It is also of interest that the patients in group 2, who had low baseline cytokines and greater disease duration, had increased EDSS while having decreased relapse rates.
We found no statistical differences in the frequency of patients switching off IFN-β therapy or developing NAb against IFN-β between the different groups (table e-13).
Assessing baseline IL-7, IL-17F, and IL-17A in extreme MS phenotypes.
In a smaller study,20 we observed that IL-7 and IL-17F levels were inversely correlated, that patients with high IL-17F levels were nonresponders and that patients with elevated IL-7 levels were responders. Here, we found that groups 3 and 6 were 2 populations of nonresponders (figure 3). We observed that group 6 had significantly higher IL-17F levels, whereas group 3 had significantly higher IL-17A levels compared to the other groups (figures 2 and 4, A and B). Both IL-17A and IL-17F are signature TH17 cytokines,21,22 indicating that groups 3 and 6 have extreme clinical phenotypes of nonresponsiveness with a strong TH17 response. Groups 1 and 5 represent the other end of the clinical extreme because almost all patients in these 2 groups responded well to IFN-β. When comparing IL-7 levels between groups 1 and 5 to the rest of the patients with MS, we found that IL-7 was significantly higher in group 5 compared to all other patients (figure 4C). In our previous study,20 we selected patients with extreme clinical responses to IFN-β. It is likely that many patients selected for that study would cluster in groups 5 and 6. We also assessed soluble IL-7R at baseline in the clustered groups and found no statistical differences (data not shown). The new data from this larger cohort reinforce the hypothesis that strong TH17 response is a characteristic of nonresponders and that high IL-7 is a characteristic of a subset of responders.
Figure 4. Interleukin (IL)–17F, IL-17A, and IL-7 concentrations in responders and nonresponders returned by baseline cytokine profile.

(A) Comparison of pretreatment concentrations of IL-17F in patients in group 3, group 6 (nonresponders), and all other patients. (B) Comparison of pretreatment concentrations of IL-17A in patients in group 3, group 6 (nonresponders), and all other patients. (C) Comparison of pretreatment concentrations of IL-7 in patients in group 1, group 5 (responders), and all other patients. p Values were determined using a Kruskal-Wallis test with Dunn multiple comparisons. The data represent the mean ± SEM.
Differential biological effects of IFN-β treatment in the 6 clustered patient groups.
MxA and CXCL10/IP10 were significantly elevated in all groups at 3 months, demonstrating IFN-β bioavailability (figures 5A and e-6). However, we found many differential changes in other cytokines (table e-14). Groups 2, 4, and 5 had significant increases in CCL2/MCP1, whereas groups 3 and 6 had no significant increase in this cytokine (figure 5B). sTRAIL, which has been previously associated with good response,23 was also differentially affected by IFN-β treatment. sTRAIL was significantly increased in groups 1 and 2, and, conversely, significantly reduced in group 3 (figure 5C). There were also differential changes in IL-8, CD40L, and IL-1α within the groups (figure 5, D–F).
Figure 5. Change of serum cytokines during early interferon (IFN)–β treatment in patients with multiple sclerosis segregated by cluster analysis.

Changes in serum concentrations of (A) CXCL10/IP10, (B) CCL2/monocyte chemoattractant protein-1 (MCP1), (C) soluble tumor necrosis factor-related apoptosis-inducing ligand (sTRAIL), (D) interleukin (IL)–8, (E) CD40L, and (F) IL-1α from baseline (BL) to month 3 (3M) in the patient groups defined by baseline cytokine profiles. p Values were determined using a paired t test. The data represent the mean ± SEM.
DISCUSSION
Traditionally, biomarker studies assess molecular differences in patient groups with clinically defined outcomes. There are confounders that arise by using this approach for biomarker studies. One confounder is how a study defines treatment response. We found that defining response by relapse rates or by EDSS progression results in different sets of biomarkers that predict treatment response. Another confounder is that MS is heterogeneous and many biomarkers are highly variable in the MS population. Here we utilized the variability in serum cytokines to stratify patients into subsets and learned that these subsets have distinct outcomes to therapy.
Our first observation was that patients with low cytokines (group 2) had longer disease duration and greater EDSS before starting therapy compared to the other patients (roughly 7 vs 4 years). This patient group had significant reduction in relapses while having increasing EDSS scores while on therapy. This observation suggests that these patients, who are in a later disease stage, have fewer inflammatory events and are closer to the progressive disease phase.24 The other groups were in an early disease phase, where relapses may be an informative measure.
Our second observation was that there are 2 distinct baseline cytokine profiles that correlate with nonresponsiveness defined by a lack of reduction in relapses. One subset of nonresponders (group 3) had high levels of IL1β, IL-8, CXCL1/Gro-α, CCL2/MCP1, and IL-17A. This confirms a recent report that increased baseline levels of IL-1β is observed in IFN-β nonresponders.7 IL-1β is required for differentiation of human TH1725,26 cells and, furthermore, IL-8, CXCL1/Gro-α, and CCL2/MCP1 are chemokines induced by IL-17 signaling, indicating that the TH17 pathway is elevated in these patients.27,28 The second subset of nonresponders (group 6) had elevated IL-17F, which is similar to a subset of nonresponders described in a smaller cohort of patients.20,29 Other authors reported that IL-17F levels were not statistically different in responders and nonresponders5,6; however, they did not consider heterogeneity of MS for their analyses. These studies predefined treatment response by combining the 2 clinical measures relapse rate and EDSS score and simply compared cytokine concentrations. This might be the reason why the influence of IL-17F on treatment response was not seen.6 There is a striking similarity between these 2 subsets of nonresponders; both have characteristics of a TH17 response. One subset of nonresponders has high levels of IL-17A and chemokines induced during a TH17 response. The other subset of nonresponders has high IL-17F, a signature cytokine of TH17 cells.21,22 It is unclear if these 2 subsets of nonresponders represent different TH17 pathways, or indicate different stages of a TH17 response or whether the TH17 cells are transdifferentiating into either TH1 or Tregs.30,31 It has been shown that TH17 cytokines and granulocyte chemokines are elevated in patients with neuromyelitis optica (NMO), a neuroinflammatory disease not responding to IFN-β therapy,32–35 suggesting that the disease processes in these subsets of IFN-β nonresponding patients with MS are similar to NMO and in future studies perhaps such patients should be tested for NMO immunoglobulin.
Our third observation was that there are 2 distinct profiles that distinguish IFN-β responders: patients with a reduction in relapse rates and no worsening in EDSS. One subset of responders, group 1, had low concentrations of all cytokines except CD40L. We speculate that this subset has a milder inflammatory response that is effectively treated with IFN-β. The other subset of responders (group 5) had high levels of IL-7 compared to the other MS subsets. The protective effects of IL-7 during IFN-β treatment are not entirely clear. However, we and others have found that IL-7 promotes TH1 differentiation and IFN-γ expression.20,36 These data suggest the hypothesis that IFN-β has anti-inflammatory effects during an IL-7-driven TH1 response and conversely has inflammatory effects during a TH17 response.
Finally, each patient group upregulated MxA and CXCL10/IP10 at 3 months after start of IFN-β therapy, demonstrating its bioavailability.17,18 This shows that all patient subsets responded biologically to IFN-β but that the differential characteristics of the subsets influence therapeutic efficacy.
There are potential limitations of this study. Approximately 10% of patients were previously on other therapies. This is only a small proportion and the number of pretreated patients did not differ between groups. Therefore, it is unlikely that this affects our findings. This similarly applies to the low proportion of patients with CIS, which also did not differ between the groups. Furthermore, a certain imbalance of baseline clinical data between groups, e.g., higher prior relapse rate in relapse nonresponders, is likely not relevant as we used the change of cytokine profile between baseline and 3 months while on therapy for evaluation of treatment response. As group 2 showed overall low cytokine levels, possibly due to the longer disease duration, one might speculate that a certain differential regulation of these cytokines is obscured. Finally, some samples were collected over 5 years prior to analysis. Therefore, there is potential for protein degradation. However, we did not see differences in cytokine profiles related to the long-term storage.
The heterogeneous nature of RRMS was notably described previously where subgroups of patients with RRMS were identified with differences in immunopathologic patterns in lesions.1,2 Our data from serum cytokines provide more evidence for immunologically distinct subgroups of MS and suggest these subgroups can stratify treatment response. Such stratification provides evidence that biomarkers can be informative for prognosis and treatment response. Further studies that include additional longitudinal samples as well as MRI data are warranted.
Supplementary Material
GLOSSARY
- CIS
clinically isolated syndrome
- CV
coefficient of variation
- DMT
disease-modifying therapy
- EDSS
Expanded Disability Status Scale
- HDMP
high-dose methylprednisolone
- IFN
interferon
- IL
interleukin
- MCP1
monocyte chemoattractant protein-1
- MIP
macrophage inflammatory protein
- MS
multiple sclerosis
- MxA
myxovirus protein A
- NAb
neutralizing antibodies
- NMO
neuromyelitis optica
- RRMS
relapsing-remitting multiple sclerosis
- SC
subcutaneous
- sTRAIL
soluble tumor necrosis factor-related apoptosis-inducing ligand
- sVCAM
soluble vascular cell adhesion molecule
Footnotes
Supplemental data at Neurology.org/nn
AUTHOR CONTRIBUTIONS
H.H.: study design, analysis and interpretation of the data, drafting the manuscript. I.A.: analysis of the data and drafting the manuscript. C.J.L.: analysis of the data and drafting the manuscript. A.M.: revising the manuscript for intellectual content. A.B.: study design, revising the manuscript for intellectual content. M.C.: study design, revising the manuscript for intellectual content. G.G.: study design, revising the manuscript for intellectual content. M.G.: revising the manuscript for intellectual content. M.H.: revising the manuscript for intellectual content. M.K.: revising the manuscript for intellectual content. F.F.: revising the manuscript for intellectual content. J.K.: study design, revising the manuscript for intellectual content. R.L.P.L.: study design, revising the manuscript for intellectual content. S.M.: revising the manuscript for intellectual content. M.M.: revising the manuscript for intellectual content. X.M.: revising the manuscript for intellectual content. D.R.: revising the manuscript for intellectual content. F. Schautzer: revising the manuscript for intellectual content. F. Sellebjerg: revising the manuscript for intellectual content. P.S.S.: study design, revising the manuscript for intellectual content. F.D.: study design, revising the manuscript for intellectual content. L.S.: study design, interpretation of data, and drafting the manuscript. R.C.A.: study design, analysis and interpretation of data, drafting the manuscript.
STUDY FUNDING
Supported by a grant from Pfizer to L.S., NIH/NIGMS P30GM110766 to I.A., European Union Grant LSHB-CT-2005-018926 to F.D., and NIH/NINDS R00 NS075099-04 to R.C.A.
DISCLOSURE
H. Hegen has participated in meetings sponsored by, received speaker honoraria or travel funding from Bayer Schering, Biogen, Merck Serono and Novartis, and received honoraria for acting as consultant for Teva Pharmaceuticals Europe. I. Adrianto received research support from NIH/NIGMS. C. Lessard and A. Millonig report no disclosures. Antonio Bertolotto served on the scientific advisory boards of Almirall, Bayer, BiogenIdec, and Genzyme; received speaker honoraria from BiogenIdec, Genzyme, Novartis, and Teva; his institution has received grant support from Bayer, BiogenIdec, Merck, Novartis, Teva, the Italian Multiple Sclerosis Society, Fondazione Ricerca Biomedica ONLUS, and San Luigi ONLUS; received speaker honoraria from BiogenIdec, Genzyme, Novartis, Sanofi-Aventis, and Teva; is on the editorial board of Multiple Sclerosis International, Progress in Neuroscience, Dataset Papers in Neuroscience, Journal of Multiple Sclerosis, Neurology and Therapy, and Multiple Sclerosis and Demyelinating Disorders; and received research support from Regione Piemonte, Italian Multiple Sclerosis Society, Associazione Ricerca Biomedica ONLUS, and San Luigi ONLUS. M. Comabella received speaking honoraria from Bayer Schering Pharma, Merck Serono, Biogen-Idec, Teva Pharmaceuticals, Sanofi-Aventis, Genzyme, and Novartis. G. Giovannoni serves on the scientific advisory board for Biogen-Idec, Fiveprime, Genzyme, GW Pharma, Ironwood, Merck-Serono, Novartis, Roche, Sanofi-Aventis, Synthon BV, Teva, Vertex Pharmaceutical, Abbvie, and Canbex; received speaker honoraria from Biogen-Idec, Genzyme, GW Pharma, Merck-Serono, Novartis, Roche, and Teva; is an editor for Multiple Sclerosis and Related Disorders; has consulted for Biogen-Idec, Fiveprime, Genzyme, GW Pharma, Ironwood, Merck-Serono, Novartis, Roche, Sanofi-Aventis, Synthon BV, Teva, Vertex Pharmaceuticals, Abbvie, and Canbex; is on the speaker's bureau for Novartis and Teva; and received research support from Genzyme and Merck. M. Guger is on the scientific advisory board for Bayer, Biogen, Genzyme, Merck, Novartis, and Teva Ratiopharm; received travel funding and/or speaker honoraria from Bayer, Biogen, Genzyme, Merck, Novartis, Teva Ratiopharm, and Almirall; and received research support from Biogen. M. Hoelzl reports no disclosures. M. Khalil received funding for travel and speaker honoraria from Bayer Schering Pharma, Novartis, Genzyme, Merck Serono, Biogen Idec, and Teva Pharmaceutical Industries. F. Fazekas served on the scientific advisory board for Bayer-Schering, Biogen-Idec, Advisor, Bristol-Myers Squibb, Advisor, Genzyme, Advisor, 2012–2015 Informatics, Scientific Advisor, Pfizer, Teva-Aventis, Advisor, Teva Pharmaceuticals, D-Pharm, data safety monitoring board for Actelion, and data safety monitoring board for University of Zurich (Nano-CL IV); received travel funding and/or speaker honoraria from Almirall, Bristol-Myers-Squibb, Merck-Serono, Novartis, Sanofi-Aventis, Shire, and Teva Pharmaceuticals; is on the editorial board of Cerebrovascular Diseases, Journal of Neurology, Polish Journal of Neurology and Neurosurgery, Stroke, and Multiple Sclerosis; is on an international advisory board for Swiss Archives of Neurology and Psychiatry; is on the speakers' bureau for Merck-Serono; and there is an unrestricted research grant to his institution by Novartis starting 2014. S.M. Malucchi reports no disclosures. M. Mehling has consulted for Action and Novartis and received research support from the University of Basel. J. Killestein has consulted for Novartis, Merck-Serono, Biogen Idec, Genzyme, and Teva; received speaker honoraria from Biogen, Novartis, Teva, and Merck-Serono; and has been involved in clinical trials of companies that market drugs for MS (Schering AG, Biogen Idec, Merck-Serono, Teva, Genzyme, and Novartis). R.LP. Lindberg received research support from Swiss Multiple Sclerosis Society, Swiss National Science Foundation, and Roche Postdoctoral Fellowship Program. X. Montalban served on the scientific advisory board for Actelion, Almirall, Bayer, Biogen, Genzyme, Merck, Novartis, Receptos, F. Hoffmann-La Roche, Sanofi-Genzyme, and Teva Pharmaceutical; received travel funding from Novartis, Teva Pharmaceutical, Merck, Biogen, Bayer, Almirall, Actelion, Genzyme, Receptos, F. Hoffmann-La Roche, and Sanofi; is on the editorial board of Multiple Sclerosis, Journal of Neurology, The International MS Journal, Revista Neurología, and Therapeutic Advances in Neurological Disorders; has consulted for Bayer, Biogen, Genzyme, Merck, Novartis, F. Hoffmann-La Roche, Sanofi-Genzyme, and Teva Pharmaceuticals; and received research support from Multiple Sclerosis Foundation of Barcelona (FEM). D. Rudzki and F. Schautzer report no disclosures. F. Sellebjerg served on the scientific advisory board for Biogen-Idec, Genzyme, Merck Serono, Sanofi-Aventis, Teva, and Novo Nordisk; received travel funding and/or speaker honoraria from Bayer Schering, Biogen Idec, Genzyme, Merck Serono, Novartis, Sanofi-Aventis, Schering-Plough, and Teva; has consulted for Biogen Idec; and received research support from Biogen Idec, Sanofi-Aventis, Novartis Danish Strategic Research Council, Danish Multiple Sclerosis Society, and Lounkær Foundation. P.S. Sorensen served on the scientific advisory board for Biogen Idec, Merck Serono, Novartis, Genmab, Teva Pharmaceutical Industries, and GlaxoSmithKline; has been on steering committees or independent data monitoring boards in clinical trials sponsored by Merck Serono, Genmab, Teva Pharmaceutical Industries, GlaxoSmithKline, and Bayer Schering; received travel funding and/or speaker honoraria from Biogen Idec, Merck Serono, Teva Pharmaceutical Industries, Bayer Schering, sanofi-aventis, Genzyme, and Novartis; was a previous Editor-in-Chief of European Journal of Neurology; is on the editorial board for European Journal of Neurology, Therapeutic Advances in Neurological Disorders, and Multiple Sclerosis Journal; and received research support from Danish Multiple Sclerosis Society, Danish Medical Research Council, and European Union Sixth Framework Programme: Life Sciences, Genomics and Biotechnology for Health. F. Deisenhammer served on the scientific advisory board for Biogen-Idec, Novartis Pharma, Genzyme, Merck, and Teva; received travel funding and speaker honoraria from Bayer Healthcare, Biogen Idec, Novartis Pharma, and Genzyme; is a section editor for Multiple Sclerosis and Related Disorders; and received research support from Biogen-Idec and Innovative Medicine Initiatives. L. Steinman is on the scientific advisory board for Novartis, Receptos, Atreca, Tolerion, and Teva; received travel funding and/or speaker honoraria from Biogen, Bayhill, Bayer, Celgene, and Receptos; is on the editorial board for MS Journal and Proceedings of the National Academy of Sciences; has a patent pending on cytokines and type 1 interferons; is on the speaker's bureau for EMD Serono; and received research support from JT Pharma, Pfizer, Biogen, and NIH. R.C. Axtell holds a patent for Biomarkers for prognosis of Multiple Sclerosis, has consulted for Biogen Idec, is on the speaker's bureau for EMD Serono, and received research support from NIH/NINDS. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Han MH, Hwang SI, Roy DB, et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature 2008;451:1076–1081. [DOI] [PubMed] [Google Scholar]
- 2.Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol 2000;47:707–717. [DOI] [PubMed] [Google Scholar]
- 3.Polman CH, Bertolotto A, Deisenhammer F, et al. Recommendations for clinical use of data on neutralising antibodies to interferon-beta therapy in multiple sclerosis. Lancet Neurol 2010;9:740–750. [DOI] [PubMed] [Google Scholar]
- 4.Rudick RA, Lee JC, Simon J, Ransohoff RM, Fisher E. Defining interferon beta response status in multiple sclerosis patients. Ann Neurol 2004;56:548–555. [DOI] [PubMed] [Google Scholar]
- 5.Hartung HP, Steinman L, Goodin DS, et al. Interleukin 17F level and interferon beta response in patients with multiple sclerosis. JAMA Neurol 2013;70:1017–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bushnell SE, Zhao Z, Stebbins CC, et al. Serum IL-17F does not predict poor response to IM IFNbeta-1a in relapsing-remitting MS. Neurology 2012;79:531–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malhotra S, Rio J, Urcelay E, et al. NLRP3 inflammasome is associated with the response to IFN-beta in patients with multiple sclerosis. Brain 2015;138:644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bustamante MF, Nurtdinov RN, Rio J, Montalban X, Comabella M. Baseline gene expression signatures in monocytes from multiple sclerosis patients treated with interferon-beta. PLoS One 2013;8:e60994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rudick RA, Rani MR, Xu Y, et al. Excessive biologic response to IFNbeta is associated with poor treatment response in patients with multiple sclerosis. PLoS One 2011;6:e19262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Comabella M, Lunemann JD, Rio J, et al. A type I interferon signature in monocytes is associated with poor response to interferon-beta in multiple sclerosis. Brain 2009;132:3353–3365. [DOI] [PubMed] [Google Scholar]
- 11.Byun E, Caillier SJ, Montalban X, et al. Genome-wide pharmacogenomic analysis of the response to interferon beta therapy in multiple sclerosis. Arch Neurol 2008;65:337–344. [DOI] [PubMed] [Google Scholar]
- 12.Hegen H, Millonig A, Bertolotto A, et al. Early detection of neutralizing antibodies to interferon-beta in multiple sclerosis patients: binding antibodies predict neutralizing antibody development. Mult Scler 2014;20:577–587. [DOI] [PubMed] [Google Scholar]
- 13.Polman CH, Reingold SC, Edan G, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria”. Ann Neurol 2005;58:840–846. [DOI] [PubMed] [Google Scholar]
- 14.Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology 1983;33:1444–1452. [DOI] [PubMed] [Google Scholar]
- 15.Millonig A, Rudzki D, Holzl M, et al. High-dose intravenous interferon beta in patients with neutralizing antibodies (HINAbs): a pilot study. Mult Scler 2009;15:977–983. [DOI] [PubMed] [Google Scholar]
- 16.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA 1998;95:14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.von Wussow P, Jakschies D, Hochkeppel HK, Fibich C, Penner L, Deicher H. The human intracellular Mx-homologous protein is specifically induced by type I interferons. Eur J Immunol 1990;20:2015–2019. [DOI] [PubMed] [Google Scholar]
- 18.Buttmann M, Merzyn C, Rieckmann P. Interferon-beta induces transient systemic IP-10/CXCL10 chemokine release in patients with multiple sclerosis. J Neuroimmunol 2004;156:195–203. [DOI] [PubMed] [Google Scholar]
- 19.Conrady CD, Zheng M, Mandal NA, van Rooijen N, Carr DJ. IFN-alpha-driven CCL2 production recruits inflammatory monocytes to infection site in mice. Mucosal Immunol 2013;6:45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee LF, Axtell R, Tu GH, et al. IL-7 promotes T(H)1 development and serum IL-7 predicts clinical response to interferon-beta in multiple sclerosis. Sci Transl Med 2011;3:93ra68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haak S, Croxford AL, Kreymborg K, et al. IL-17A and IL-17F do not contribute vitally to autoimmune neuro-inflammation in mice. J Clin Invest 2009;119:61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seiderer J, Elben I, Diegelmann J, et al. Role of the novel Th17 cytokine IL-17F in inflammatory bowel disease (IBD): upregulated colonic IL-17F expression in active Crohn's disease and analysis of the IL17F p.His161Arg polymorphism in IBD. Inflamm Bowel Dis 2008;14:437–445. [DOI] [PubMed] [Google Scholar]
- 23.Wandinger KP, Lunemann JD, Wengert O, et al. TNF-related apoptosis inducing ligand (TRAIL) as a potential response marker for interferon-beta treatment in multiple sclerosis. Lancet 2003;361:2036–2043. [DOI] [PubMed] [Google Scholar]
- 24.Sand IK, Krieger S, Farrell C, Miller AE. Diagnostic uncertainty during the transition to secondary progressive multiple sclerosis. Mult Scler 2014;20:1654–1657. [DOI] [PubMed] [Google Scholar]
- 25.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol 2007;8:942–949. [DOI] [PubMed] [Google Scholar]
- 26.Wilson NJ, Boniface K, Chan JR, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol 2007;8:950–957. [DOI] [PubMed] [Google Scholar]
- 27.Watanabe H, Kawaguchi M, Fujishima S, et al. Functional characterization of IL-17F as a selective neutrophil attractant in psoriasis. J Invest Dermatol 2009;129:650–656. [DOI] [PubMed] [Google Scholar]
- 28.Shahrara S, Pickens SR, Mandelin AM, II, et al. IL-17-mediated monocyte migration occurs partially through CC chemokine ligand 2/monocyte chemoattractant protein-1 induction. J Immunol 2010;184:4479–4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Axtell RC, de Jong BA, Boniface K, et al. T helper type 1 and 17 cells determine efficacy of interferon-beta in multiple sclerosis and experimental encephalomyelitis. Nat Med 2010;16:406–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gagliani N, Vesely MC, Iseppon A, et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature 2015;523:221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harbour SN, Maynard CL, Zindl CL, Schoeb TR, Weaver CT. Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proc Natl Acad Sci USA 2015;112:7061–7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herges K, de Jong BA, Kolkowitz I, et al. Protective effect of an elastase inhibitor in a neuromyelitis optica-like disease driven by a peptide of myelin oligodendroglial glycoprotein. Mult Scler 2012;18:398–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsushita T, Tateishi T, Isobe N, et al. Characteristic cerebrospinal fluid cytokine/chemokine profiles in neuromyelitis optica, relapsing remitting or primary progressive multiple sclerosis. PLoS One 2013;8:e61835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Palace J, Leite MI, Nairne A, Vincent A. Interferon beta treatment in neuromyelitis optica: increase in relapses and aquaporin 4 antibody titers. Arch Neurol 2010;67:1016–1017. [DOI] [PubMed] [Google Scholar]
- 35.Uzawa A, Mori M, Hayakawa S, Masuda S, Kuwabara S. Different responses to interferon beta-1b treatment in patients with neuromyelitis optica and multiple sclerosis. Eur J Neurol 2010;17:672–676. [DOI] [PubMed] [Google Scholar]
- 36.Davis CC, Marti LC, Sempowski GD, Jeyaraj DA, Szabolcs P. Interleukin-7 permits Th1/Tc1 maturation and promotes ex vivo expansion of cord blood T cells: a critical step toward adoptive immunotherapy after cord blood transplantation. Cancer Res 2010;70:5249–5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.