

Abstract
Recurrent hypoglycemia is common in infants and children. In developing rat models, recurrent moderate hypoglycemia leads to neuronal injury in the medial prefrontal cortex. To understand the effects beyond neuronal injury, three-week-old male rats were subjected to five episodes of moderate hypoglycemia (blood glucose concentration, approximately 30 mg/dl for 90 min) once daily from postnatal day 24 to 28. Neuronal injury was determined using Fluoro-jade B histochemistry on postnatal day 29. The effects on brain-derived neurotrophic factor (BDNF) and its cognate receptor, tyrosine kinase B (TrkB) expression, which is critical for prefrontal cortex development, were determined on postnatal day 29 and at adulthood. The effects on prefrontal cortex-mediated function were determined by assessing prepulse inhibition of the acoustic startle reflex on postnatal day 29 and two weeks later, and by testing for fear-potentiated startle at adulthood. Recurrent hypoglycemia led to neuronal injury confined primarily to the medial prefrontal cortex. BDNF and TrkB expression in the prefrontal cortex was suppressed on postnatal day 29 and was accompanied by lower prepulse inhibition, suggesting impaired sensorimotor gating. Following the cessation of recurrent hypoglycemia, prepulse inhibition had recovered at two weeks. BDNF/TrkB expression in the prefrontal cortex had normalized and fear-potentiated startle was intact at adulthood. Recurrent moderate hypoglycemia during development has significant adverse effects on the prefrontal cortex in the post-hypoglycemia period.
Keywords: brain-derived neurotrophic factor, prefrontal cortex, developing brain, prepulse inhibition, recurrent hypoglycemia, young rat
1. Introduction
Recurrent hypoglycemia is common in infants and children with congenital hyperinsulinism and insulin therapy for type 1 diabetes [1]. Recurrent severe hypoglycemia alters brain structure and leads to cognitive deficits in these children [2–4]. Even mild and asymptomatic hypoglycemia is associated with attention and learning deficits, especially in the immediate post-hypoglycemia period [5,6].
Most hypoglycemia episodes during development are of mild to moderate severity and are not associated with neurological signs, such as altered consciousness or seizures. Studies in developing rats demonstrate that the medial prefrontal cortex (PFC) is highly vulnerable to injury during moderate hypoglycemia [7–10]. The effects beyond neuronal injury are not well characterized. In both humans and rats, the peak development of PFC occurs in the postnatal period [11,12] and is characterized by increased glucose delivery to the region for supporting the energy requirement of synaptogenesis and neurotransmission [13]. Frequent disruptions in glucose supply due to recurrent hypoglycemia may compromise energy production in the PFC and alter its development and function.
The aim of the present study was to characterize the acute and long-term effects of recurrent moderate hypoglycemia on the expression of brain-derived neurotrophic factor (BDNF) and its receptors in the PFC and PFC-regulated behaviors in three-week-old rats. Rats of this age are used to model the effects of hypoglycemia in young children due to similarities in the stage of brain development and substrate utilization [7,9,11,14,15]. BDNF plays a critical role in the normal development of PFC [16–18]. It is a complex gene that generates multiple splice variants containing a common coding exon. BDNF-II, -IV and -VI variants are predominantly expressed in the cerebral cortex [19]. Binding of BDNF to the full-length isomer of tyrosine kinase receptor B (TrkBFL) promotes neuronal growth and survival [20]. Conversely, binding of BDNF to the pan-neurotropin p75 (p75NTR) receptor leads to cell cycle arrest, reduced neurite growth and apoptosis [20]. We have recently demonstrated that an acute episode of moderate hypoglycemia does not alter BDNF, TrkB and p75NTR expression in the PFC of three-week-old rats [14]. The effects of recurrent moderate hypoglycemia are not known. Other conditions that perturb energy metabolism in the postnatal period, such as recurrent hypoxia, are associated with suppressed BDNF expression in the PFC [21,22].
We assessed the functional effects of recurrent hypoglycemia using behavioral tests of prepulse inhibition (PPI) of the acoustic startle reflex and fear conditioning. PPI measures sensorimotor gating, the ability of a weak sensory stimulus to inhibit the startle response to a subsequent intense stimulus. PPI is regulated by the medial PFC, hippocampus, nucleus accumbens, amygdala and pontine tegmental area [23] and is sensitive to imbalances in BDNF/TrkB expression in the PFC [24–27]. Fear was assessed as potentiation of the acoustic startle reflex (fear-potentiated startle; FPS) at adulthood. Two forms of Pavlovian fear conditioning paradigms were used that differ only in the relative timing of presentation of the conditioned and unconditioned stimuli: “trace” conditioning, which is dependent on the integrity of PFC and dorsal hippocampus, and “delay” conditioning, which is not. Both forms of fear conditioning are dependent on the amygdala [28].
Materials and Methods
Animal Preparation
The Institutional Animal Care and Use Committee at University of Minnesota approved the study and its guidelines for care and use of animals were followed. Pregnant Sprague Dawley rats were purchased (Charles River Laboratories, Raleigh, NC) on gestational day 2 and allowed to deliver spontaneously. Litter size was culled to 8 (6 males 2 females per litter) on P3. Pups were weaned on P21. Same-sex littermates were group-housed until P28 and then in same-sex pairs until adulthood. Only male rats (N = 64) were used due to the known sex-specific differences in post-insult BDNF expression and PPI regulation [25,29]. Rats were maintained under standard laboratory conditions with 12hour light-and-dark cycles.
Induction of Hypoglycemia
Rats were randomly assigned to the Control and Recurrent Hypoglycemia (RH) groups. To control for between-versus within-litter variance, equal numbers of rats in each litter were assigned to the two groups. RH group was subjected to insulin-induced hypoglycemia from P24 to P28, once daily between 8 AM and 10 AM (total, 5 episodes). This model has been described previously [30]. Briefly, regular insulin (Novo Nordisk Inc., Clayton, NC) was injected in a dose of 10 IU/kg ip without prior fasting. The target blood glucose concentration was 20–40 mg/dl, a value considered moderate hypoglycemia, as it is not associated with coma or seizures in rats of comparable age [9,30]. This severity and frequency of hypoglycemia is associated with cognitive and behavioral deficits in human infants and adults [31–33]. Littermates in the Control group were injected with an equivalent volume of 0.9% saline (Control group). Food was withheld (water available) in both groups and the ambient temperature was maintained at 34.0±1.0°C [30]. Blood glucose concentration was determined in tail vein samples every 15 min. Hypoglycemia was terminated 120 min after the insulin administration by injecting 10% dextrose (200 mg/kg ip). Blood glucose concentration was determined 2 hours later in randomly selected rats from each litter to confirm resolution of hypoglycemia. Rats were left undisturbed until the day of additional experiments.
Behavioral Testing
For both PPI and FPS, rats were trained and tested in four identical 7.5 cm × 8.5 cm × 17.0 cm stabilimeter devices. Each stabilimeter consisted of a Plexiglas cage resting on four compression springs and located within a ventilated, sound-attenuating chamber. The ventilation fans elevated background noise in the chamber to 65 dB. Cage movement resulted in displacement of a piezoelectric accelerometer (Model ACH-01, Measurement Specialties, Valley Forge, PA) attached to each cage. Voltage output from the accelerometer was filtered and amplified using a custom-built signal processor, digitized on a scale of arbitrary units (au) ranging from 0–1000 (National Instruments SCB100 and PCI-6071E boards) and recorded using Matlab (The MathWorks, Natick, MA). Startle amplitude was defined as the peak accelerometer voltage during the first 200 ms after onset of the startle stimulus. High frequency speakers (Radio Shack Supertweeters, range: 5–40 kHz) located 10 cm from each cage delivered the startle stimuli, which were bursts of filtered white noise (low pass: 22 kHz, rise-decay <5 ms).
PPI tests were given on P29 (i.e., 24 hours after the last hypoglycemia episode) and repeated two weeks later on P42 (n=16 on each day) using previously reported methods [34,35]. After a 5-min acclimation period, six 115-dB, 40-ms startle stimuli were presented at 30-sec intervals to habituate the startle response. Then, 15 prepulse-startle pairings were intermixed with 5 startle-alone trials, with a variable 8–23 sec intertrial interval. For prepulse-startle pairings, onset of the prepulse (68-, 71-, or 77-dB) preceded onset of the 115-dB startle stimulus by 100 ms. Percent PPI was calculated as [(startle-alone) − (prepulse + startle)]/startle-alone × 100.
FPS training began on P56 (i.e., 4 weeks after the termination of recurrent hypoglycemia). The foot shock unconditioned stimulus (US) was a 0.5-s, 0.8-mA constant current scrambled shock, delivered by a shock generator (no. SGS-004; BRS-LVE, Bellsville, ME, USA) through the four bars of the stabilimeter. The conditioned stimulus (CS) was a 7.5-s, 70-dB band-pass filtered noise with high and low cutoffs set at 4 kHz and 24 dB per octave attenuation. The noise was delivered through a low-frequency speaker (Radio Shack woofer, Model no. 40-1024A) situated 15 cm from the cage.
To measure levels of baseline startle, rats (n=16) underwent 2 days of startle testing prior to fear conditioning. On each day, after a 5-min acclimation period, they were presented with 40 startle stimuli (20 each at 95 and 105 dB intermixed pseudo randomly; 30 sec interstimulus interval). For fear conditioning on days 3–5, after a 5 min acclimation period, different groups of rats were exposed to delay or trace fear conditioning. For delay fear conditioning, the rats were presented with 12 trials consisting of the 7.5 sec tone CS co-terminating with the 0.5 sec shock US (90–180-s variable intertrial interval). For trace fear conditioning, onset of the 0.5 sec shock US occurred 3 sec after offset of a 4 sec tone CS. Hence the interval between onsets of the two stimuli in the two conditioning paradigms was the same. FPS was tested on day 6 (i.e. P62; young adulthood). After a 5 min acclimation period, 30 startle stimuli (95 and 105 dB) were followed by 10 startle stimuli presented 3.5 sec after the onset of the CS and 10 presented 7.0 sec after CS onset (i.e., at the time shock would have been presented during training). Average potentiation across these two time points provides a reliable measure of FPS. These CS-startle stimulus pairings were intermixed with 10 additional startle stimulus presentations in the absence of the CS. FPS was calculated as [(CS + startle) − (startle-alone)]/startle-alone × 100%.
Tissue Preparation
Rats were killed on either P29, 24 hour after the last hypoglycemia episode, or P65 (young adulthood) using sodium pentobarbital (100 mg/kg, ip.). The brain was removed within 10 min of pentobarbital administration and the PFC was dissected on ice, flash-frozen using liquid nitrogen and stored at −80°C. Rats used for histochemistry underwent in situ perfusion-fixation before brain removal. Serial 20-μm coronal brain sections were obtained using a cryostat, mounted on glass slides and stored at −20°C until histochemistry.
Assessment of Regional Injury
Regional injury was determined on P29 using FJB histochemistry using published methods [7,8] (n=8). Pilot studies demonstrated that injury was primarily limited to rostral brain sections (3.5 mm to 8 mm anterior to the zero plane of the stereotaxic coordinates at P21). Digital photomicrographs of 4 to 6 sections from this region were collected and all FJB-stained cells in the brain sections were manually counted in a blinded fashion [7,8].
Quantitative RT-PCR
The transcript expression of BDNF (Bdnf; variants II, IV and VI), TrkB (Ntrk2), p75NTR (Ngfr), and the downstream targets of BDNF/TrkB signaling, early growth response-1 and -2 (Egr1 and Egr2), 3-hydroxy-3-methylglutaryl-CoA reductase (Hmgcr) and profilin-1 and -2 (Pfn1 and Pfn2) that regulate brain development and plasticity [36–38] was determined using previously described methods [8,14] (n=6–10 on P29 and P65).. Each sample was assayed in duplicate and normalized against ribosomal protein S18.
Western Blot Analysis
BDNF and TrkB (full-length [TrkBFL] and truncated [TrkBTR] isomers) protein levels were determined using primary antibodies against BDNF (1:1000; Abcam, Cambridge, MA), TrkB (1:1000; Cell Signaling, Danvers, MA) and β-actin (1:5000; Sigma Chemical Co., St. Louis, MO), and fluorescent secondary antibodies (1:2500; Life Technologies, Grand Island, NY) using published methods [8] (n=4–6 on P29 and P65). The target protein intensity relative to β-actin was determined.
Data Analysis
The effects of recurrent hypoglycemia on neuronal injury, mRNA and protein expression, and trace- and delay-fear conditioning were determined using unpaired t tests. The acute and long-term effects on PPI were determined using ANOVA. To assess for any differences in startle between the groups, an analysis of startle magnitude during the startle habituation period (“baseline” startle) was conducted. PPI was evaluated using a 2 × 2 × 3 (Group × Age × prepulse intensity) mixed-model ANOVA, with age and prepulse intensity as repeated measures, followed by post hoc comparisons using Tukey’s HSD test. Data are presented as mean±SEM. Statistical significance was set at p<0.05.
RESULTS
Blood Glucose Concentration
The target blood glucose concentration (20–40 mg/dl) was reached within 30 min of insulin administration in the RH group and was maintained in this range until the 10% dextrose administration at 120 min (i.e., 90 min of hypoglycemia) on each of the five days (Table 1). Animals were less active, but conscious and seizure-free during the period of hypoglycemia. The blood glucose concentration 2 hours post-hypoglycemia was comparable in the two groups (Control group, 139±7 mg/dl, RH group, 130±31 mg/dl). Recurrent hypoglycemia did not affect body weight on either P29 (Control group, 106±2 g; RH group, 109±2 g) or P65 (Control group, 345±8 g; RH group, 359±11 g).
Table 1.
Mean±SEM Blood Glucose Concentrations in the Control and Recurrent Hypoglycemia Groups
| Postnatal Day | Control Group | RH Group |
|---|---|---|
| 24 | 137± 2 | 33± 2* |
| 25 | 129± 3 | 35± 4* |
| 26 | 133± 4 | 21± 2* |
| 27 | 125± 9 | 21± 4* |
| 28 | 127± 3 | 30± 2* |
| Overall | 132±1 | 31±1* |
Three-week-old rats were subjected to insulin-induced hypoglycemia, once daily from postnatal day 24 to 28. Littermates in the Control group were injected with equal volume of normal saline. Blood glucose concentration was measured in the tail vein sample every 15 min during the 120 min of observation. N = 31–84 blood samples from a total of 64 rats on each day.
p<0.001 vs. Control Group.
Abbreviation: RH, recurrent hypoglycemia.
Neuronal Injury Post-hypoglycemia
Twenty-four hours post-hypoglycemia, FJB-positive cells, indicative of cell injury were present in the RH group, but not in the Control group (FJB positive cells/brain section: Control group, 0±0; RH group, 86±21, p<0.01). FJB positive cells were visualized primarily in the cingulate and orbital regions of the medial PFC (46% of all FJB positive cells, Figure 1) and layers III and IV of the parietal (35%) and temporal (19%) cortices (not shown). FJB positive cells were absent in other brain regions.
Figure 1.
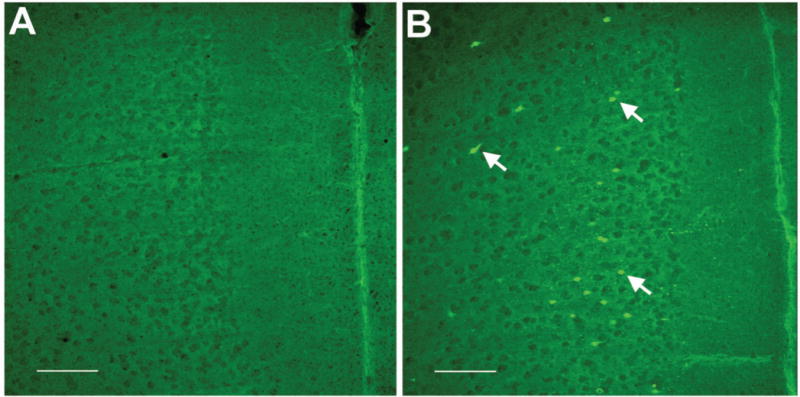
Neuronal injury after recurrent hypoglycemia in young rats. Three-week-old male rats were subjected to insulin-induced moderate hypoglycemia, once daily from postnatal day 24 to 28. Littermates in the Control group were injected with equal volume of normal saline. Coronal brain sections of the prefrontal cortex collected 24 hours after the last hypoglycemia episode from representative rats from the Control group (A) and Recurrent Hypoglycemia group (B) and stained for injured cells using Fluoro-Jade B histochemistry demonstrate Fluoro-Jade B positive cells in the medial prefrontal cortex in the Recurrent Hypoglycemia group (arrows in B). Bar = 100 μm.
Effects of Recurrent hypoglycemia on BDNF Expression in the PFC
On P29, Bdnf-IV expression was 35% lower in the RH group (p <0.001, Figure 2A). Bdnf-II (Control group, 1.00±0.08; RH group, 0.89±0.10) and Bdnf-VI (Figure 2A) expression was not altered. BDNF protein level was 16% lower in the RH group (p=0.03, Figure 2B) and was accompanied by 21% lower TrkBFL protein level (p=0.03, Figure 2B). TrkBTR protein level was not altered (Figure 2B). The expression of downstream targets of BDNF/TrkB signaling, Erg1, Erg2, Hmgcr, Pfn1 and Pfn2 was 16% to 34% lower in the RH group (p<0.05, Table 2).
Figure 2.
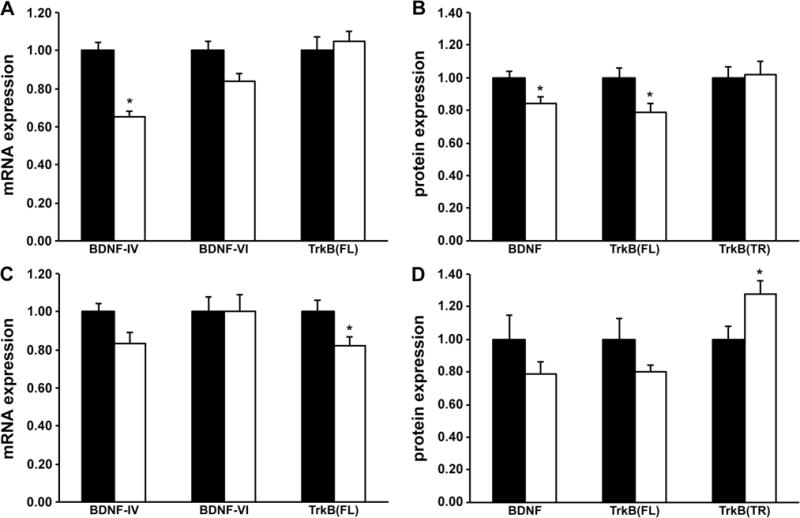
Acute and long-term effects of recurrent hypoglycemia on brain-derived neurotrophic factor (BDNF) and tyrosine kinase B (TrkB) receptor mRNA and protein expressions in the prefrontal cortex of young rats. Three-week-old male rats were subjected to insulin-induced moderate hypoglycemia, once daily from postnatal day 24 to 28. Littermates in the Control group were injected with equal volume of normal saline. BDNF and TrkB transcript and protein expression in the prefrontal cortex was determined 24 hours post-hypoglycemia (A and B) and at adulthood (C and D). Black bars = Control group, white bars = Recurrent Hypoglycemia group. Values are mean ± SEM normalized to the Control group; n = 6–10 for mRNA expression and n = 4–6 for protein expression. *p<0.05 vs. Control group (unpaired t tests). Abbreviations: TrkB(FL), full-length isoform of TrkB; TrkB(TR), truncated isoform of TrkB.
Table 2.
Acute and Long-term Effects of Recurrent Hypoglycemia on the Transcript Expression Downstream of BDNF/TrkB Signaling in the Prefrontal Cortex of Young Rats
| Transcript | Postnatal Day 29 | Postnatal Day 65 | ||
|---|---|---|---|---|
| Control Group | RH Group | Control Group | RH Group | |
| Egr1 | 1.00±0.04 | 0.81±0.05* | 1.00±0.05 | 0.99±0.08 |
| Egr2 | 1.00±0.12 | 0.66±0.10* | 1.00±0.12 | 1.23±0.15 |
| Hmgcr | 1.00±0.05 | 0.84±0.04* | 1.00±0.10 | 1.12±0.14 |
| Pfn1 | 1.00±0.07 | 0.78±0.06* | 1.00±0.06 | 0.90±0.05 |
| Pfn2 | 1.00±0.09 | 0.77±0.05* | 1.00±0.08 | 0.79±0.07 |
Three-week-old rats were subjected to insulin-induced moderate hypoglycemia, once daily from postnatal day 24 to 28. Littermates in the Control group were injected with equal volume of normal saline. Brain tissue was harvested on either postnatal day 29 or 65 for determining transcript expression in the prefrontal cortex. Values are mean±SEM normalized to the Control group at each age; n=6–10.
p<0.05 vs. Control group of corresponding age (unpaired t tests).
Abbreviations: Egr, early growth response; Hmgcr, 3-hydroxy-3-methylgluaryl-CoA reductase; Pfn, profilin; RH, recurrent hypoglycemia.
On P65, the expression of Bdnf-II (Control group, 1.00±0.10; RH group, 1.03±0.12), Bdnf-IV and Bdnf-VI (Figure 2C), as well as BDNF protein (Figure 2D) were comparable in the Control and RH groups. TrkBFL protein level was not altered (Figure 2D), while TrkBTR protein level was 28% higher (p<0.05; Figure 2D) in the RH group. Erg1, Erg2, Hmgcr, Pfn1 and Pfn2 expression was not altered (Table 2).
Recurrent hypoglycemia did not alter p75NTR receptor mRNA (Ngfr) expression at either age (not shown).
Effects of Recurrent Hypoglycemia on Behavioral Tests
Baseline startle amplitude was comparable in the two groups on P29 (Control group, 2.5±0.2 au; RH group, 2.7±0.2 au) and P42 (Control group, 2.1±0.3 au; RH group, 2.0±0.2 au). PPI was stronger on P42 than on P29 in both groups (Figure 3). There were significant main effects of Group (F=4.2, p<0.05), Age at test (F=66.2, p<0.01) and Group × Age interaction (F=4.9, p<0.05) on PPI. As expected, PPI was positively related to prepulse intensity (F=17.3, p<0.01), but there were no significant interactions as a function of this factor (Fs<1.3). PPI was significantly lower in the RH group, compared with the Control group at P29, and with either group at P42 (p<0.05 for all, Tukey’s HSD tests; Figure 3). Recurrent hypoglycemia did not significantly alter the magnitude of trace and delay FPS on P65 (Supplemental Figure).
Figure 3.
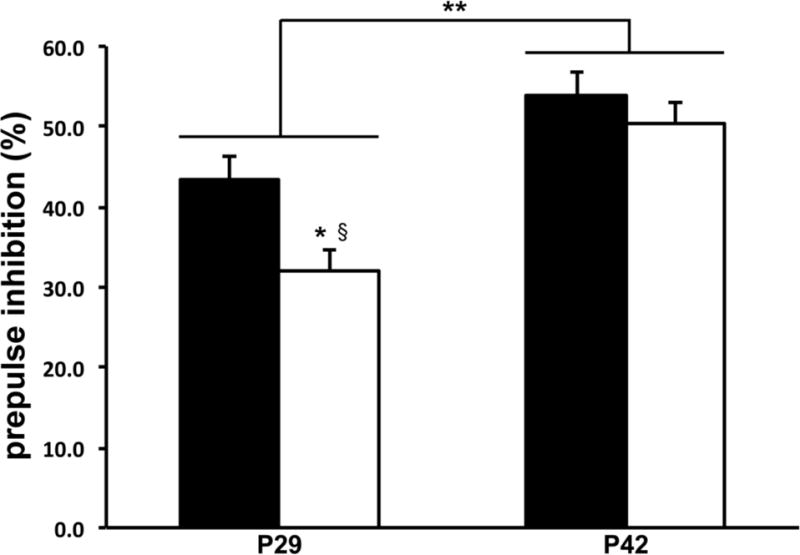
Acute and long-term effects of recurrent hypoglycemia on prepulse inhibition of acoustic stimulus in young rats. Three-week-old male rats were subjected to insulin-induced moderate hypoglycemia, once daily from postnatal day 24 to 28. Littermates in the Control group were injected with equal volume of normal saline. Prepulse inhibition was tested 24 hours (on P29) and two weeks (on P42) post-hypoglycemia. Black bars = Control group, white bars = Recurrent Hypoglycemia group. Values are mean ± SEM; n = 16. There are significant main effects of Group, Age at test and Group × Age interaction (p<0.05 for each; ANOVA). *p<0.05 vs. P29 Control group; §p<0.05 vs. P42 Control group and P42 Recurrent Hypoglycemia group, both (Tukey’s HSD tests). **p<0.05; P29 vs. P42 (ANOVA). Abbreviation: P, postnatal day.
DISCUSSION
Recurrent moderate hypoglycemia led to injury in the medial PFC as evident by the presence of FJB-positive cells in the region. Although not characterized in the present study, previous hypoglycemia studies in developing and adult rats have established that FJB-positive cells represent neuronal injury [7–9,14,39]. As in those studies, FJB-positive cells were primarily seen in the cingulate and orbital regions of the PFC, highlighting the vulnerability of the medial PFC to injury during moderate hypoglycemia. The reasons for the increased vulnerability are not well understood. In adult humans, acute hypoglycemia leads to neuronal activation in the medial PFC that fails to normalize after the correction of hypoglycemia [40,41]. Persistent activation in the setting of neuroglycopenia and compromised energy metabolism [42–44] may have predisposed these neurons to injury.
In addition to neuronal injury, recurrent hypoglycemia suppressed BDNF and TrkB expression in the PFC. Although pentobarbital is known to attenuate BDNF and TrkB mRNA induction in the brain regions [45,46], the short interval (<10 min) between pentobarbital administration and tissue collection rules out this possibility in the present study. It takes several hours for BDNF and TrkB suppression to occur after pentobarbital administration or in other conditions (for example, stress) [45–47]. BDNF and TrkB suppression in the present study differs from the absence of a similar effect following a single episode of moderate hypoglycemia in rats of comparable age [14]. A similarly disparate pattern has been demonstrated with postnatal hypoxia in mice, where exposure to a single day of hypoxia does not alter BDNF expression in the PFC, while exposure to recurrent (3–10 days) hypoxia leads to progressive BDNF suppression in the region [21]. Thus, BDNF and TrkB suppression likely reflects the cumulative effect of recurrent hypoglycemia in the present study. The downregulation of Egr1, Egr2, Hmgcr, Pfn1 and Pfn2 transcripts suggests that BDNF/TrkB signaling also may be suppressed in the post-hypoglycemia period [48]. However, this possibility remains speculative in the absence of confirmatory protein assay or histochemical analysis. It is noteworthy that recurrent moderate hypoglycemia leads to dampening of PFC activation in the post-hypoglycemia period in adult rats [49,50].
The observed functional effects paralleled the BDNF/TrkB changes in PFC. Decreased PPI on P29 suggests impaired sensorimotor gating in the post-hypoglycemia period. This finding differs from the intact PPI reported in a previous study of recurrent hypoglycemia in younger rats [10]. Differences in timing of hypoglycemia in the two studies may explain the discrepant results. Recurrent hypoglycemia occurred during the period of peak PFC development (P24 to P30) [12] in the present study, whereas hypoglycemia episodes (from P9 to P20) had ceased prior to this period in the Moore et al study [10]. Lower BDNF/TrkB expression in the PFC may explain the impaired PPI in the present study. PPI is lower in BDNF heterozygous mice that have 40–50% lower BDNF expression in the frontal cortex than the wild-type mice [24,25]. Intracerebral BDNF administration rapidly normalizes PPI deficits in DBA/2J mice [26]. Decreased or disrupted glutamatergic and GABAergic neurotransmission in the PFC also may have had a role in the lower PPI in the present study [27]. Glutamate and GABA concentrations decrease in the brain during hypoglycemia and the depleted concentrations are not restored to the prehypoglycemia levels after the termination of hypoglycemia [42,43]. Additionally, along with the medial PFC, the hippocampus, nucleus accumbens, amygdala and pontine tegmental area regulate PPI [23]. Although we did not find FJB-stained cells in these brain regions, we cannot rule out subtle injury that may have negatively influenced PPI. Finally, hypoglycemia is a potent stressor and leads to robust corticosterone response in three-week-old rats [51]. Early life stress negatively influences regional gene expression and behavioral performance, and is known to suppress BDNF expression in the PFC [52–55]. Future studies are necessary to address all these possibilities.
PPI had normalized in the recurrent hypoglycemia group on P42. As we did not repeat the test between P29 and P42, it is not possible to know how rapidly PPI normalized following the cessation of hypoglycemia. PPI on P42 was stronger than on P29 in both groups, which is consistent with normal development [56]. Antecedent hypoglycemia also did not affect fear conditioning at adulthood, suggesting intact functional integrity of PFC, dorsal hippocampus and amygdala by this time point and ruling out new-onset behavioral deficits beyond P42. BDNF and TrkBFL expression also had normalized at adulthood. Although TrkBTR protein expression was upregulated, its functional relevance is unclear. It has been postulated that TrkBTR upregulation may negatively modulate TrkB kinase activity by forming TrkB heterodimers [57]. Such an effect is unlikely in the present study, since the expression of downstream targets of BDNF/TrkB signaling were not affected. The absence of long-term behavioral deficits in the present study is similar to the data from adult rats [58]. This is not surprising given that three-week-old rats and adult rats have comparable vulnerability to hypoglycemia-induced brain injury [7,14]. It is possible that testing under hypoglycemia conditions [59] or testing for more complex cognitive or affective behaviors [10] would have uncovered additional functional deficits.
In summary, recurrent hypoglycemia during the rapid phase of PFC development led to neuronal injury and suppressed BDNF and TrkB expression in the region and impaired sensorimotor gating in three-week-old rats. Although these effects were short-term, they were seen after a relatively small number of moderately severe and short-duration hypoglycemia episodes. From the clinical perspective, the functional deficits in the present study parallel the functional deficits observed in the post-hypoglycemia period in children with type 1 diabetes [5]. Even though these functional impairments are transient, typically lasting less than 24 hours [5,60], they interfere with the ability to learn efficiently and have important academic and social implications for the child [61]. The potential for injury and functional deficits suggests the importance of preserving brain energy metabolism using alternative substrates during insulin therapy [14] and exploring novel neuroprotective strategies, such as enhancing BDNF levels through physical activity [62], in this population.
Supplementary Material
Acknowledgments
The authors acknowledge Bruce Kennedy in the Graduate Program in Neuroscience at University of Minnesota for assistance with the behavioral data analysis.
Statement of Financial Support
Funded by NIH/NICHD HD47276 and Viking Children’s Fund, Department of Pediatrics, University of Minnesota, Minneapolis, MN 55455.
Abbreviations
- BDNF
brain-derived neurotrophic factor
- Egr-1
early growth response-1
- Egr-2
early growth response-2
- FJB
Fluoro-jade B
- FPS
fear-potentiated startle
- HMGCR
3-hydroxy-3-methylglutaryl-CoA reductase
- p75NTR
pan-neurotropin receptor
- P
postnatal day
- PFC
prefrontal cortex
- PPI
prepulse inhibition
- Pfn-1
profilin-1
- Pfn-2
profilin-2
- RH group
Recurrent Hypoglycemia group
- TrkB
tyrosine kinase receptor B
- TrkBFL
full-length isomer of tyrosine kinase receptor B
- TrkBTR
truncated isomer of tyrosine kinase B
Footnotes
Statement of Conflict of Interest
None of the authors have conflict of interest to declare.
References
- 1.Rao R, Hershey T. The impact of hypoglycemia on the developing brain. In: Seaquist ER, Robertson PR, editors. Translational endocrinology and metabolism: Hypoglycemia in diabetes update. 4. Vol. 3. Chevy Chase, MD: Endocrine Society; 2012. pp. 137–159. [Google Scholar]
- 2.Stenninger E, Flink R, Eriksson B, Sahlen C. Long-term neurological dysfunction and neonatal hypoglycaemia after diabetic pregnancy. Arch Dis Child Fetal Neonatal Ed. 1998;79:F174–179. doi: 10.1136/fn.79.3.f174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hershey T, Perantie DC, Warren SL, Zimmerman EC, Sadler M, White NH. Frequency and timing of severe hypoglycemia affects spatial memory in children with type 1 diabetes. Diabetes Care. 2005;28:2372–2377. doi: 10.2337/diacare.28.10.2372. [DOI] [PubMed] [Google Scholar]
- 4.Blasetti A, Chiuri RM, Tocco AM, Di Giulio C, Mattei PA, Ballone E, Chiarelli F, Verrotti A. The effect of recurrent severe hypoglycemia on cognitive performance in children with type 1 diabetes: A meta-analysis. J Child Neurol. 2011;26:1383–1391. doi: 10.1177/0883073811406730. [DOI] [PubMed] [Google Scholar]
- 5.Reich JN, Kaspar JC, Puczynski MS, Puczynski S, Cleland JW, Dell’Angela K, Emanuele MA. Effect of a hypoglycemic episode on neuropsychological functioning in diabetic children. J Clin Exp Neuropsychol. 1990;12:613–626. doi: 10.1080/01688639008401005. [DOI] [PubMed] [Google Scholar]
- 6.Topitsch D, Schober E, Wurst E, Kryspin-Exner I. Changes in attention with hypo- and hyperglycaemia in children with insulin dependent diabetes mellitus. Eur J Pediatr. 1998;157:802–805. doi: 10.1007/s004310050939. [DOI] [PubMed] [Google Scholar]
- 7.Ennis K, Tran PV, Seaquist ER, Rao R. Postnatal age influences hypoglycemia-induced neuronal injury in the rat brain. Brain Res. 2008;1224:119–126. doi: 10.1016/j.brainres.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rao R, Sperr D, Ennis K, Tran P. Postnatal age influences hypoglycemia-induced poly(adp-ribose) polymerase-1 activation in the brain regions of rats. Pediatr Res. 2009;66:642–647. doi: 10.1203/PDR.0b013e3181bbce69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamada KA, Rensing N, Izumi Y, De Erausquin GA, Gazit V, Dorsey DA, Herrera DG. Repetitive hypoglycemia in young rats impairs hippocampal long-term potentiation. Pediatr Res. 2004;55:372–379. doi: 10.1203/01.PDR.0000110523.07240.C1. [DOI] [PubMed] [Google Scholar]
- 10.Moore H, Craft TK, Grimaldi LM, Babic B, Brunelli SA, Vannucci SJ. Moderate recurrent hypoglycemia during early development leads to persistent changes in affective behavior in the rat. Brain Behav Immun. 2010;24:839–849. doi: 10.1016/j.bbi.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rice D, Barone S., Jr Critical periods of vulnerability for the developing nervous system: Evidence from humans and animal models. Environ Health Perspect. 2000;108(Suppl 3):511–533. doi: 10.1289/ehp.00108s3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Eden CG, Uylings HB. Postnatal volumetric development of the prefrontal cortex in the rat. J Comp Neurol. 1985;241:268–274. doi: 10.1002/cne.902410303. [DOI] [PubMed] [Google Scholar]
- 13.Nehlig A. Cerebral energy metabolism, glucose transport and blood flow: Changes with maturation and adaptation to hypoglycaemia. Diabetes Metab. 1997;23:18–29. [PubMed] [Google Scholar]
- 14.Ennis K, Dotterman H, Stein A, Rao R. Hyperglycemia accentuates and ketonemia attenuates hypoglycemia-induced neuronal injury in the developing rat brain. Pediatr Res. 2015;77:84–90. doi: 10.1038/pr.2014.146. [DOI] [PubMed] [Google Scholar]
- 15.Nehlig A, Pereira de Vasconcelos A. Glucose and ketone body utilization by the brain of neonatal rats. Prog Neurobiol. 1993;40:163–221. doi: 10.1016/0301-0082(93)90022-k. [DOI] [PubMed] [Google Scholar]
- 16.McAllister AK. Neurotrophins and cortical development. Results Probl Cell Differ. 2002;39:89–112. doi: 10.1007/978-3-540-46006-0_5. [DOI] [PubMed] [Google Scholar]
- 17.Pandya CD, Pillai A. Trkb interacts with erbb4 and regulates nrg1-induced nr2b phosphorylation in cortical neurons before synaptogenesis. Cell Commun Signal. 2014;12:47. doi: 10.1186/s12964-014-0047-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ninan I. Synaptic regulation of affective behaviors; role of bdnf. Neuropharmacology. 2014;76(Pt C):684–695. doi: 10.1016/j.neuropharm.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aid T, Kazantseva A, Piirsoo M, Palm K, Timmusk T. Mouse and rat bdnf gene structure and expression revisited. J Neurosci Res. 2007;85:525–535. doi: 10.1002/jnr.21139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang EJ, Reichardt LF. Trk receptors: Roles in neuronal signal transduction. Ann Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- 21.Tian X, Hua F, Sandhu HK, Chao D, Balboni G, Salvadori S, He X, Xia Y. Effect of delta-opioid receptor activation on bdnf-trkb vs. Tnf-alpha in the mouse cortex exposed to prolonged hypoxia. Int J Mol Sci. 2013;14:15959–15976. doi: 10.3390/ijms140815959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.von Arnim CA, Hellweg R, Buchner M, Huber R, Riepe MW. Regional selectivity of amyloid mrna expression and neurotrophins on repetitive inhibition of oxidative phosphorylation. J Neural Transm. 2005;112:491–498. doi: 10.1007/s00702-005-0277-5. [DOI] [PubMed] [Google Scholar]
- 23.Swerdlow NR, Geyer MA, Braff DL. Neural circuit regulation of prepulse inhibition of startle in the rat: Current knowledge and future challenges. Psychopharmacology (Berl) 2001;156:194–215. doi: 10.1007/s002130100799. [DOI] [PubMed] [Google Scholar]
- 24.Hill RA, van den Buuse M. Sex-dependent and region-specific changes in trkb signaling in bdnf heterozygous mice. Brain Res. 2011;1384:51–60. doi: 10.1016/j.brainres.2011.01.060. [DOI] [PubMed] [Google Scholar]
- 25.Manning EE, van den Buuse M. Bdnf deficiency and young-adult methamphetamine induce sex-specific effects on prepulse inhibition regulation. Front Cell Neurosci. 2013;7:92. doi: 10.3389/fncel.2013.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Naumenko VS, Bazovkina DV, Morozova MV, Popova NK. Effects of brain-derived and glial cell line-derived neurotrophic factors on startle response and disrupted prepulse inhibition in mice of dba/2j inbred strain. Neurosci Lett. 2013;550:115–118. doi: 10.1016/j.neulet.2013.06.056. [DOI] [PubMed] [Google Scholar]
- 27.Grottick AJ, Bagnol D, Phillips S, McDonald J, Behan DP, Chalmers DT, Hakak Y. Neurotransmission- and cellular stress-related gene expression associated with prepulse inhibition in mice. Brain Res Mol Brain Res. 2005;139:153–162. doi: 10.1016/j.molbrainres.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 28.Burman MA, Gewirtz JC. Timing of fear expression in trace and delay conditioning measured by fear-potentiated startle in rats. Learn Mem. 2004;11:205–212. doi: 10.1101/lm.66004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chavez-Valdez R, Martin LJ, Razdan S, Gauda EB, Northington FJ. Sexual dimorphism in bdnf signaling after neonatal hypoxia-ischemia and treatment with necrostatin-1. Neuroscience. 2014;260:106–119. doi: 10.1016/j.neuroscience.2013.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lehmann AE, Ennis K, Georgieff MK, Rao R, Tran PV. Evidence for a hyporesponsive limbic-hypothalamic-pituitary-adrenal axis following early-life repetitive hypoglycemia in adult male rats. Am J Physiol Regul Integr Comp Physiol. 2011;301:R484–490. doi: 10.1152/ajpregu.00678.2010. [DOI] [PubMed] [Google Scholar]
- 31.Lucas A, Morley R, Cole TJ. Adverse neurodevelopmental outcome of moderate neonatal hypoglycaemia. BMJ. 1988;297:1304–1308. doi: 10.1136/bmj.297.6659.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duvanel CB, Fawer CL, Cotting J, Hohlfeld P, Matthieu JM. Long-term effects of neonatal hypoglycemia on brain growth and psychomotor development in small-for-gestational-age preterm infants. J Pediatr. 1999;134:492–498. doi: 10.1016/s0022-3476(99)70209-x. [DOI] [PubMed] [Google Scholar]
- 33.Langan SJ, Deary IJ, Hepburn DA, Frier BM. Cumulative cognitive impairment following recurrent severe hypoglycaemia in adult patients with insulin-treated diabetes mellitus. Diabetologia. 1991;34:337–344. doi: 10.1007/BF00405006. [DOI] [PubMed] [Google Scholar]
- 34.Pisansky MT, Wickham RJ, Su J, Fretham S, Yuan LL, Sun M, Gewirtz JC, Georgieff MK. Iron deficiency with or without anemia impairs prepulse inhibition of the startle reflex. Hippocampus. 2013;23:952–962. doi: 10.1002/hipo.22151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gewirtz JC, Davis M. Habituation of prepulse inhibition of the startle reflex using an auditory prepulse close to background noise. Behav Neurosci. 1995;109:388–395. doi: 10.1037//0735-7044.109.3.388. [DOI] [PubMed] [Google Scholar]
- 36.Alder J, Thakker-Varia S, Bangasser DA, Kuroiwa M, Plummer MR, Shors TJ, Black IB. Brain-derived neurotrophic factor-induced gene expression reveals novel actions of vgf in hippocampal synaptic plasticity. J Neurosci. 2003;23:10800–10808. doi: 10.1523/JNEUROSCI.23-34-10800.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeSteno DA, Schmauss C. Induction of early growth response gene 2 expression in the forebrain of mice performing an attention-set-shifting task. Neuroscience. 2008;152:417–428. doi: 10.1016/j.neuroscience.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murk K, Wittenmayer N, Michaelsen-Preusse K, Dresbach T, Schoenenberger CA, Korte M, Jockusch BM, Rothkegel M. Neuronal profilin isoforms are addressed by different signalling pathways. PLoS One. 2012;7:e34167. doi: 10.1371/journal.pone.0034167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tkacs NC, Pan Y, Raghupathi R, Dunn-Meynell AA, Levin BE. Cortical fluoro-jade staining and blunted adrenomedullary response to hypoglycemia after noncoma hypoglycemia in rats. J Cereb Blood Flow Metab. 2005;25:1645–1655. doi: 10.1038/sj.jcbfm.9600152. [DOI] [PubMed] [Google Scholar]
- 40.Teves D, Videen TO, Cryer PE, Powers WJ. Activation of human medial prefrontal cortex during autonomic responses to hypoglycemia. Proc Natl Acad Sci U S A. 2004;101:6217–6221. doi: 10.1073/pnas.0307048101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Teh MM, Dunn JT, Choudhary P, Samarasinghe Y, Macdonald I, O’Doherty M, Marsden P, Reed LJ, Amiel SA. Evolution and resolution of human brain perfusion responses to the stress of induced hypoglycemia. Neuroimage. 2010;53:584–592. doi: 10.1016/j.neuroimage.2010.06.033. [DOI] [PubMed] [Google Scholar]
- 42.Rao R, Ennis K, Long JD, Ugurbil K, Gruetter R, Tkac I. Neurochemical changes in the developing rat hippocampus during prolonged hypoglycemia. J Neurochem. 2010;114:728–738. doi: 10.1111/j.1471-4159.2010.06797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sutherland GR, Tyson RL, Auer RN. Truncation of the krebs cycle during hypoglycemic coma. Med Chem. 2008;4:379–385. doi: 10.2174/157340608784872235. [DOI] [PubMed] [Google Scholar]
- 44.Jiang L, Herzog RI, Mason GF, de Graaf RA, Rothman DL, Sherwin RS, Behar KL. Recurrent antecedent hypoglycemia alters neuronal oxidative metabolism in vivo. Diabetes. 2009;58:1266–1274. doi: 10.2337/db08-1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dragunow M, Hughes P, Mason-Parker SE, Lawlor P, Abraham WC. Trkb expression in dentate granule cells is associated with a late phase of long-term potentiation. Brain Res Mol Brain Res. 1997;46:274–280. doi: 10.1016/s0169-328x(97)00021-1. [DOI] [PubMed] [Google Scholar]
- 46.Castren E, da Penha Berzaghi M, Lindholm D, Thoenen H. Differential effects of mk-801 on brain-derived neurotrophic factor mrna levels in different regions of the rat brain. Exp Neurol. 1993;122:244–252. doi: 10.1006/exnr.1993.1124. [DOI] [PubMed] [Google Scholar]
- 47.Greenwood BN, Strong PV, Foley TE, Thompson RS, Fleshner M. Learned helplessness is independent of levels of brain-derived neurotrophic factor in the hippocampus. Neuroscience. 2007;144:1193–1208. doi: 10.1016/j.neuroscience.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tran PV, Fretham SJ, Carlson ES, Georgieff MK. Long-term reduction of hippocampal brain-derived neurotrophic factor activity after fetal-neonatal iron deficiency in adult rats. Pediatr Res. 2009;65:493–498. doi: 10.1203/PDR.0b013e31819d90a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hurst P, Garfield AS, Marrow C, Heisler LK, Evans ML. Recurrent hypoglycemia is associated with loss of activation in rat brain cingulate cortex. Endocrinology. 2012;153:1908–1914. doi: 10.1210/en.2011-1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jahagirdar V, Ramcharitar J, Cotero VE, McNay EC. Moderate recurrent hypoglycemia markedly impairs set-shifting ability in a rodent model: Cognitive and neurochemical effects. Open Diabetes J. 2012;5:1–7. doi: 10.2174/1876524601205010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rao R. Hypothalamic-pituitary-adrenal axis programming after recurrent hypoglycemia during development. J Clin Med. 2015;4:1729–1740. doi: 10.3390/jcm4091729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cohen JL, Glover ME, Pugh PC, Fant AD, Simmons RK, Akil H, Kerman IA, Clinton SM. Maternal style selectively shapes amygdalar development and social behavior in rats genetically prone to high anxiety. Dev Neurosci. 2015;37:203–214. doi: 10.1159/000374108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kleiber ML, Laufer BI, Stringer RL, Singh SM. Third trimester-equivalent ethanol exposure is characterized by an acute cellular stress response and an ontogenetic disruption of genes critical for synaptic establishment and function in mice. Dev Neurosci. 2014;36:499–519. doi: 10.1159/000365549. [DOI] [PubMed] [Google Scholar]
- 54.Blaze J, Scheuing L, Roth TL. Differential methylation of genes in the medial prefrontal cortex of developing and adult rats following exposure to maltreatment or nurturing care during infancy. Dev Neurosci. 2013;35:306–316. doi: 10.1159/000350716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gourley SL, Kedves AT, Olausson P, Taylor JR. A history of corticosterone exposure regulates fear extinction and cortical nr2b, glur2/3, and bdnf. Neuropsychopharmacology. 2009;34:707–716. doi: 10.1038/npp.2008.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lipska BK, Swerdlow NR, Geyer MA, Jaskiw GE, Braff DL, Weinberger DR. Neonatal excitotoxic hippocampal damage in rats causes post-pubertal changes in prepulse inhibition of startle and its disruption by apomorphine. Psychopharmacology (Berl) 1995;122:35–43. doi: 10.1007/BF02246439. [DOI] [PubMed] [Google Scholar]
- 57.Merlio JP, Ernfors P, Kokaia Z, Middlemas DS, Bengzon J, Kokaia M, Smith ML, Siesjo BK, Hunter T, Lindvall O, et al. Increased production of the trkb protein tyrosine kinase receptor after brain insults. Neuron. 1993;10:151–164. doi: 10.1016/0896-6273(93)90307-d. [DOI] [PubMed] [Google Scholar]
- 58.McNay EC, Williamson A, McCrimmon RJ, Sherwin RS. Cognitive and neural hippocampal effects of long-term moderate recurrent hypoglycemia. Diabetes. 2006;55:1088–1095. doi: 10.2337/diabetes.55.04.06.db05-1314. [DOI] [PubMed] [Google Scholar]
- 59.McNay EC, Sherwin RS. Effect of recurrent hypoglycemia on spatial cognition and cognitive metabolism in normal and diabetic rats. Diabetes. 2004;53:418–425. doi: 10.2337/diabetes.53.2.418. [DOI] [PubMed] [Google Scholar]
- 60.Bischoff LG, Warzak WJ, Maguire KB, Corley KP. Acute and chronic effects of hypoglycemia on cognitive and psychomotor performance. Nebr Med J. 1992;77:253–262. [PubMed] [Google Scholar]
- 61.Northam EA, Rankins D, Cameron FJ. Therapy insight: The impact of type 1 diabetes on brain development and function. Nat Clin Pract Neurol. 2006;2:78–86. doi: 10.1038/ncpneuro0097. [DOI] [PubMed] [Google Scholar]
- 62.Tonoli C, Heyman E, Roelands B, Buyse L, Piacentini F, Berthoin S, Bailey S, Pattyn N, Meeusen R. Bdnf, igf-i, glucose and insulin during continuous and interval exercise in type 1 diabetes. Int J Sports Med. 2015 Jul 24; doi: 10.1055/s-0035-1548886. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.