

Abstract
The cysteine cathepsins are a group of 11 proteases whose function was originally believed to be the degradation of endocytosed material with a high degree of redundancy. However, it has become clear that these enzymes are also important regulators of both health and disease. Thus, selective tools that can discriminate between members of this highly related class of enzymes will be critical to further delineate the unique biological functions of individual cathepsins. Here we present the design and synthesis of a near-infrared quenched activity-based probe (qABP) that selectively targets cathepsin S which is highly expressed in immune cells. Importantly, this high degree of selectivity is retained both in vitro and in vivo. In combination with a new green-fluorescent pan-reactive cysteine cathepsin qABP we performed dual color labeling studies in bone marrow derived immune cells and identified vesicles containing exclusively cathepsin S activity. This observation demonstrates the value of our complimentary cathepsin probes and provides evidence for the existence of specific localization of cathepsin S activity in dendritic cells.
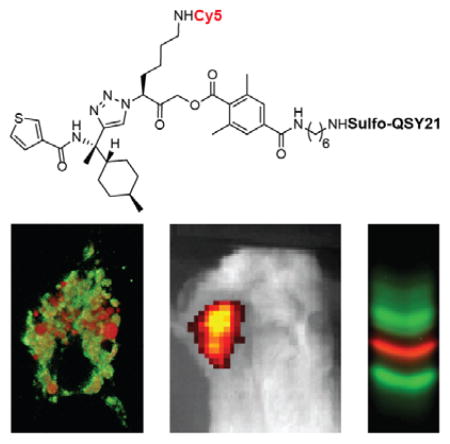
Graphical abstract
INTRODUCTION
Cysteine cathepsins are part of the C1a family of clan CA proteases.1 These include cathepsins B, L, S, F, H, O, W, X (also known as Z or P), C (or dipeptidyl-peptidase 1), K (or O2), and V (with the alternative name L2).2 While their main function was originally believed to be primarily the degradation of endocytosed and intracellular protein waste, it is now clear that cysteine cathepsins are important players in regulating aspects of both normal physiology and disease pathology. This includes involvement in cancer and inflammatory diseases such as rheumatoid arthritis, chronic obstructive pulmonary disease (COPD), inflammatory bowel disease, asthma, sepsis and cystic fibrosis. Although initially believed to be highly redundant, recent studies have identified specific functions for individual members of the cysteine cathepsin family. While the majority of the cysteine cathepsins demonstrate a ubiquitous expression profile, cathepsin S expression is mainly restricted to antigen-presenting cells (APCs) where it contributes to antigen presenting capacity by controlling trafficking and maturation of major histocompatibility complex (MHC) class II molecules.3 This includes professional APCs such as dendritic cells, macrophages and B cells4 as well as nonprofessional APCs, such as intestinal epithelial cells.5 Although primarily localized to the endolysomal system, cathepsin S has been linked to functions outside the cell, where it contributes to matrix degradation6 and dendritic cell motility.7 Furthermore, an increase in cathepsin S expression is associated with several diseases that are characterized by the infiltration of myeloid-derived immune cell types.
In mouse models of cancer, cathepsin S activity within the tumor microenvironment is primarily supplied by tumor-associated macrophages.8, 9 Therefore, cathepsin S is expected to be associated with any cancer with macrophage infiltration. However, cancer cell derived cathepsin S also contributes to neovascularization and tumor growth10, 11 and depletion or therapeutic inhibition of cathepsin S in both breast cancer and stromal cells significantly reduced metastasis specifically to the brain.11 This finding was also linked to clinical outcome in breast cancer patients, where high cathepsin S expression at the primary site correlated with decreased brain metastasis-free survival. A recent study revealed cathepsin S as a major player in tumor-initiating cell development12 and the finding that cathepsin S is required for pro-tumor, M2 polarization of tumor-associated macrophages highlights yet another mechanism by which cathepsin S impacts tumor development and clinical outcome. 13
In addition to cancer, cathepsin S has recently been linked to pathology of TRPV4-mediated inflammatory pain through proteolytic cleavage of PAR213, 14 and cystic fibrosis where increased cathepsin S activity is found in the patients’ bronchoalveolar lavage (BAL) fluid.15 Although it is clear that cathepsin S is a highly relevant clinical biomarker and potential therapeutic target, many of the biological and molecular questions regarding the contribution of cathepsin S to this wide array of pathologies remain to be answered. Most of the current mechanistic knowledge of cathepsin S is based on studies with genetic knockout animal models. However, knockout of one cathepsin has been shown to influence the expression of other members of the family.16–18 Furthermore, proteases often function in enzymatic networks,19 complicating direct assessment of protease function from genetic knockout data. To get a better understanding of the function, dynamics and localization of cathepsin S activity, there is a need for tools that allow direct assessment of activity levels in living cells and in complex animal models.
One approach to tracking and imaging protease activity is to use substrate reporters that are cleaved by a protease to produce a fluorescent signal. These types of fluorogenic substrate probes have been developed for in vivo assessment of cathepsin S activity. In one example an auto-quenched dendrimeric structure equipped with a protease cleavable site (Leu-Arg)20 was used to determine the role of cathepsin S in arterial and aortic valve calcification in a mouse model of chronic renal disease.21 In another example, a strategy termed ‘reverse design’ was used to generate small molecule fluorogenic substrate probes based on the structure of cathepsin S selective inhibitors. These substrates were then used for noninvasive imaging of inflammation and tumor margins.22, 23 Although such substrate probes are useful tools for the assessment of protease activity, they do not provide precise information about the localization of the target protease as the cleavage products are imaged rather than the protease that produced them. Furthermore, it remains difficult to confirm the in vivo selectivity of most substrates as many proteases may be capable of processing any given peptide substrate.
As an alternative to substrate based probes, activity-based probes (ABPs) are small molecule reporters of enzymatic activity that are designed to covalently and irreversibly attach to the active-site nucleophile of the target enzyme in a mechanism dependent manner.24–27 One sub-family of ABPs are the quenched activity-based probe (qABPs), which are intrinsically fluorescently quenched or “dark” but fluorescently label the target upon mechanism-based nucleophilic displacement of the quencher group.28 This trait makes qABPs optimal for live cell and noninvasive optical imaging and for the quantitative assessment of proteolytically active protease pools in live cells. Importantly, because qABPs covalently and irreversibly label the target protease, probe selectivity can be validated biochemically.
Recently, a coumarin-labeled fluorescent vinyl sulfone ABP was reported to have cathepsin S selectivity in tissue homogenates29 and a cell-permeable, radioiodinated, irreversible diazomethyl ketone containing ABP was used to probe cathepsin S activity in whole blood.30 However the overall selectivity of these probes remains questionable as they were shown to label other cathepsin targets in certain cell populations. Thus, more selective probes will be required to begin to analyze specific localization of cathepsin S activity in live cells and organisms.
In this work, we describe the design, synthesis and characterization of a potent, cathepsin S-selective near-infrared qABP. Our non-peptidic qABP proved to be highly selective for cathepsin S in cells as well as in various mouse organs in vivo and we were able to noninvasively image syngeneic mammary tumors in living mice. Furthermore, by synthesizing a complementary green-fluorescent pan-reactive qABP we were able to perform dual-color live cell activity localization studies to define the location of proteolytically active cathepsin S in vesicular compartments in primary mouse bone marrow-derived dendritic cells (BMDCs). The exact mechanisms and pathways by which endolysosmal proteolysis impacts antigen presentation, signal transduction and the interplay between the different proteases and other enzymes remain only partly understood.31 We believe that the chemical tools presented here will help to answer some of these remaining questions.
RESULTS AND DISCUSSION
Probe Synthesis and In Vitro Characterization
For the development of a cathepsin S selective qABP, we started with the scaffold from our recently reported non-peptidic qABP BMV083 (Figure 1a),8 which has improved in vivo properties compared to the peptidic qABP GB137.32 Substrate activity screening (SAS) efforts demonstrated that improved cathepsin S selectivity can be obtained by increasing the bulkiness of the P2 substrate substituent.33 We therefore designed the qABP BMV117 (Figure 1a) containing a trans-4-methylcyclohexyl at the P2 position of the probe (synthetic details can be found in the Supporting Information). Indeed, when we compared the labeling profiles of the prior probe BMV083 to the new BMV117 probe in live RAW 264.7 cells (mouse leukemic monocyte macrophage cell line) we found that BMV117 showed exclusive labeling of cathepsin S, whereas BMV083 also labeled a substantial amount of cathepsin B and, at higher concentrations, cathepsin L (Figure 1b and Figure S1A).8 However, the gain in selectivity by introducing increased steric bulk in the P2 position came at the cost of a 10-fold decrease in potency towards cathepsin S. We recently reported the optimized near-infrared pan-reactive cysteine cathepsin qABP BMV109 that contains a more reactive phenoxymethyl ketone (PMK) electrophile (also known as the “warhead”), a shorter spacer and a more hydrophilic sulfo-QSY21 quencher. This probe targets a broad panel of cysteine cathepsin targets and has better aqueous solubility and improved in vivo properties compared to the previous generation acyloxymethyl ketone (AOMK) probes.34 In an effort to increase the potency of BMV117, while retaining the remarkable cathepsin S selectivity, we synthesized three analogues by introducing the sulfo-QSY21 in place of the more hydrophobic QSY21 (BMV157), incorporating the tetrafluoro-substituted PMK electrophile (LO219) and shortening the spacer tethering the quencher to the electrophile (LO143; Figure 1a). We compared the labeling profile of each of the new probes to the pan-reactive qABP BMV109 and the lead cathepsin S selective qABP BMV117 in intact RAW 264.7 cells (Figure 1c). Substituting the QSY21 quencher in BMV117 for sulfo-QSY21 resulted in a dramatic increase in potency toward cathepsin S, while retaining extremely high cathepsin S selectivity (Figure 1c and Figure S1B–D). The labeling profiles of LO219 and LO143 revealed that introduction of the PMK electrophile and the combination with a shorter spacer reduces cathepsin S selectivity. This suggests that the more bulky nature of the 2,6-dimethylbenzoic acid-derived AOMK electrophile, in combination with the optimal trans-4-methylcyclohexyl at the P2 position and the hydrophilic sulfo-QSY21 may contribute to the superior cathepsin S selectivity and potency of BMV157. Thus, BMV157 represents our most potent and selective cathepsin S probe to date, based on cellular studies. We therefore moved forward with applications in vivo to further assess its selectivity.
Figure 1. The development of a cathepsin S selective qABP.

A) Structures of the qABP BMV083 and the novel probes synthesized and characterized in this work. B) Concentration-dependent labeling profile of BMV083 compared to BMV117 in live RAW cells. C) Concentration-dependent labeling profile of BMV109 compared to BMV117, BMV157, LO219 and LO143 in live RAW cells.
Noninvasive Optical Imaging of Syngeneic Orthotopic Mouse Breast Tumors
Given the fact that tumor-associated macrophages are the key contributors of cathepsin S in the tumor microenvironment,8, 9 we set out to characterize the tumor imaging capacity, the bio-distribution and cathepsin S-selectivity of BMV157 in a syngeneic orthotopic mouse model of breast cancer.35 10 days after tumor implantation, we injected mice with equimolar amounts of BMV157 or BMV109 (20 nmol) via the tail vein and then performed noninvasive optical imaging of Cy5 fluorescence eight hours post injection. This data confirmed that qABP BMV157 was able to delineate tumor margins with substantial contrast (Figure 2a).
Figure 2. In vivo characterization of the cathepsin S selective qABP BMV157 in a syngeneic orthotopic mouse breast cancer model.

A) Noninvasive optical imaging of 4T1 tumor-bearing Balb/c mice injected with BMV157. B) Ex vivo tumor fluorescence (top panel) and in vivo fluorescently labeled tumor-associated proteins visualized after SDS-PAGE by in-gel fluorescence scanning (lower panel) of tumor-bearing mice treated with vehicle, BMV157 or BMV109. C) Quantification of tumor cysteine cathepsin labeling intensity of the ex vivo IVIS measurements and fluorescence SDS-PAGE. D) In vivo tissue specific labeling profile of BMV157 compared to the pan-reactive qABP BMV109 visualized after SDS-PAGE by in-gel fluorescence scanning.
After live-animal imaging, we measured tumor fluorescence ex vivo, followed by analysis of the fluorescently labeled proteins by SDS-PAGE (Figure 2b). Analysis of tumor fluorescence intensity ex vivo showed not surprisingly that the BMV109 fluorescence was on average ~6 times brighter than the signal for the cathepsin S specific probe BMV157 (Figures 2b and 2c). This difference in intensity was attributed to the fact that BMV109 labeled cathepsins X, B, S and L,34 while BMV157 exclusively labeled cathepsin S, consistent with the high selectivity of this qABP observed in cells (Figure 2c). We also analyzed liver, kidney, spleen, heart and lung tissues and labeling profiles showed similar bio-distribution for the pan-reactive and the cathepsin S selective qABPs (Figure 2d and S2). In all cases, whole organ fluorescence intensities correlated well with total cysteine cathepsin labeling intensities determined by SDS-PAGE (Figure S2c). Most importantly, BMV157 showed remarkable cathepsin S selectivity in all organs analyzed, even in organs with relatively low cathepsin S activity compared to cathepsin B and L, such as the liver and kidney. The selectivity of any probe, inhibitor or substrate based reporter depends highly on concentrations used, labeling time, the relative activity levels of the (off-)targets and the type of proteome, with in vivo experiments being the most challenging.
Dual-Color Live Cell Cysteine Cathepsin Activity Imaging
Since cysteine cathepsin activity (and protease activity in general) is highly regulated by environmental cues, proteolytic activation and endogenous inhibitors, the assessment of protein levels and localization does not provide direct evidence for proteolytic activity. Specific cysteine cathepsin functions may be reflected by distinct localization of activity. Specific compartmentalization of cathepsin S has been reported in the human thyroid gland, where it is localized to distinct vesicular compartments lacking cathepsins B, K and L based on immunostaining.36 Unlike most other cysteine cathepsins that are optimally active at acidic pH and mostly inactive at neutral pH, cathepsin S possesses activity over a broad pH range, retaining most of its activity at neutral and even slightly alkaline pH.37 This means that even when localized to the same sub-cellular location, levels of activity of the different cathepsin family members is controlled by local environmental factors such as pH, redox potential and the specific localization of inhibiting factors. To image the localization of cathepsin S activity with respect to the activity of other members of the cysteine cathepsin family, we synthesized a complementary green-fluorescent qABP based on the broad-spectrum BMV109 probe scaffold (EM053, Figure 3a). We found that the combination of the green fluorescent BODIPY-FL and the quencher BHQ-10 gave the most optimal quenching efficiency. Labeling live primary human monocyte-derived dendritic cells with increasing concentrations of EM053 confirmed a dose dependent labeling of multiple cysteine cathepsin targets, including cathepsin X, B, S and L, which was similar to the labeling profile observed with the pan-reactive qABP BMV109 (Figure 3b, lane 1,2 and 8). We next aimed to specifically label proteolytically active cathepsin S with BMV157 (Cy5) and simultaneously label the remaining active members with EM053 (BODIPY-FL). Titration of human monocyte-derived dendritic cells with BMV157 followed by immediate addition of EM053 resulted in concentration dependent Cy5 labeling of cathepsin S and concomitant competition of the BODIPY FL labeling of cathepsin S by EM053 (Figure 3b, lanes 3–6). The Cy5 cathepsin S labeling as well as the EM053 cathepsin S competition saturated at a concentration of 5 μM of BMV157 (Figure 3b, lane 4).
Figure 3. Complementary cysteine cathepsin labeling.

A) Structures of the pan-reactive cysteine cathepsin qABPs BMV109 and EM053. B) Labeling profile and simultaneous multi-color labeling of cathepsin S with the near infra-red labeled BMV157 and the green pan-reactive EM053 in living human monocyte-derived dendritic cells.
We therefore used this optimized labeling condition to perform multispectral live cell imaging experiments to determine the spatial and quantitative distribution of cathepsin S activity relative to the activity of other members of the cysteine cathepsin family, such as cathepsin X, B and L in primary mouse bone marrow-derived macrophages (BMMs) and BMDCs. Co-labeling of the cells with BMV157 and EM053 for 2 hours prior to imaging resulted in robust Cy5 and BODIPY FL labeling primarily in vesicular compartments throughout the cells (Figure 4 and Figure S3). Pre-incubation of the cells with the pan-cysteine cathepsin inhibitor JPM-OEt abolished all fluorescence activation thereby confirming the activity of cysteine cathepsins as the source of the fluorescence signals (Figure S3). The slight heterogeneity observed in fluorescence intensity upon qABP labeling of the BMDCs and BMMs generated with the in vitro bone marrow differentiation methods confirms the heterogeneity described for these two experimental cell populations (Figure S3).38 To be able to perform multispectral imaging on living cells, each fluorophore must be detected simultaneously, since the motility of the vesicular compartments containing the majority of the labeled cysteine cathepsins is very high (Video S1). In the labeled BMMs analyzed, cathepsin S activity co-localized with the activity of the cysteine cathepsins labeled with EM053, although the relative cathepsin S activity per vesicle varies (Figure 4a). After completion of the imaging experiment, we harvested the cells, lysed them and determined protein content by fluorescent SDS-PAGE (Figure 4b). This confirmed the selective labeling of cathepsin S by BMV157 and revealed cathepsins X, B and L as the primary targets of EM053. We obtained similar results when BMDCs were imaged, with one major difference: we observed vesicular compartments that had highly elevated and seemingly exclusive cathepsin S activity (Figure 4c,d). Macrophages and dendritic cells (DCs) have been shown to have differences in the quantity and activity of proteolytic enzymes in their endolysosomal pathways35, as well as differences in pH regulation resulting in overall less acidic endolysosomal environments in DCs.39 Indeed, cysteine cathepsin activity profiling with BMV109 showed higher activity of cathepsins B and L in BMMs compared to BMDCs (Figure S4). Contrary to the reported major difference in expression levels of cathepsin S40, we find little difference in cathepsin S activity between the two experimental immune cell types. Recently it was shown that individual phagosomes behave autonomously, with respect to cargo degradation and antigen presentation as a result of differential phagosome maturation.41 This finding could explain the differential cathepsin compartmentalization in specialized vesicular compartments. The confirmation and further understanding of this phenomenon will be the basis for further investigation. Overall, these observations highlight the potential of the near-infrared cathepsin S selective qABP in combination with complementary pan-reactive cysteine cathepsin qABPs for activity localization studies in living cells and animal models.
Figure 4. Live cell co-localization of cathepsin S and other cysteine cathepsin activity using the novel complementary set of qABPs.

A) Live cell confocal microscopy and B) biochemical characterization of BMV157 (5 μM) and EM053 (5 μM) co-treated mouse bone marrow-derived macrophages. C, D) Confocal microscopy of mouse bone marrow-derived dendritic cells exposed to BMV157 (5 μM) and EM053 (5 μM). Three-dimensional reconstruction of confocal microscopy images of BMV157 labeled cathepsin S (red) and pan-cathepsin labeling with EM053 (green) (C). Orthogonal projection of the confocal images showing co-localization and lack thereof of BMV157 and EM053 labeled cathepsins (D). Scale bar 10μm.
CONCLUSIONS
The cysteine cathepsin family members, once thought to only be responsible for degradation within lysosomal compartments, are slowly revealing their diverse functional roles in numerous physiological processes. As their role in early development and progression of various human pathologies is becoming more evident, the cysteine cathepsins are increasingly being validated as viable therapeutic targets. Due to its distinctive expression profile and direct involvement in antigen presentation, cathepsin S has attracted significant attention from scientists, both in the pharmaceutical industry and in academia. In order to investigate the mechanisms important for cathepsin S-mediated pathology, reliable molecular tools that can monitor cathepsin S activity are needed. These tools are important not only to demonstrate the value of cathepsin S activity for diagnosis and as a biomarker for disease progression, but also to develop and characterize novel therapeutic inhibitors or disease modulators. Several cathepsin S inhibitors are currently in various phases of clinical trials.42 (Eli Lilly Inc., Virobay Inc.) Building on our experience in the design and synthesis of cysteine protease ABPs, we describe here a highly selective, non-peptidic cathepsin S near-infrared qABP BMV157. This probe can be used for sensitive, selective and quantitative assessment of cathepsin S activity in cells and even in whole organisms. Complementary to this near-infrared cathepsin S selective imaging tool, we synthesized and characterized a green-fluorescent pan-cysteine cathepsin qABP EM053, which we used in combination with BMV157 to visualize localization of cathepsin S activity relative to the activity of cathepsin X, B and L in living cells. Importantly, the power of our complimentary chemical toolkit was demonstrated by the observation that, unlike BMMs, BMDCs contain a pool of vesicles that exclusively possess cathepsin S activity. This work therefore provides valuable tools that can be used to determine the characteristics of these distinct sub-cellular compartments. While it remains unclear how and why such cathepsin S specific vesicles exist, they may play important roles in dendritic cell immunology and provide insight into how this professional APC functions in vivo. Overall, our results highlight the potential of the near-infrared cathepsin S selective qABP and its combination with complementary pan-reactive cysteine cathepsin qABPs for in depth activity localization studies.
Supplementary Material
Acknowledgments
This work was supported by NIH grants R01 EB005011 R01 HL116307 (to M.B.), The Netherlands Organization for Scientific Research (NWO) Rubicon fellowship (to M.V. and W.A.v.d.L.), NIH grant R01 GM054051 (to J.A.E.), and NIH Re-entry into Biomedical Sciences Supplement 3R01EB005011-06S1 (to K.O.B). C.G.F is recipient of an NWO Spinoza award, ERC advanced grant PATHFINDER (269019) and KWO award KUN2009-4402 from the Dutch Cancer Society. We thank the members of the Bogyo lab and the Figdor lab for insightful discussions, T. Doyle at the Stanford Small Animal Facility and S. Lynch at the Stanford NMR Facility. We thank Ben Joosten for assistance with CLSM, Josh Lichtman and Josh Elias for recording HRMS data, A. Chien and T. McLaughlin at the Stanford Mass Spectrometry Facility for their technical assistance.
Footnotes
The authors declare no competing financial interest.
Experimental details of the synthesis of all probes and biological experiments, supplemental figures and NMR spectra.
References
- 1.Rawlings ND, Barrett AJ. Nucleic Acids Res. 1999;27:325–331. doi: 10.1093/nar/27.1.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turk B, Turk D, Turk V. EMBO J. 2012;31:1630–1643. doi: 10.1038/emboj.2012.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Conus S, Simon HU. Swiss Med Wkly. 2010;140:w13042. doi: 10.4414/smw.2010.13042. [DOI] [PubMed] [Google Scholar]
- 4.Driessen C, Bryant RA, Lennon-Dumenil AM, Villadangos JA, Bryant PW, Shi GP, Chapman HA, Ploegh HL. J Cell Biol. 1999;147:775–790. doi: 10.1083/jcb.147.4.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beers C, Burich A, Kleijmeer MJ, Griffith JM, Wong P, Rudensky AY. J Immunol. 2005;174:1205–1212. doi: 10.4049/jimmunol.174.3.1205. [DOI] [PubMed] [Google Scholar]
- 6.Fonovic M, Turk B. Biochim Biophys Acta. 2014;1840:2560–2570. doi: 10.1016/j.bbagen.2014.03.017. [DOI] [PubMed] [Google Scholar]
- 7.Faure-Andre G, Vargas P, Yuseff MI, Heuze M, Diaz J, Lankar D, Steri V, Manry J, Hugues S, Vascotto F, Boulanger J, Raposo G, Bono MR, Rosemblatt M, Piel M, Lennon-Dumenil AM. Science. 2008;322:1705–1710. doi: 10.1126/science.1159894. [DOI] [PubMed] [Google Scholar]
- 8.Verdoes M, Edgington LE, Scheeren FA, Leyva M, Blum G, Weiskopf K, Bachmann MH, Ellman JA, Bogyo M. Chem Biol. 2012;19:619–628. doi: 10.1016/j.chembiol.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gocheva V, Wang HW, Gadea BB, Shree T, Hunter KE, Garfall AL, Berman T, Joyce JA. Genes Dev. 2010;24:241–255. doi: 10.1101/gad.1874010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Small DM, Burden RE, Jaworski J, Hegarty SM, Spence S, Burrows JF, McFarlane C, Kissenpfennig A, McCarthy HO, Johnston JA, Walker B, Scott CJ. Int J Cancer. 2013;133:2102–2112. doi: 10.1002/ijc.28238. [DOI] [PubMed] [Google Scholar]
- 11.Sevenich L, Bowman RL, Mason SD, Quail DF, Rapaport F, Elie BT, Brogi E, Brastianos PK, Hahn WC, Holsinger LJ, Massague J, Leslie CS, Joyce JA. Nat Cell Biol. 2014;16:876–888. doi: 10.1038/ncb3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee TK, Cheung VC, Lu P, Lau EY, Ma S, Tang KH, Tong M, Lo J, Ng IO. Hepatology. 2014;60:179–191. doi: 10.1002/hep.27070. [DOI] [PubMed] [Google Scholar]
- 13.Yang M, Liu J, Shao J, Qin Y, Ji Q, Zhang X, Du J. Mol Cancer. 2014;13:43-4598-13-43. doi: 10.1186/1476-4598-13-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao P, Lieu T, Barlow N, Metcalf M, Veldhuis NA, Jensen DD, Kocan M, Sostegni S, Haerteis S, Baraznenok V, Henderson I, Lindstrom E, Guerrero-Alba R, Valdez-Morales EE, Liedtke W, McIntyre P, Vanner SJ, Korbmacher C, Bunnett NW. J Biol Chem. 2014;289:27215–27234. doi: 10.1074/jbc.M114.599712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weldon S, McNally P, McAuley DF, Oglesby IK, Wohlford-Lenane CL, Bartlett JA, Scott CJ, McElvaney NG, Greene CM, McCray PB, Jr, Taggart CC. Am J Respir Crit Care Med. 2014;190:165–174. doi: 10.1164/rccm.201311-1986OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sevenich L, Schurigt U, Sachse K, Gajda M, Werner F, Muller S, Vasiljeva O, Schwinde A, Klemm N, Deussing J, Peters C, Reinheckel T. Proc Natl Acad Sci U S A. 2010;107:2497–2502. doi: 10.1073/pnas.0907240107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gopinathan A, Denicola GM, Frese KK, Cook N, Karreth FA, Mayerle J, Lerch MM, Reinheckel T, Tuveson DA. Gut. 2012;61:877–884. doi: 10.1136/gutjnl-2011-300850. [DOI] [PubMed] [Google Scholar]
- 18.Tamhane T, Arampatzidou M, Gerganova V, Tacke M, Illukkumbura R, Dauth S, Schaschke N, Peters C, Reinheckel T, Brix K. Biol Chem. 2014;395:1201–1219. doi: 10.1515/hsz-2014-0151. [DOI] [PubMed] [Google Scholar]
- 19.Mason SD, Joyce JA. Trends Cell Biol. 2011;21:228–237. doi: 10.1016/j.tcb.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galande AK, Hilderbrand SA, Weissleder R, Tung CH. J Med Chem. 2006;49:4715–4720. doi: 10.1021/jm051001a. [DOI] [PubMed] [Google Scholar]
- 21.Aikawa E, Aikawa M, Libby P, Figueiredo JL, Rusanescu G, Iwamoto Y, Fukuda D, Kohler RH, Shi GP, Jaffer FA, Weissleder R. Circulation. 2009;119:1785–1794. doi: 10.1161/CIRCULATIONAHA.108.827972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caglic D, Globisch A, Kindermann M, Lim NH, Jeske V, Juretschke HP, Bartnik E, Weithmann KU, Nagase H, Turk B, Wendt KU. Bioorg Med Chem. 2011;19:1055–1061. doi: 10.1016/j.bmc.2010.10.028. [DOI] [PubMed] [Google Scholar]
- 23.Hu HY, Vats D, Vizovisek M, Kramer L, Germanier C, Wendt KU, Rudin M, Turk B, Plettenburg O, Schultz C. Angew Chem Int Ed Engl. 2014;53:7669–7673. doi: 10.1002/anie.201310979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Serim S, Haedke U, Verhelst SH. Chem Med Chem. 2012;7:1146–1159. doi: 10.1002/cmdc.201200057. [DOI] [PubMed] [Google Scholar]
- 25.Kam CM, Abuelyaman AS, Li Z, Hudig D, Powers JC. Bioconjug Chem. 1993;4:560–567. doi: 10.1021/bc00024a021. [DOI] [PubMed] [Google Scholar]
- 26.Liu Y, Patricelli MP, Cravatt BF. Proc Natl Acad Sci U S A. 1999;96:14694–14699. doi: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greenbaum D, Medzihradszky KF, Burlingame A, Bogyo M. Chem Biol. 2000;7:569–581. doi: 10.1016/s1074-5521(00)00014-4. [DOI] [PubMed] [Google Scholar]
- 28.Edgington LE, Verdoes M, Bogyo M. Curr Opin Chem Biol. 2011;15:798–805. doi: 10.1016/j.cbpa.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mertens MD, Schmitz J, Horn M, Furtmann N, Bajorath J, Mares M, Gutschow M. Chem biochem. 2014;15:955–959. doi: 10.1002/cbic.201300806. [DOI] [PubMed] [Google Scholar]
- 30.Veilleux A, Black WC, Gauthier JY, Mellon C, Percival MD, Tawa P, Falgueyret JP. Anal Biochem. 2011;411:43–49. doi: 10.1016/j.ab.2010.11.022. [DOI] [PubMed] [Google Scholar]
- 31.van Kasteren SI, Overkleeft HS. Curr Opin Chem Biol. 2014;23C:8–15. doi: 10.1016/j.cbpa.2014.08.011. [DOI] [PubMed] [Google Scholar]
- 32.Blum G, von Degenfeld G, Merchant MJ, Blau HM, Bogyo M. Nat Chem Biol. 2007;3:668–677. doi: 10.1038/nchembio.2007.26. [DOI] [PubMed] [Google Scholar]
- 33.Patterson AW, Wood WJ, Hornsby M, Lesley S, Spraggon G, Ellman JA. J Med Chem. 2006;49:6298–6307. doi: 10.1021/jm060701s. [DOI] [PubMed] [Google Scholar]
- 34.Verdoes M, Oresic Bender K, Segal E, van der Linden WA, Syed S, Withana NP, Sanman LE, Bogyo M. J Am Chem Soc. 2013;135:14726–14730. doi: 10.1021/ja4056068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lelekakis M, Moseley JM, Martin TJ, Hards D, Williams E, Ho P, Lowen D, Javni J, Miller FR, Slavin J, Anderson RL. Clin Exp Metastasis. 1999;17:163–170. doi: 10.1023/a:1006689719505. [DOI] [PubMed] [Google Scholar]
- 36.Jordans S, Jenko-Kokalj S, Kuhl NM, Tedelind S, Sendt W, Bromme D, Turk D, Brix K. BMC Biochem. 2009;10:23-2091-10-23. doi: 10.1186/1471-2091-10-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kirschke H, Wiederanders B, Bromme D, Rinne A. Biochem J. 1989;264:467–473. doi: 10.1042/bj2640467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Masurier C, Pioche-Durieu C, Colombo BM, Lacave R, Lemoine FM, Klatzmann D, Guigon M. Immunology. 1999;96:569–577. doi: 10.1046/j.1365-2567.1999.00728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, Lennon-Dumenil AM, Seabra MC, Raposo G, Amigorena S. Cell. 2006;126:205–218. doi: 10.1016/j.cell.2006.05.035. [DOI] [PubMed] [Google Scholar]
- 40.Delamarre L, Pack M, Chang H, Mellman I, Trombetta ES. Science. 2005;307:1630–1634. doi: 10.1126/science.1108003. [DOI] [PubMed] [Google Scholar]
- 41.Hoffmann E, Kotsias F, Visentin G, Bruhns P, Savina A, Amigorena S. Proc Natl Acad Sci U S A. 2012;109:14556–14561. doi: 10.1073/pnas.1203912109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Payne CD, Deeg MA, Chan M, Tan LH, LaBell ES, Shen T, DeBrota DJ. Br J Clin Pharmacol. 2014;78:1334–1342. doi: 10.1111/bcp.12470. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.