

Abstract
Objective:
To assess in an observational study whether serum peptide antibody reactivities may distinguish aquaporin-4 (AQP4) antibody (Ab)–positive and -negative neuromyelitis optica spectrum disorders (NMOSD) and relapsing-remitting multiple sclerosis (RRMS).
Methods:
We screened 8,700 peptides that included human and viral antigens of potential relevance for inflammatory demyelinating diseases and random peptides with pooled sera from different patient groups and healthy controls to set up a customized microarray with 700 peptides. With this microarray, we tested sera from 66 patients with AQP4-Ab-positive (n = 16) and AQP4-Ab-negative (n = 19) NMOSD, RRMS (n = 11), and healthy controls (n = 20).
Results:
Differential peptide reactivities distinguished NMOSD subgroups from RRMS in 80% of patients. However, the 2 NMOSD subgroups were not well-discriminated, although those patients are clearly separated by their antibody reactivities against AQP4 in cell-based assays. Elevated reactivities to myelin and Epstein-Barr virus peptides were present in RRMS and to AQP4 and AQP1 peptides in AQP4-Ab-positive NMOSD.
Conclusions:
While AQP4-Ab-positive and -negative NMOSD subgroups are not well-discriminated by peptide antibody reactivities, our findings suggest that peptide antibody reactivities may have the potential to distinguish between both NMOSD subgroups and MS. Future studies should thus concentrate on evaluating peptide antibody reactivities for the differentiation of AQP4-Ab-negative NMOSD and MS.
Neuromyelitis optica (NMO) is a rare, often severe, autoimmune CNS disease that predominantly targets the optic nerves and the spinal cord. The discovery of a specific serum biomarker, an autoantibody directed against the astrocytic water channel aquaporin-4 (AQP4-Ab),1 strongly suggests that NMO is distinct from classic multiple sclerosis (MS), and has also led to the recognition of an expanded spectrum of clinical manifestations of NMO, termed NMO spectrum disorders (NMOSD).2,3
About 20%–40% of patients with clinical signs of NMOSD do not have AQP4-Abs and the underlying pathogenesis in these patients is unclear.4 The recent detection of anti–myelin oligodendrocyte glycoprotein (MOG) antibodies in some of the AQP4-Ab-seronegative patients with clinical signs of NMOSD suggests that autoantigens other than AQP4 may play a role in this condition.5
The most important differential diagnosis of NMOSD remains MS. Indeed, initially misdiagnosing NMOSD as MS occurs frequently (40%).6 A correct distinction between NMOSD and MS is crucial as the prognosis in NMOSD is usually worse, and some MS therapies such as interferon-β, natalizumab, or fingolimod are probably ineffective or harmful in NMOSD.7
Therefore, the identification of biomarkers to differentiate between NMOSD and MS is highly desirable for improved diagnosis and appropriate treatment. Peptide microarrays were previously used to distinguish between different disease courses and neuropathologic subtypes of MS, as well as acute disseminated encephalomyelitis and relapsing-remitting MS (RRMS).8,9 The aim of this study was to clarify whether serum peptide antibody reactivities may distinguish AQP4-Ab-positive and -negative NMOSD and RRMS.
METHODS
We developed a customized peptide microarray containing more than 700 peptides that represent human and viral antigens potentially relevant for inflammatory demyelinating CNS diseases as well as random peptides. With this selection microarray, we tested sera from 66 patients with AQP4-Ab-positive and AQP4-Ab-negative NMOSD, RRMS, and healthy controls.
Standard protocol approvals, registrations, and patient consent.
The study was approved by the institutional review boards of the Charité Universitätsmedizin Berlin (EA1/182/10 and EA1/131/09), the University of Düsseldorf (3419), the University of Heidelberg (S-405/2008), and the University Medical Center Göttingen (19/09/10). All participants gave written informed consent.
Patients and healthy controls.
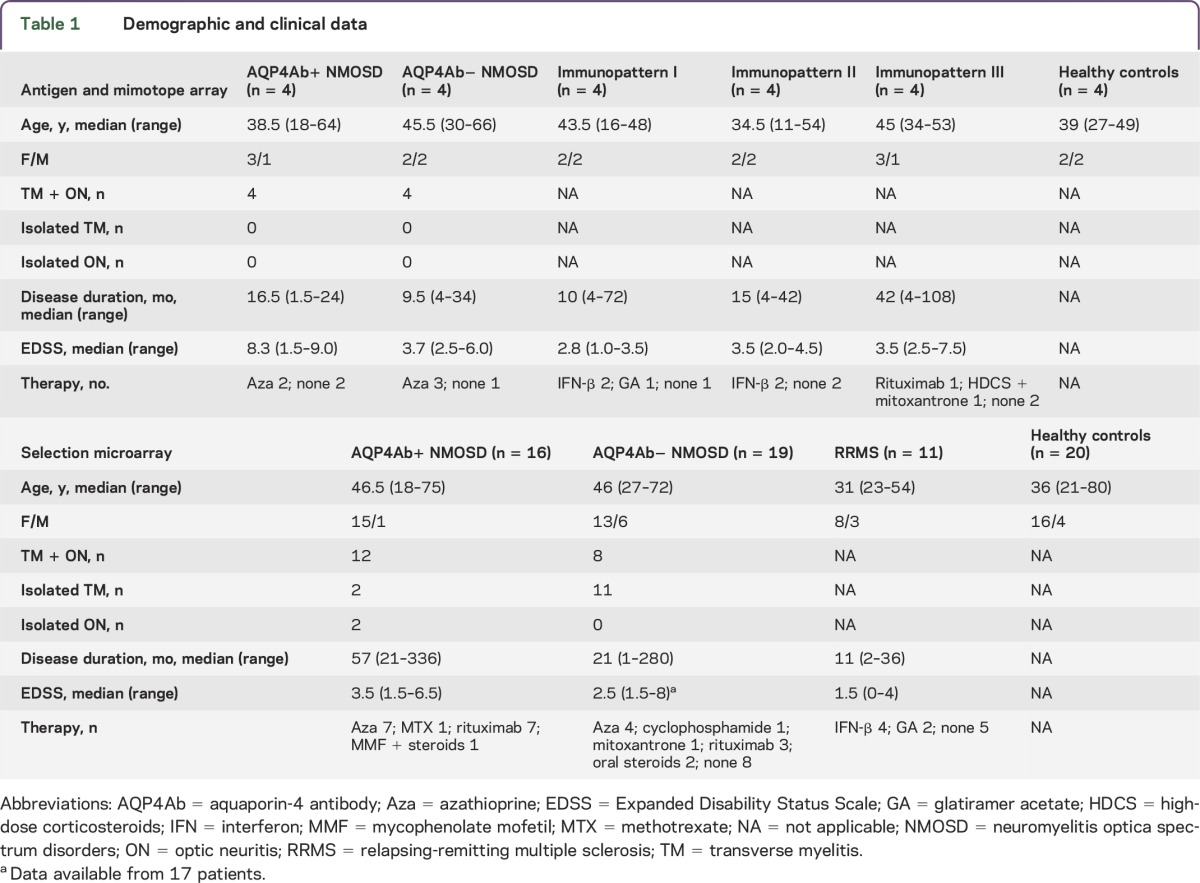
Blood samples included 2 nonoverlapping sets of sera. For a preselection of peptides with the antigen microarray and mimotope microarray (see below), we analyzed serum pools from 4 patients each with NMOSD negative for AQP4-Abs (AQP4Ab− NMOSD), NMOSD positive for AQP4-Abs (AQP4Ab+ NMOSD), MS with histopathologically defined immunopatterns of demyelination I, II, and III,10 and healthy controls. For the final microarray analysis (selection microarray, see below), blood samples were obtained from patients with AQP4Ab− NMOSD (n = 19), patients with AQP4Ab+ NMOSD (n = 16), patients with RRMS (n = 11), and healthy controls (n = 20). All available patients with AQP4− NMOSD were included in the study as well as patients from other groups randomly selected with similar group size. Patients with NMOSD were recruited at the Department of Neurology and NeuroCure Clinical Research Center, Charité-Universitätsmedizin Berlin, the Department of Neurology, University of Heidelberg, and the Department of Neurology, Heinrich-Heine University Düsseldorf. Patients with MS with histopathologically defined immune patterns were recruited at the Department of Neuropathology, University Medical Center Göttingen, and patients with RRMS (non-biopsied) as well as healthy controls at the Department of Neurology and NeuroCure Clinical Research Centre, Charité-Universitätsmedizin Berlin. The group of patients with AQP4Ab+ and AQP4Ab− included patients diagnosed with NMOSD according to the revised Wingerchuk 2006 criteria,11 as well as patients with NMOSD.12 RRMS was diagnosed according to the revised McDonald criteria.13 All sera analyzed with the selection microarray were tested by cell-based assays for antibodies to AQP414 and to MOG (MOG-Abs) (EUROIMMUN, Lübeck, Germany). AQP4-Abs were present in sera from all patients in the AQP4Ab+ NMOSD group and absent in all other groups. MOG-Abs were not detected in any serum samples. Demographics, clinical data, and treatments of the different groups are summarized in table 1.
Table 1.
Demographic and clinical data
Microarray staining and reading.
Peptide arrays were carried out as previously described in detail (appendix e-1 at Neurology.org/nn). In setup experiments, a prestaining with the secondary DyLight 549 conjugated goat anti-human immunoglobulin G (IgG) antibody (diluted 1:1,000 in staining buffer; incubation for 30 minutes; anti-human IgG [H&L] goat antibody; Rockland, Gilbertsville, PA) was performed and showed that all background interactions were negligible compared to subsequent serologic assays. The arrays were then incubated overnight with serum samples diluted 1:1,000 in staining buffer (phosphate-buffered saline, pH 7.4, 0.05% Tween 20 with 10% Rockland Blocking Buffer MB-070). Next, the DyLight 549 conjugated goat anti-human IgG was used as secondary antibody. Microarrays were read with a Fujifilm Life Science (Stamford, CT) FLA-5100 imaging system using a second harmonic generation 532 nm (green) diode laser and a LPG filter. HA and FLAG epitopes were printed on the microarrays in a square surrounding the other peptides and stained in a final step with the corresponding antibodies (mouse anti-HA-Cy5 11801; mouse anti-FLAG M2-Cy3 11802; provided by PEPperPRINT, Heidelberg, Germany). An example of a stained microarray is given in figure e-1. Quantification of spot intensities and peptide annotation were done with PepSlide Analyzer (PEPperPRINT). Results are expressed as fluorescence intensity units, which reflect the extent of antibody binding to the selected peptides.
Peptide microarrays.
Three different types of peptide microarrays were used: antigen microarray, mimotope microarray, and selection microarray. Peptide microarrays were produced with an innovative method using a peptide laser printer and amino acid particles for a combinatorial synthesis of peptides (PEPperPRINT).15 All peptide probes were printed in duplicates on the array. In a first step, the antigen and mimotope microarrays were incubated with 6 different pools of sera (5 pools of patient sera, 1 pool of healthy controls) for a preselection of reactive peptides. In a second step, selection microarrays were printed with the selected peptides. These selection microarrays were used for screening for peptide antibody reactivities in the sera from the 66 patients and controls.
Antigen microarray.
Based on a literature search, we composed a set of 46 (auto) antigens of potential relevance for the pathology of inflammatory demyelinating diseases (table e-1). The full-length amino acid sequences of these antigens were synthesized as 15-mer peptides (offset 5, overlap 10 amino acids), resulting in a peptide microarray containing a total of 4,418 different peptide probes.
Mimotope microarray.
Additionally, 4,290 15-mer random peptides (mimotopes) were generated by a computer algorithm and printed on a microarray (PepSlide Designer; Sicasys GmbH, Heidelberg, Germany).
Selection microarray.
Based on peptide reactivities identified in the screening with the antigen and mimotope microarray and including peptides of special interest as well as control peptides, 702 peptides were selected for the selection microarray (see Results and table e-2). This selection microarray was used to analyze whether different peptide antibody reactivities may distinguish between AQP4Ab+ and AQP4Ab− NMOSD as well as RRMS.
Statistics and bioinformatic analysis.
Statistical analysis of the microarray data was performed with R statistical computing software (version 3.1.2; www.r-project.org). Median signal intensities were log2 transformed and the means between the 2 replicate spots of the same peptide were calculated. Correlation between replicate spots was used as a quality control. To reduce technical variability between the different measurements of patient sera, a normalization was performed by subtracting the average of all 39 control peptides per patient from each measurement (normalized intensity = log2 signal to noise ratio). Peptides with either consistently high or consistently low reactivities in the antigen and mimotope microarrays were selected as controls (see list in table e-2). Three peptides were excluded due to cross-reactivities with anti-HA antibodies used to stain control peptides. To test for pairwise differences among AQP4Ab+ NMOSD, AQP4Ab− NMOSD, RRMS, and controls, the R package limma was used, an empirical Bayes method for the analysis of microarray data using linear models for the assessment of differential expression.16 For the analyses, only noncontrol peptides were used to which at least 5% of the study participants had normalized intensity measurements greater than log2(1,000). p Values were adjusted for multiple hypothesis testing using the method of Benjamini and Hochberg to control for a false discovery rate. Peptides found to be most significant by limma as well as AQP4 peptides were visualized in a heat map from the R package gplots to further classify subgroups of patients. For this, individual peptide data were standardized using z scores (i.e., x−μ/σ) based on the data from the healthy controls. To focus on groups of peptides instead of individual peptides, we used global tests from the R package RepeatedHiDim17 to test for significant interactions, while main effects in mean expression of the groups were evaluated by the Welch test. MA plots (i.e., Bland-Altman plots) were used to visualize differential peptide reactivities between pairs of patient groups. Classification of patient groups based on the peptide reactivities was performed using nearest shrunken centroids with the R package PAMR. PAMR uses an internal feature selection and 10-fold cross-validation.18 Significance thresholds were set at 5%.
RESULTS
A customized microarray for inflammatory demyelinating CNS diseases.
To set up a customized microarray for inflammatory demyelinating diseases (selection microarray), we first screened 8,708 peptides represented on the antigen and mimotope microarrays for differential peptide reactivities with 6 pools of serum samples of AQP4Ab− NMOSD, AQP4Ab+ NMOSD, MS with histopathologically defined immunopatterns of demyelination I, II, and III,10 or controls. Based on the results of this screening, 439 peptides were selected according to one of the following criteria (table e-2A): peptides showed differential binding in any of the pairwise comparisons between serum pools (>2-fold and intensity >2,048 arbitrary intensity units) or peptides had very high reactivity in at least one serum pool (intensity >4,096 arbitrary intensity units). Furthermore, 39 control peptides and 224 peptides of special interest (Epstein-Barr nuclear antigen-1 [EBNA-1] peptides; viral peptides with reactivities >1,024 arbitrary intensity units; selected AQP4, AQP1, MOG, and KIR4.1 peptides; and peptides with differential binding of MS immunopatterns as published by Quintana et al.8) were assembled on the microarray. A list of all 702 peptides is provided in table e-2B.
Differential peptide antibody reactivities are most frequent in RRMS.
For identification of peptide antibody reactivities that may distinguish among AQP4Ab+ and AQPAb− NMOSD, RRMS, and controls, we screened a total of 66 sera from those 4 groups with the selection microarray. To select for robust peptide reactivities, we included only those 225 peptides in the further pairwise comparisons to which at least 5% of study participants had normalized intensity values greater than log2(1,000) (see Statistics and bioinformatic analysis; complete list of results in table e-3). We report differentially bound peptides with a p value <0.05 with peptides remaining significant after adjustment for multiple testing given in brackets. The highest number of differentially bound peptides were found in the comparisons of RRMS with AQP4Ab+ NMOSD (n = 69 [34]), controls (n = 60 [16]), or AQP4Ab− NMOSD (n = 46 [22]). Lower numbers of differentially bound peptides were evident in comparisons between the 2 NMOSD subgroups (n = 13 [0]) and between controls and AQP4Ab+ NMOSD (n = 9 [0]) or AQP4Ab− NMOSD (n = 19 [0]). When comparing all patients with NMOSD with patients with RRMS, we identified 66 (33) differentially bound peptides.
Antibody reactivities to the majority of differentially bound peptides were higher in patients with RRMS and included Epstein-Barr virus (EBV) peptides, myelin peptides (PLP, MAG), oligodendrocytic peptides (NOGO-A, AN-2), astrocytic peptides (AQP4, S100b), axonal/neuronal peptides (neurofascin, neurofilament, contactin-2), and several mimotopes. Peptides derived from type 1 interferon, amyloid-β, hypoxia-inducible factor 1-α, transferrin receptor protein 1, and chitinase-3-like protein 1 also showed higher reactivities.
AQP4Ab+ NMOSD sera did not show higher reactivities to AQP4 peptides than AQP4Ab− NMOSD sera when analyzing individual AQP4 peptides.
Antibody signatures differentiate patient subgroups.
We generated heat maps to compare multiple antibody reactivities (antibody signatures) between the different patient groups. The 68 peptides used in this analysis comprised of the 20 most significant peptides from each pairwise subgroup comparison (table e-3) as well as an additional 4 AQP4 peptides that were detected in at least 5% of study participants with normalized intensity measurements above log2(1,000) (figure 1). Numerous reactivities against single peptides were higher in patients than in controls, and reached up to 5 standard deviations, indicating clear increases in antibody reactivities. Heat maps showed a clustering of RRMS and AQP4Ab+ NMOSD (figure 1A), indicating different antibody signatures in these patient groups. Such a clustering was also seen in RRMS compared to AQP4Ab– NMOSD (figure 1B). However, antibody signatures did not clearly separate the 2 NMOSD subgroups (figure 1C).
Figure 1. Neuromyelitis optica spectrum disorder (NMOSD) subgroups and patients with relapsing-remitting multiple sclerosis (RRMS) show different antibody signatures.

Heat maps (A: RRMS [blue] vs aquaporin-4 antibody [AQP4Ab]+ NMOSD [red]; B: RRMS [blue] vs AQP4Ab− NMOSD [purple]; C: AQP4Ab+ NMOSD [red] vs AQP4Ab− NMOSD [purple]). The top 20 peptides from all pairwise subgroup comparisons as well as all AQP4 peptides are shown as rows (see key for peptides). Patients from the different subgroups are shown as columns (see key for patient groups). The color (see key for z score) indicates the intensity of the peptides standardized to the healthy controls, with blue indicating an upregulation and red a downregulation of peptide reactivities, given in standard deviations. Heat maps show a clustering of RRMS and AQP4Ab+ NMOSD (A) as well as RRMS and AQP4Ab− NMOSD (B), suggesting different antibody signatures in these patient groups. In contrast, such a clustering is not evident comparing the 2 NMOSD subgroups (C). CMV = cytomegalovirus; EBV = Epstein-Barr virus; VZV = varicella-zoster virus.
Elevated reactivities to myelin and EBV peptides in RRMS.
In the next step, groups of related peptides, rather than individual peptides, were compared between AQP4Ab+ NMOSD, AQP4Ab− NMOSD, and RRMS. All peptides that belonged to one protein (e.g., AQP1, AQP4, NOGO-A), a certain virus (EBV, cytomegalovirus, varicella-zoster virus), or a group of proteins (all myelin proteins) were combined. For this analysis, we report all highly significant differences in group comparisons (p ≤ 0.01).
Patients with RRMS showed higher reactivities against myelin peptides than patients with AQP4Ab+ NMOSD (p = 0.003) and patients with AQP4Ab− NMOSD (p = 0.0002) (figure 2, A and B). Myelin peptide reactivities were increased at most to approximately 150% in patients with RRMS compared to patients with AQP4Ab+ NMOSD and to 133% compared to patients with AQP4Ab− NMOSD. Values on the x-axis in figure 2, A–D, represent the average reactivities of peptides for the investigated groups.
Figure 2. Elevated antibody reactivities against myelin and Epstein-Barr virus (EBV) peptides in relapsing-remitting multiple sclerosis (RRMS) and higher anti–aquaporin-4 (AQP4) peptide reactivities in neuromyelitis optica spectrum disorders positive for AQP4−Abs (AQP4Ab+ NMOSD).

Comparison of selected peptide groups in different patient subsets shown with MA plots. (A) Myelin peptides RRMS vs AQP4Ab+ NMOSD. (B) Myelin peptides RRMS vs AQP4Ab− NMOSD. (C) EBV peptides RRMS vs AQP4Ab+ NMOSD. (D) AQP4 peptides AQP4Ab+ vs AQP4Ab− NMOSD. Each dot represents one peptide. The x-axis displays the average normalized signal to show the general level of peptide binding. The y-axis indicates the difference between the patient groups given as a percentage. Greater reactivities in the first mentioned group are found above the 100% horizontal line, and lower reactivities below this line. Patients with RRMS show higher antibody reactivities against myelin peptides and EBV-EBNA 1 peptides than do patients with NMOSD. Higher antibody reactivities against AQP4 peptides are evident in the AQP4Ab+ compared to the AQP4Ab− NMOSD group.
Higher antibody reactivities to EBV peptides were observed in RRMS than in AQP4Ab+ NMOSD (p = 0.0012). Such a difference was not evident when comparing patients with RRMS with patients with AQP4Ab− NMOSD (p = 0.1097). Reactivities against EBV peptides in RRMS were up to 230% of those found in AQP4Ab+ NMOSD (figure 2C).
Elevated reactivities to AQP4 and AQP1 peptides in AQP4Ab+ NMOSD.
We next analyzed antibody reactivities to the group of AQP4 peptides in the 2 NMOSD subgroups. Although single AQP4 peptides had not shown significant differences in antibody binding, group comparisons demonstrated higher AQP4 peptide reactivities in AQP4Ab+ NMOSD than in AQP4Ab− NMOSD (p = 0.0001) (figure 2D). Reactivities of AQP4Ab+ NMOSD increased at most to approximately 133%. Patients with AQP4Ab+ NMOSD also showed higher reactivities than patients with RRMS (p = 0.0110). In addition, higher reactivities against AQP1 peptides were found in the AQP4Ab+ NMOSD group compared to AQP4Ab− NMOSD (p = 0.0052).
Correct patient subgroup classification by peptide reactivities is expected in 61%–81%.
We tested whether antibody signatures permit subgroup classification into AQP4Ab+ and AQP4Ab− NMOSD as well as RRMS. The predicted correct subgroup classification was 78% for the differentiation of AQP4Ab+ NMOSD and RRMS and 80% for the differentiation of AQP4Ab− NMOSD and RRMS. Differentiation of subgroups was based on 17 (AQP4Ab+ NMOSD vs RRMS) or 33 peptides (AQP4Ab− NMOSD and RRMS). In contrast, the predicted correct subgroup classification based on 70 peptides was only 66% for the comparison between AQP4Ab+ and AQP4Ab− NMOSD and based on 33 peptides 61% for the comparison of all NMOSD vs RRMS.
DISCUSSION
We report a systematic serum antibody screening in patients with NMOSD with and without AQP4-Abs, in patients with RRMS, and in healthy controls, employing a customized peptide microarray specifically designed for inflammatory demyelinating CNS diseases. The key results of this study are that NMOSD and RRMS show differential antibody reactivities to a number of human and viral antigens as well as to random peptides (mimotopes), and that based on these antibody reactivities a correct patient subgroup classification to distinguish RRMS and NMOSD can be achieved in approximately 80% of patients. However, NMOSD subgroups, although clearly differentiated by the positivity or negativity for AQP4-Abs in cell-based assays, are not well discriminated by multiple peptide antibody reactivities.
Transfer of AQP4-Abs to animals produces NMO-like lesions, indicating that AQP4-Abs are pathogenic.19–21 The detection of AQP4-Abs by cell-based assays transfected with full-length, conformational AQP4 protein represents the diagnostic gold standard for the diagnosis of AQP4Ab+ NMOSD with a higher sensitivity in the detection of anti-AQP4-Abs compared to peptide-based assays.22 Several studies reported antibodies against linear AQP4 peptides,23,24 showing that antibody reactivities directed against AQP4 peptides can be detected using peptide microarrays. Consistent with this, we observed higher reactivities to the group of AQP4 peptides in AQP4Ab+ vs AQP4Ab− NMOSD. However, when comparing antibody reactivities to single AQP4 peptides between patients with AQP4Ab+ vs AQP4Ab− NMOSD and patients with RRMS, we were unable to identify specific reactivities, and thus no specific epitopes within the AQP4 protein that were preferentially targeted by AQP4-Abs were detected. Interestingly, we found higher reactivities to AQP1 peptides in the AQP4Ab+ NMOSD group compared to the AQP4Ab− NMOSD group. Whether the water channel AQP1 may be an additional autoimmune target in patients with NMOSD is a matter of controversy.25,26
A total of 20%–40% of patients showing typical NMOSD clinical symptoms do not have AQP4-Abs according to the applied assay,4 and only some of these patients have MOG-Abs.27 Our results showed that antibody signatures of AQP4Ab+ and AQP4Ab− NMOSD are more similar, but can be well-differentiated from RRMS. Although studies based on a small number of samples suggested that AQP4Ab+ and AQP4Ab− NMOSD do not share the same pathophysiology,19,28 other in vitro studies showed that sera from AQP4Ab− patients induced complement-dependent astrocyte cell death, as did AQP4Ab+ sera, although to a lower extent.29 One explanation could be that patients with AQP4Ab− NMOSD do indeed have AQP4-Abs that are undetectable even by highly sensitive cell-based assays. Thus, we cannot rule out the possibility of a false assignment of true seropositives to the seronegative group. Another possibility would be that other not yet identified antibodies acting in a manner similar to AQP4-Abs (e.g., targeting astrocytes) could play a pathogenetic role. Finally, a T-B-cell cooperation is necessary in NMO lesion formation30,31 and the role of T-cell pathogenicity may differ between AQP4Ab+ and AQP4Ab− NMOSD.
With optimized autoantibody detection, a mean sensitivity for AQP4-Ab detection of 76.7% is found.2 Thus, compared to the AQP4-Ab testing with cell-based assays, the selection microarray analysis might not to be helpful for differentiating NMOSD in general (including AQP4+ NMOSD) from RRMS, yet may be beneficial for patients with NMOSD without AQP4-Abs, with a predicted correct subgroup classification of 80%. Future studies may thus concentrate on evaluating peptide antibody reactivities for the differentiation of AQP4-Ab-negative NMOSD and MS.
EBV is a risk factor for MS, and EBV seroprevalence in patients with MS reaches nearly 100%. Patients with MS are known to have an elevated antibody response to EBV proteins and peptides.32,33 Significantly higher anti-EBNA-1 IgG antibody titers in patients with MS than in patients with NMOSD were found,34 which is in line with our results. In addition, patients with RRMS had higher anti-myelin peptide reactivities than patients in both NMOSD subgroups. The suspected antigenic target in MS is the oligodendrocyte/myelin unit and not the astrocyte as in NMOSD. Antibody reactivities against myelin proteins in MS have been studied extensively,35 but the specificity and pathogenicity of these antibodies is unclear. MOG-Abs have been associated with clinical syndromes of AQP4− NMOSD, acute disseminated encephalomyelitis, and MS5 and it is suggested that MOG-Ab-associated diseases may represent a separate disease entity.36,37 For this reason, patients with MOG-Abs as measured in cell-based assays were excluded in this work.
A limitation of our study is the small number of participants in each group. Our results are therefore preliminary and need confirmation with larger and independent patient cohorts. In addition, we cannot rule out that prior immunosuppressive/immunomodulatory treatment might have influenced the results. However, we employed strict criteria for allotting patients to the different patient groups and controlled for multiple testing. Our peptide microarray contains linear but not conformational epitopes. It is assumed that successful identification of pathogenic autoantibodies relies on expression systems that display conformational epitopes with all modifications that occur in vivo.38 Nonetheless, antibody reactivities against peptides, although with a lower frequency, may also be detectable in diseases in which antibodies against conformational epitopes are thought to be pathogenic, as shown for AQP4 antibodies.23,24 In addition, peptides may also induce autoimmune disease, as is evidenced by the induction of experimental allergic encephalomyelitis, an experimental model of MS, with MOG peptides. Finally, although it is unlikely that it will be possible to detect single pathogenic autoantibodies using our approach, the peptide microarray technique has the advantage of analyzing multiple antibody reactivities involved in autoimmune diseases. Antigen arrays have been used successfully in autoimmune diseases for disease diagnosis, monitoring of disease progression and response to therapy, and discovering mechanisms of pathogenesis.39,40 Our results revealed antibody signatures that may be a valuable tool in differentiating NMOSD and RRMS.
Future research should focus on reproducing the results in different cohorts and may also be applied to patients with RRMS stratified according to their histopathologic characteristics.10 Different peptide antibody signatures have been shown previously in different MS stages and histologically classified MS,8 results that we extended by showing that differential antibody signatures might distinguish NMOSD and RRMS. Better diagnostic tools in inflammatory demyelinating diseases are an important basis for targeted and individualized patient care.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the study participants for donating blood as well as Bibiane Seeger, Mareike Gloth, Sven Müller, Anna Eschlbeck, and the Nikon Imaging Center at the University of Heidelberg for technical assistance.
GLOSSARY
- AQP4-Ab
aquaporin-4 antibody
- EBNA-1
Epstein-Barr nuclear antigen-1
- EBV
Epstein-Barr virus
- IgG
immunoglobulin G
- MOG
myelin oligodendrocyte glycoprotein
- MS
multiple sclerosis
- NMO
neuromyelitis optica
- NMOSD
neuromyelitis optica spectrum disorders
- RRMS
relapsing-remitting multiple sclerosis
Footnotes
Supplemental data at Neurology.org/nn
AUTHOR CONTRIBUTIONS
The statistical analysis was performed by D.E., T.B., and T.F. (academic affiliation). I.M., T.F., W.B., K.R., and F. Paul designed the study. I.M., T.B., D.E., F. Pache, L.S., M.R., O.A., S.J., B.W., H.D., K.R., and F. Paul were involved in acquisition and/or analysis of the data, T.F. and W.B. additionally in interpretation of data for the work. I.M., T.B., D.E., W.B., K.R., and F. Paul drafted the manuscript; all authors revised it critically for important intellectual content. All authors approved the final version to be published. I.M. and W.B. had access to all the data and take responsibility for the data, accuracy of the data analysis, and the conduct of the study.
STUDY FUNDING
This work was supported by grants from the German Ministry for Education and Research (BMBF, “German Competence Network Multiple Sclerosis” [KKNMS], Pattern MS/NMO), the DFG Transregional Collaborative Research Group (TRR-SFB) 43 and EU-FP7 (Eugène Devic European [EDEN] initiative); and by the Open Access Publication Funds of the Göttingen University. B.W. was supported by grants from Merck Serono and the Dietmar-Hopp-Stiftung.
DISCLOSURE
I. Metz received speaker honoraria and/or travel funding from BiogenIdec, Bayer Healthcare, Teva, Serono, and Novartis; and received research support from BiogenIdec, German Ministry for Education and Research. T. Beiβbarth and D. Ellenberger report no disclosures. F. Pache received travel funding from Genzyme, Bayer, Biogen Idec, and ECTRIMS; and received research support from KKNMS and Novartis. L. Stork reports no disclosures. M. Ringelstein received travel funding and/or speaker honoraria from Novartis, Bayer Schering, Biogen Idec, and Genzyme. M. Aktas served on the scientific advisory board for Medimmune; received travel funding and/or speaker honoraria from Bayer, Novartis, Biogen, Merck, Serono, Teva, and Geynzme; has been an academic editor for PLoS ONE; consulted for Novartis; and received research support from Novartis, Bayer, Biogen, German Research Foundation, German Ministry of Science, Heinrich and Erna Schaufler-Foundation, and Walter and Ilse Rose-Foundation. Sven Jarius reports no disclosures. B.T. Wildemann served on the scientific advisory board for Novartis; received travel funding and/or speaker honoraria from Bayer Healthcare, Biogen, Merck Serono, Genzyme, Novartis, and Teva; and received research support from Biogen, Biotest, Merck Serono, Novartis, Teva Dietmar-Hopp-Foundation, and German Ministry of Education and Research. H. Dihazi reports no disclosures. T. Friede served on the scientific advisory boards for Biogen, Novartis, Bayer, Janssen, and Grunenthal; was an editor for Biometrical Journal; is an Associate Editor for Biometrical Journal; is on the editorial board for MBC Medical Research Methodology; is an academic editor for PLoS ONE; is a section editor for PLoS ONE; has consulted for Novartis, Pharmalog Institut fur Klinische Forschung GmbH, Biogen Idec, and Bayer; and received research support from Deutsche Forschungsgemeinschaft, Bundesministerium fur Bildung und Forschung, and Executive Agency for Health and Consumers. Wolfgang Brück served on the scientific advisory board for Genzyme, Novartis, Biogen Idec, and Teva; received speaker honoraria from Teva, Sanofi, Genzyme, Novartis, Merck-Serono, Biogen, and Bayer; is an editorial board member for Acta Neuropathologica, Therapeutic Advances in Neurological Disorders, Multiple Sclerosis International, and Neuropathology and Applied Neurobiology; and received research support from German Research Foundation, German Ministry for Science and Education, Tschira Foundation. K. Ruprecht served on the scientific advisory board for Sanofi-Aventis/Genzyme, Novartis, and Roche; received travel funding and/or speaker honoraria from Bayer Healthcare, Biogen Idec, Merck Serono, Sanofi-Aventis/Genzyme, Teva Pharmaceuticals, Novartis, and Guthy Jackson Foundation; is an academic editor for PLoS ONE; receives publishing royalties from Elsevier; and received research support from Novartis and German Ministry of Education and Research. F. Paul received speaker honoraria and travel funding from Bayer, Novartis, Biogen Idec, Teva, Sanofi-Aventis/Genzyme, and Merck Serono; is an Associate Editor for Neurology® Neuroimmunology & Neuroinflammation; is an Academic Editor for PLoS ONE; has consulted for SanofiGenzyme, BiogenIdec, and MedImmune; and received research support from Bayer, Novartis, Biogen Idec, Teva, Sanofi-Aventis/Genzyme, Merck Serono, German Research Council, Werth Stiftung of the City of Cologne, German Ministry of Education and Research, Arthur Arnstein Stiftung Berlin, Guthy Jackson Foundation, and National Multiple Sclerosis Society. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med 2005;202:473–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015;85:177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol 2007;6:805–815. [DOI] [PubMed] [Google Scholar]
- 4.Kitley J, Leite MI, Kuker W, et al. Longitudinally extensive transverse myelitis with and without aquaporin 4 antibodies. JAMA Neurol 2013;70:1375–1381. [DOI] [PubMed] [Google Scholar]
- 5.Waters P, Woodhall M, O'Connor KC, et al. MOG cell-based assay detects non-MS patients with inflammatory neurologic disease. Neurol Neuroimmunol Neuroinflamm 2015;2:e89. 10.1212/NXI.0000000000000089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jarius S, Ruprecht K, Wildemann B, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflammation 2012;9:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trebst C, Jarius S, Berthele A, et al. Update on the diagnosis and treatment of neuromyelitis optica: recommendations of the Neuromyelitis Optica Study Group (NEMOS). J Neurol 2014;261:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quintana FJ, Farez MF, Viglietta V, et al. Antigen microarrays identify unique serum autoantibody signatures in clinical and pathologic subtypes of multiple sclerosis. Proc Natl Acad Sci USA 2008;105:18889–18894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van HK, Tomooka BH, Kidd BA, et al. Serum autoantibodies to myelin peptides distinguish acute disseminated encephalomyelitis from relapsing-remitting multiple sclerosis. Mult Scler 2013;19:1726–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lucchinetti C, Brück W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol 2000;47:707–717. [DOI] [PubMed] [Google Scholar]
- 11.Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006;66:1485–1489. [DOI] [PubMed] [Google Scholar]
- 12.Wingerchuk DM, Weinshenker BG. Neuromyelitis optica (Devic's syndrome). Handb Clin Neurol 2014;122:581–599. [DOI] [PubMed] [Google Scholar]
- 13.Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 2011;69:292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jarius S, Probst C, Borowski K, et al. Standardized method for the detection of antibodies to aquaporin-4 based on a highly sensitive immunofluorescence assay employing recombinant target antigen. J Neurol Sci 2010;291:52–56. [DOI] [PubMed] [Google Scholar]
- 15.Beyer M, Nesterov A, Block I, et al. Combinatorial synthesis of peptide arrays onto a microchip. Science 2007;318:1888. [DOI] [PubMed] [Google Scholar]
- 16.Smyth GK, Yang YH, Speed T. Statistical issues in cDNA microarray data analysis. Methods Mol Biol 2003;224:111–136. [DOI] [PubMed] [Google Scholar]
- 17.Jung K, Becker B, Brunner E, Beissbarth T. Comparison of global tests for functional gene sets in two-group designs and selection of potentially effect-causing genes. Bioinformatics 2011;27:1377–1383. [DOI] [PubMed] [Google Scholar]
- 18.Tibshirani R, Hastie T, Narasimhan B, Chu G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc Natl Acad Sci USA 2002;99:6567–6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bradl M, Misu T, Takahashi T, et al. Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann Neurol 2009;66:630–643. [DOI] [PubMed] [Google Scholar]
- 20.Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, Papadopoulos MC. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain 2010;133:349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wrzos C, Winkler A, Metz I, et al. Early loss of oligodendrocytes in human and experimental neuromyelitis optica lesions. Acta Neuropathol 2013;127:523–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jarius S, Wildemann B. Aquaporin-4 antibodies (NMO-IgG) as a serological marker of neuromyelitis optica: a critical review of the literature. Brain Pathol 2013;23:661–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu X, Green M, Gilden D, Lam C, Bautista K, Bennett JL. Identification of peptide targets in neuromyelitis optica. J Neuroimmunol 2011;236:65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marnetto F, Hellias B, Granieri L, et al. Western blot analysis for the detection of serum antibodies recognizing linear Aquaporin-4 epitopes in patients with Neuromyelitis Optica. J Neuroimmunol 2009;217:74–79. [DOI] [PubMed] [Google Scholar]
- 25.Long Y, Zheng Y, Shan F, et al. Development of a cell-based assay for the detection of anti-aquaporin 1 antibodies in neuromyelitis optica spectrum disorders. J Neuroimmunol 2014;273:103–110. [DOI] [PubMed] [Google Scholar]
- 26.Schanda K, Waters P, Holzer H, et al. Antibodies to aquaporin-1 are not present in neuromyelitis optica. Neurol Neuroimmunol Neuroinflamm 2015;2:e160. 10.1212/NXI.0000000000000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kitley J, Woodhall M, Waters P, et al. Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology 2012;79:1273–1277. [DOI] [PubMed] [Google Scholar]
- 28.Marignier R, Nicolle A, Watrin C, et al. Oligodendrocytes are damaged by neuromyelitis optica immunoglobulin G via astrocyte injury. Brain 2010;133:2578–2591. [DOI] [PubMed] [Google Scholar]
- 29.Sabater L, Giralt A, Boronat A, et al. Cytotoxic effect of neuromyelitis optica antibody (NMO-IgG) to astrocytes: an in vitro study. J Neuroimmunol 2009;215:31–35. [DOI] [PubMed] [Google Scholar]
- 30.Pohl M, Fischer MT, Mader S, et al. Pathogenic T cell responses against aquaporin 4. Acta Neuropathol 2011;122:21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mitsdoerffer M, Kuchroo V, Korn T. Immunology of neuromyelitis optica: a T cell-B cell collaboration. Ann N Y Acad Sci 2013;1283:57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Larsen PD, Bloomer LC, Bray PF. Epstein-Barr nuclear antigen and viral capsid antigen antibody titers in multiple sclerosis. Neurology 1985;35:435–438. [DOI] [PubMed] [Google Scholar]
- 33.Ruprecht K, Wunderlich B, Giess R, et al. Multiple sclerosis: the elevated antibody response to Epstein-Barr virus primarily targets, but is not confined to, the glycine-alanine repeat of Epstein-Barr nuclear antigen-1. J Neuroimmunol 2014;272:56–61. [DOI] [PubMed] [Google Scholar]
- 34.Masuda S, Mori M, Arai K, et al. Epstein-Barr virus persistence and reactivation in neuromyelitis optica. J Neurol Neurosurg Psychiatry 2015;86:1137–1142. [DOI] [PubMed] [Google Scholar]
- 35.Fraussen J, Claes N, de BL, Somers V. Targets of the humoral autoimmune response in multiple sclerosis. Autoimmun Rev 2014;13:1126–1137. [DOI] [PubMed] [Google Scholar]
- 36.Reindl M, Rostasy K. MOG antibody-associated diseases. Neurol Neuroimmunol Neuroinflamm 2015;2:e60. 10.1212/NXI.0000000000000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zamvil SS, Slavin AJ. Does MOG Ig-positive AQP4-seronegative opticospinal inflammatory disease justify a diagnosis of NMO spectrum disorder? Neurol Neuroimmunol Neuroinflamm 2015;2:e62. 10.1212/NXI.0000000000000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schirmer L, Srivastava R, Hemmer B. To look for a needle in a haystack: the search for autoantibodies in multiple sclerosis. Mult Scler 2014;20:271–279. [DOI] [PubMed] [Google Scholar]
- 39.Ousman SS, Tomooka BH, van Noort JM, et al. Protective and therapeutic role for alphaB-crystallin in autoimmune demyelination. Nature 2007;448:474–479. [DOI] [PubMed] [Google Scholar]
- 40.Yeste A, Quintana FJ. Antigen microarrays for the study of autoimmune diseases. Clin Chem 2013;59:1036–1044. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.