

Abstract
Atherosclerosis is the leading cause of morbidity and mortality in the U.S., and is a multifactorial disease that preferentially occurs in regions of the arterial tree exposed to disturbed blood flow. The detailed mechanisms by which d-flow induces atherosclerosis involve changes in the expression of genes, epigenetic patterns, and metabolites of multiple vascular cells, especially endothelial cells. This review presents an overview of endothelial mechanobiology and its relation to the pathogenesis of atherosclerosis with special reference to the anatomy of the artery and the underlying fluid mechanics, followed by a discussion of a variety of experimental models to study the role of fluid mechanics and atherosclerosis. Various in vitro and in vivo models to study the role of flow in endothelial biology and pathobiology are discussed in this review. Furthermore, strategies used for the global profiling of the genome, transcriptome, miRNA-nome, DNA methylome, and metabolome, as they are important to define the biological and pathophysiological mechanisms of atherosclerosis. These “omics” approaches, especially those which derive data based on a single animal model, provide unprecedented opportunities to not only better understand the pathophysiology of atherosclerosis development in a holistic and integrative manner, but also to identify novel molecular and diagnostic targets.
Keywords: Mechanosensitive, blood flow, shear stress, endothelial cells, atherosclerosis, genomics, epigenomics, miRNomics, metabolomics
Graphical abstract
1 Introduction and Overview
Although the physical transduction of force to macroscopic objects has been studied exhaustively, the study of the interplay between physical forces and living cells, known as mechanobiology, is relatively young. Furthermore, although the forces at work in the biological system are much more subtle than in more traditionally studied systems, the responses to these forces are arguably more complex. This review focuses specifically on the effects of one particular force, fluid shear stress imposed by blood flow, on the vascular endothelium in the cardiovascular system and its effects on the pathology of atherosclerosis. Finally, this review aims to present how techniques such as “omics” can be used to translate our knowledge of mechanobiology into specific targets that can be developed into therapeutics for atherosclerosis.
2. Vascular Fluid Mechanics and Disease
2.1. Hemodynamics in the arteries
The cardiovascular system is comprised of a heart pump, a low-pressure venous system which brings blood back to the heart, and a high-pressure arterial system which supplies blood to the body’s tissues. The arterial system is divided between the lower pressure pulmonary circulation to the lungs and the higher pressure systemic arterial system flowing from the left ventricle of the heart to the aorta and out to the rest of the body. This review is concerned with the arteries functioning in the systemic circulation. The basic arterial wall structure consists of three layers (Figure 1): the innermost intimal layer comprised of the endothelium, the middle medial layer mainly comprised of smooth muscle cells, and the outermost adventitial layer, comprised of fibroblastic cells. The endothelium is a monolayer of cells which function as the barrier between the blood and the rest of the vessel wall, as well as a surface to resist blood clotting. Smooth muscle cells mainly act as the mechanical strength needed to support blood pressure-induced stretch. The forces experienced by the arterial wall include the normal stress of blood pressure induced by blood flow, the circumferential stretch induced by cyclic strain driven by the pressure pulse, and the wall shear stress exerted by the blood flowing tangential to the surface of the blood vessel. The focus of this review is the effect of wall shear stress on the endothelium and its role in pathophysiology of atherosclerosis.
Figure 1. Hemodynamic forces acting on the artery wall.

The blood vessel wall is comprised of three layers: the intimal layer, medial layer, and adventitial layer. The intimal layer is mainly composed of the endothelium, the medial layer is mainly comprised of smooth muscle cells, and the outermost adventitial layer is comprised of fibroblastic cells. The forces experienced by the arterial wall include the normal stress of blood pressure induced by blood flow, the circumferential stretch induced by cyclic strain driven by the pressure pulse, and the wall shear stress exerted by the blood flowing tangential to the surface of the blood vessel. Adapted from Tarbell et al. (2014).
Wall shear stress (WSS), the frictional force between the blood and the endothelium, has emerged as a major determinant of endothelial function and gene expression. Shear stress can be mathematically described in a steady, fully developed, laminar flow through a straight tube known as Poiseuille flow. For Poiseuille flow, the shear stress is directly proportional to viscosity of the fluid and the volumetric flow rate, and inversely proportional to the cube of the radius of the vessel lumen. However, due to variabilities in the cross section within the arteries and the fact that arteries are not uniformly straight, the calculated WSS can significantly vary from the measured WSS. Physiologically, the shear stress must be maintained within a narrow range and even small deviations in magnitude or direction can greatly impact endothelial homeostasis. Although it is assumed the mean shear stress is 15 dyn/cm2 throughout the vascular system, this value is derived from an average over the values taken over the cardiac cycle in large straight arteries experiencing steady, unidirectional laminar flow [1–10]. Furthermore, the principle of minimum work as derived by Murray, which in mathematical form deduces that the cube of the radius of the mother vessel equals the sum of cubes of the radii of the daughter vessels [11–13] also supports this value. However, more recently, it has emerged that the actual mean wall shear stress varies along the arterial tree [14–17]. As shown in Figure 2A, in healthy humans, the common carotid artery ranges from 9.5–15 dyn/cm2 with an average of 11.6 dyn/cm2 whereas the brachial artery, common femoral artery, superficial femoral artery, infrarenal aorta, and suprarenal aorta are much lower, with averages of 6.5, 4.3, 4.4, .2, and 7.3 dyn/cm2 respectively [14–16, 18–31]. From these studies, it was concluded by previous reviewers that the wall shear stress values are dependent on the distance from the aortic root in that the more downstream vessels have lower shear stress values [32, 33].
Figure 2. WSS in humans and mice.

Average shear stress values along the arterial tree in healthy humans. Adapted from Cheng et al. (2007) (A). Average shear stress values in the healthy carotid artery of a healthy male. Adapted from Tarbell (2014) (B) Average shear stress values in the left anterior descending coronary artery. Adapted from Samady et al. (2011) (C). Average wall shear stress in the murine aortic arch Adapted from Suo et al. (2006) (D). Average wall shear stress in the carotid arteries following partial ligation Adapted from Nam et al. (2010) (E).
Not only does the shear stress vary widely with location, there are also differences in shear stress among species due to anatomical differences [30] [27–29]. In the common carotid arteries of dogs, the WSS is 15.8 dyn/cm2, whereas in rabbits the values in shear stress varied widely but the average was 23.3 dyn/cm2. In rats and mice, the values also varied widely, but the average values for the common carotid were 46.6 and 64.8 dyn/cm2 respectively. In the femoral artery, the dogs had a value of 9.8 dyn/cm2, whereas for rabbits it was 156.8, and for rats it was 65.9 [27, 28, 30, 34–40]. These values varied widely due to the various types of methods and anesthetics used for measurements. However, despite the variability, these studies indicate that the wall shear stress is roughly correlated with animal size. Larger animals, such as humans, have the lowest WSS when comparing the same artery to smaller animals such as mice. The nonuniformity in shear stress among arteries within the same animal and across species can be explained by differences in the vessel lumen diameter, as WSS has been found to inversely correlate with lumen diameter [32, 33].
Furthermore, the geometry of the vessel plays a major role in the WSS. The human carotid can be used as an example of the changes in the shear stress profile due to geometric changes (Figure 2B). The carotid artery bifurcates into the external and the internal carotid artery, respectively. Figure 2B shows the wall shear stress in a healthy male. Although the time-averaged wall shear stress is approximately 8 dyn/cm2 through the majority of the artery, the carotid sinus experiences a much lower shear stress than the rest of the artery, thus indicating that shear stress is not uniform throughout the whole artery. Another example of WSS variation within the artery is shown in Figure 2C. In the left anterior descending coronary artery, the WSS widely varies between 5 dyn/cm2 in small pockets, to 20 dyn/cm2 in large sections of the artery, to some sections reaching up to 60 dyn/cm2 [41]. This heterogeneity has also been studied in mice. In the murine aortic arch, the WSS only reaches up to 150 dyn/cm2 in the inner curvature (lesser curvature), whereas the straighter greater curvature reaches up to 600 dyn/cm2. Furthermore, in the greater curvature (GC), the velocity vectors are generally in the same direction, whereas the lesser curvature (LC) has velocity vectors in multiple directions [42]. Thus, not only does the magnitude of shear stress widely vary within the artery, but the directionality also varies due to the geometry of the artery. This heterogeneity in shear stress among the arteries arises in areas of differential gene expression. This can be clearly seen in a murine model of atherosclerosis known as the partial carotid ligation (PCL) model [43]. Figure 2E shows that when the left carotid artery (LCA) is ligated, the WSS drops from approximately 110 dyn/cm2 to 30 dyn/cm2, and thus contributes to pathogenesis of atherosclerosis.
2.2. Atherosclerosis and Localization of Plaques
Although atherosclerosis is the most common cause of death in the world [44] and thus is widely studied, it is a complex disease and the mechanisms by which it occurs are still being elucidated. What is known is that atherosclerosis is an inflammatory disease occurring in the arterial wall. Atherosclerosis is initiated by inflammation in the endothelial layer, which allows the endothelium to become more permeable. After the barrier of the endothelium is compromised, bloodborne lipids such as those associated with low density lipoproteins (LDL), accumulate under in the intima. Once these lipids are present, immune cells, particularly monocytes, transmigrate into the intima as well with the aid of the endothelium and upon contact with the LDL, become foam cells to create lesions. These initial lesions then become progressively larger with age. The lesions, or plaques, are what characterizes atherosclerosis [45]. It is generally regarded that there are distinct stages of plaque progression (Figure 3). In brief, these include LDL accumulation in the intima, oxidation by resident macrophages and smooth muscle cells, recruitment of circulating monocytes by cytokines and transmigration by binding to endothelial cell adhesion molecules, foam cell formation, smooth muscle migration to the intimal layer, smooth muscle proliferation, and finally the formation of a necrotic core. Although plaques can be heterogeneous in nature, the consequences of plaque formation end in occlusion of the artery and a loss of blood flow and oxygen to downstream regions and organs. This can occur by either the development of a plaque so large that it becomes occlusive, or the formation of a smaller plaque which is vulnerable and erodes so that the endothelium is denuded and an occlusive thrombus forms [46–48].
Figure 3. Stages of atherosclerosis.

The development of an atherosclerotic plaque initiates with LDL infiltration into the subendothelium, where it is oxidized by macrophages and smooth muscle cells, resulting in the conversion of macrophages to foam cells (1 and 2). The release of growth factors and cytokines by the resultant dysfunctional endothelium attracts additional monocytes (3 and 4). Foam cell accumulation and smooth muscle cell proliferation result in the growth of the plaque (6–8). Adapted from Faxon et al. (2004).
Interestingly, it has been observed that atherosclerotic plaques develop in specific regions of the vasculature. Specifically, as early as the 1960s, it was observed that plaques develop at sites of curvature, branching, or cross-sectional expansion and these sites experience flow separation [49]. At these regions, the flow departs from pulsatile, unidirectional flow to create flow-separation zones including flow reversal, oscillatory flow, and turbulence [50–53]. These sites include the abdominal aorta, the carotid bifurcation, and the lesser curvature of the aorta [49]. However, Caro and colleagues were the first to show that lesions develop directly upstream of these flow dividers in regions of low wall shear stress [54, 55]. Following this initial study, Caro and Nerem perfused the common carotid arteries of dogs and studied cholesterol uptake, which is one of the initiating steps of atherosclerosis [56]. They found that the uptake of lipids in arteries could not be correlated with fluid phase mass transport rates, which lead the investigators to conclude that the blood flow directly on the arterial wall affected the cholesterol transport. In conjunction with Caro’s initial study, the hypothesis that atherosclerosis is localized to areas of low wall shear stress due to its effect on the arterial wall was validated by many other investigators [57–63]. Particularly, Ku and colleagues noted that not only is low wall shear stress an indicator of a site of plaque development, but also in these regions there is a reversal of the flow during the pulsatile flow cycle. Taken together, the flow pattern associated with atherosclerotic plaques corresponds to low, oscillating, shear stress known as disturbed flow (d-flow or OS).
3. Atherosclerosis and the Endothelium
3.1. Endothelial Regulation by Flow
Although Caro and Nerem found that the direct flow of fluid on the arterial wall affected cholesterol transport, it was not shown until later that the endothelium was involved. The direct effect of fluid flow on endothelial cells (ECs) was first demonstrated by Nerem and Dewey independently [64, 65]. In these studies, these investigators found that ECs align in the direction of flow. Later, Frangos et al. and Grabowski et al. observed that application of shear stress to static cells rapidly induced the antithrombotic prostacyclin [66, 67]. Furthermore, Mo et al. and Shen et al. independently found that shear transiently induces release of intracellular Ca2+, which acts as a signaling molecule [68, 69]. Finally, Kuchan et al. and Korenaga et al. found that shear induces sustained release of the vasodilator nitric oxide (NO) [70–72]. Taken together, these studies on cultured ECs suggested that fluid shear stress directly affects the phenotype of ECs.
The main functions of the endothelium are to maintain a barrier between the blood and underlying tissues, regulation of vascular tone, recruitment of immune cells to sites of injury, and to form new blood vessels. Numerous studies have shown that each of these endothelial functions is greatly impacted by shear stress. One of the earliest functions to be studied was endothelial permeability as described above (Caro and Nerem), as well as further studies into the effects on individual transport pathways (tight junctions, adherens junctions, vesicles, and leaky junctions [56]. Specifically, endothelial permeability increases after the onset of shear stress, but decreases after prolonged exposure [73, 74]. Furthermore, it was found that the biphasic response of endothelial permeability is due to increases in NO production from shear stress, which decrease permeability [75–77]. Taken together, these studies suggest that the acute increase in permeability is most relevant to the microcirculation, which responds to needs of specific organs, whereas the chronic response, which is downregulation of permeability due to sustained shear, are protective against the formation of atherosclerosis in that LDL is not allowed to penetrate the wall [78]. Furthermore, it has been shown in a mouse model of atherosclerosis that endothelial permeability is increased in atheroprone regions due to the degradation of the endothelial extracellular matrix (ECM) by matrix metalloproteinases (MMPs) [79]. Additionally, alterations in the endothelial ECM lead to stiffening of the intimal layer, which also affects endothelial permeability [80]. Not only does d-flow negatively impact endothelial permeability and ECM integrity and stiffness, it has also been shown to increase migration and angiogenesis [81, 82].
Due to the effects of flow on the endothelium, it is unsurprising that several studies have found that there are site-specific changes in the endothelium at plaque-prone regions, particularly at curves, branches, and bifurcations. Numerous studies have shown that monocyte adhesion to the endothelium has been enhanced in these regions due to the presence of increased chemoattractants and adhesion molecules [83–85]. Furthermore, endothelial transcription profiles taken in these d-flow regions from mice [86] and pigs [87] indicate that in general, ECs exhibit a pro-inflammatory phenotype when exposed to d-flow. Hajra et al. found that the subunits of the pro-inflammatory nuclear transcription factor NFκB (p65, IκBα, IκBβ) were upregulated in areas of the proximal aortas of mice which were prone to lesion formation [86]. NFκB was only activated in a minority of the cells basally, but was highly activated when stimulated with LPS or hypercholesterolemia. Later, Passerini et al. found that in ECs isolated from either the inner aortic arch of pigs, which experiences d-flow, vs. the descending thoracic aorta, which experiences unidirectional laminar flow/laminar shear stress (LS), there was a general upregulation of several inflammatory cytokines and NFκB elements [87]. Furthermore, in cells isolated from the thoracic aorta expressed more antioxidative genes, which are generally less inflammatory. Taken together, these studies indicated that the endothelium in these regions has a pro-inflammatory phenotype.
Overall, disturbed blood on endothelium induces pro-inflammatory changes that impact endothelial leakiness, stiffness, and the ability to form new vessels.
3.2. Models of Flow and Shear Stress to Study Endothelial Function and Atherosclerosis
In vitro
There are a variety of in vitro and in vivo models to study the effects of d-flow on the endothelium specifically, as well as atherosclerosis (summarized in Figure 4). One of the first and most characterized in vitro models of shear stress is the cone-and-plate viscometer [65, 88]. In this system, shear stress is applied to cultured cells in a stationary plate by a rotating cone. A modified version was later introduced [89], which included a speed-controlled motor with variable rotational velocities. More recently, our lab has developed a modified cone-and-plate which is housed in a standard incubator and programmed shear stress profiles can be controlled by computer ([90, 91]. Another in vitro model is the parallel-plate flow chamber, developed originally by Frangos, McIntire, and colleagues [66, 92]. In this system, a flow chamber consisting of a polycarbonate plate, a rectangular Silastic gasket, and a glass slide (or cover slip) with the attached EC monolayer were held together by a vacuum maintained at the periphery of the slide. Flow was driven either by the hydrostatic pressure head between the two reservoirs to produce steady flow or via cam-driven clamps upstream of the chamber to produce pulsatile flow. Although the cone-and-plate and the parallel-plate flow chamber systems are the most commonly used in vitro shear systems, microfluidic chambers have become more recently used as they allow for high throughput experiments. This method was pioneered and commercialized by Schaff et al. [93–95].
Figure 4. Models of Flow and Shear Stress.

Cone-and-plate viscometer. Adapted from Jo et al. (1991) (A). Parallel plate flow chamber. Adapted from Lawrence et al. (1987) (B). Microfluidic flow chamber. Adapted from Schaff et al. (2007) (C). Carotid cuff model Adapted from Cheng et al. (2005) (D). Partial carotid ligation model. Adapted from Nam et al. (2010). (E).
Ex vivo and in vivo
Although in vitro models provide insight into the role of shear stress in endothelial biology, ex vivo models allow for the study of shear stress on the endothelium in conjunction with related factors, such as the ECM [96]. Early ex vivo models from the 1990s consisted of explanted artery segments cannulated at the ends. In these setups, the arteries were perfused with a controlled intraluminal pressure, flow pulsatility, and direction in a culture medium bath [97, 98]. In one such system, Gambillara et al. showed that low magnitude, oscillatory shear stress reduced endothelial nitric oxide synthase (eNOS) expression in porcine carotid arteries [98]. Lu and Kassab also used porcine arteries to show NO levels drop after exposure to flow reversal [99]. Furthermore, an ex vivo model for murine carotid arteries was developed by Gleason et al. [100]. In this model, defined mechanical stress can be applied to the arteries based on a computer controller.
There are also several in vivo models of atherosclerosis. These models are available in a variety of animals, including pigs, primates, rats, and mice [96]. However, due to the ease of genetic and physical manipulation of mice, mice are the most popular animal models of atherosclerosis. One of the first models of atherosclerosis in mice was the hypercholesterolemia model developed by Paigen et al. [101–104]. It was found that only the C57BL/6J strain could develop atherosclerosis if hypercholesterolemia was induced by genetic mutation and high-fat diet. The two most widely used mutations are the apolipoprotein E (ApoE) disruption [105–107] and the LDL receptor deletion [108]. The most common diets are Paigen’s diet, which includes cholate [102], and the Western diet, which does not include cholate and is less inflammatory [106, 109]. Other models of atherosclerosis use surgical intervention to the arteries to induce atherosclerosis. Specifically, models such as injection of foreign proteins [110, 111] in rabbits, drying models in rats [112], and the more established wire [113] and later balloon injury models [114, 115] in mice directly injured the artery. However, the constrictive perivascular cuff model in mice [116–118] and the ligation models in mice are the most popular methods to rapidly induce atherosclerosis by alterations in flow, and thus shear stress (in conjunction with the hypercholesterolemic models). In the perivascular cuff model, the constricted region experiences higher shear stress, whereas the proximal section is exposed to d-flow and thus develops atherosclerotic plaques Figure 4D. Both complete ligations and incomplete ligations are also commonly used models. In the complete ligations, the carotid artery in rodents is ligated and this induces vascular remodeling, neointimal hyperplasia, and atheroma formation [60, 118–128]. In this model, there is no wall shear stress on the endothelium. However, as this injures the artery, this model does not isolate the role of shear stress alone in atherosclerosis. Finally, among the incomplete ligations, first studied in pigs [129], we have robustly developed a partial carotid ligation model [43]. In this model, 3 of the 4 branches of the left carotid artery is ligated while the right artery serves as an internal control (Figure 4E).
4. Mechanosensing and Mechanotransduction in Atherosclerosis
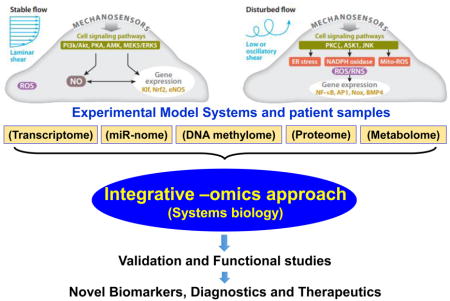
In the context of vascular biology, mechanosensitivity describes the ability of the endothelium to perceive mechanical stimuli and translate these stimuli into biological signaling events. Both mechanosensing and mechanotransduction are fundamental physiological mechanisms allowing the ECs to react to physical forces such as shear stress and cyclic stretch. A stimulus is perceived by mechanosensors on the endothelial cell surface and is translated into a cell signaling event via effector proteins known as mechanotransducers. In the last 15 years, our understanding about mechanosensors, mechanotransducers, and the process of mechanotransduction in the endothelium has increased significantly, but it is still an incomplete picture. Overall, there is no clear distinction between mechanosensing and mechanotransduction. Whether the mechanosensors themselves work as transducers or whether the mechanosensors are separate sub-cellular signaling entities remains to be discovered. Also, it is still unknown as to what are the precise biological functions of mechanotransduction in the endothelium. A variety of endothelial mechanosensors and their associated downstream pathways have already been identified [130–133]. This section summarizes the key mechanosensors and mechanotransducers in the endothelium that play a critical role in normal and pathophysiological processes.
4.1. Mechanosensors
On the luminal, junctional, and basal surfaces of ECs, there are numerous mechanoreceptors capable of detecting and responding to shear stress stimuli (summarized in Figure 5A). After activation of any one of these mechanoreceptors, a complex network of several intracellular pathways is triggered in a process known as mechanotransduction. These pathways are activated simultaneously and/or cross-talk with each other. These pathways lead to regulation of several transcription factors, which bind positive or negative shear stress responsive elements (SSREs) in the promoters of mechanosensitive genes, thus inducing or suppressing gene expression respectively. Furthermore, a shear stress stimulus could trigger a change in conformation of a membrane protein in order to expose previously hidden sites. Thus, a new binding site would be available to downstream biochemical substrates, which would initiate a cell signaling cascade. Below, we discuss the role of major mechanical sensors, including platelet endothelial cell adhesion molecule 1 (PECAM1), the glycocalyx, caveolins, cytoskeletal structures, integrins, angiotensin type 1 (AT1) receptor (AT1R), and the nucleus in the regulation of endothelial function.
Figure 5. Mechanosensors and Mechanosensitive Signaling Pathways.

The endothelial cell senses shear stress through a variety of mechanosensors, including surface mechanoreceptors (ion channels, receptor tyrosine kinases, G protein coupled receptors), cell-cell and cell-matrix adhesion complexes (PECAM1/VE-cadherin/VEGFR2 complex and focal adhesion kinases), the glycocalyx, and cytoskeletal elements. Adapted from Chatzizisis et al. (2007) (A) The shear stress stimulus leads to the activation of a variety of intracellular pathways. LS activates pathways that result in alignment in the direction of flow, increased NO production, and suppression of inflammatory cell adhesion molecules. On the other hand, OS induces high expression of cell adhesion molecules, production of inflammatory cytokines, high oxidative stress, and a leaky cell barrier Adapted from Noguchi & Jo (2011) (B).
PECAM1 is an important molecule that is present on the endothelial surface and is primarily localized to junctions between ECs. PECAM1 becomes phosphorylated at tyrosine residues in response to mechanical stimuli. Osawa et al. report that this phosphorylation causes association of PECAM1 with SHP-2, a phosphatase that activates extracellular signal-regulated kinase (ERK) [134]. ERK activation, which has been previously implicated in mechanotransduction, depends on PECAM1 phosphorylation. Therefore, it can be speculated that in response to mechanical disturbance of the plasma membrane, PECAM1 becomes phosphorylated by an unknown kinase, and subsequently recruits SHP-2, thereby activating ERK. In turn, SHP-2 can dephosphorylate PECAM1, however, persistent flow-shear stress could re-initiate the phosphorylation cycle, thus maintaining the cellular response [135]. Furthermore, PECAM1 (in complex with vascular endothelial growth factor receptor (VEGFR2) and VE-cadherin) activates phosphatidylinositol-3-OH kinase (PI3K), which is another mechanosensitive signaling kinase. After PI3K is activated, another mechanosensitve kinase which is involved in the same signaling pathway as PI3K, Akt, is also activated. Akt activation leads to cytoskeletal arrangement so that ECs exposed to anti-atherogenic LS align in the direction of flow, whereas in ECs exposed to pro-atherogenic OS, have Rac1 activation, which in turn leads to increased reactive oxygen species (ROS) and NFκB activation. Thus, in OS, the same mechanosensors lead to the expression of pro-inflammatory genes by activating NFκB. [136, 137]
The glycocalyx is also present on the surface of ECs. The glycocalyx is comprised of various macromolecules. Some of these include glycoproteins bearing oligosaccharides and terminal sialic acids, proteoglycans, and glycosaminoglycans (GAGs) [138]. The glycocalyx constantly senses the shear stress on the endothelium. The glycocalyx also interacts with other plasma proteins, enzymes, enzyme inhibitors, growth factors and cytokines through structural cationic sites, as well as cationic amino acids, in order to make a matrix of biopolyelectrolytes [138–142]. It is frequently observed that the glycocalyx undergoes structural and functional modifications when interacting with these molecules [143–146]. These properties allow the glycocalyx to function as a selectively permeable barrier for macromolecules by filtering molecules based on size and charge. Given its role as a selectively permeable barrier, its ability to sense the mechanical forces imposed by blood flow, and sequentially transduce these signals into intracellular biochemical responses with vasoregulatory properties [147–151], these qualities distinguish the glycocalyx as a vital element of a functional endothelium. Likewise, proteoglycans are proteins that contain specific sites where sulfated GAGs are covalently attached en route from the endoplasmic reticulum to the Golgi apparatus. These constituents provide a downstream signaling event upon interaction with the local fluid-shear environment [146, 152].
Caveolin-1 is also a membrane-associated element. Specifically, caveolin-1 is a protein that associates with cholesterol and sphingolipid-rich regions of the membrane and usually forms pits or caveolae. Similarly, glypicans, along with their heparin sulfate chains localize to these regions. Transmembrane syndecans are shown to cluster on the outer edge of caveolae and along with other molecules, are involved with eNOS signaling. Particularly, these molecules have been shown to inactivate eNOS [153–157]. On the other side of the plasma membrane, the cytoplasmic tails of the syndecans associate with the cytoskeleton and assist in its organization through molecules such as ezrin, tubulin, syntenin, syndesmos, dynamin and α-actinin [158–161]. Syndecans also directly associate with proteins involved in signaling, such as protein kinase C-α (PKCα), phosphatidylinositol-4,5-biphosphate, and Calcium/Calmodulin-Dependent Serine Protein Kinase (CASK), a protein containing both a calcium/calmodulin-dependent and a guanylate kinase domain [158, 162–164]. Active participation in signaling stems from the phosphorylation of certain intracytoplasmic residues, which act as switches controlling the oligomerization state and thus altering the binding properties of syndecans. Ultimately, this enables them to orchestrate events on both sides of the membrane [158, 162–164]. Finally, it is also worth noting that the secondary structures of all syndecan ectodomains are predicted to contain almost exclusively solvent-accessible loops, implying that these molecules are flexible (models by PROFsec, and YASPIN) [165, 166].
Integrins are also membrane-associated mechanosensors. Integrins are hetero dimeric transmembrane proteins comprised of non-covalent interactions between α and β subunits that serve as major molecular links between ECs and the ECM. Extracellular domains of the α subunits participate in adhesion and ligand recognition, and upon activation, the short cytoplasmic domains of β subunits, which lack kinase function, physically connect to the cytoskeleton and recruit proteins for signaling [167–170]. The specificity of integrin signaling is made possible by α and β-subunits that form the hetero dimeric pair. The α-subunit generally confers ECM specificity [170, 171], whereas the β-subunit interacts with the cytoplasmic environment. Evidence for shear stress activation of integrins is provided by both direct assessment of integrin conformational changes in response to shear stress and blockade of the shear-induced responses by monoclonal antibodies or via the use of the integrin binding Arg-Gly-Asp (RGD) peptide. When cells are plated on ECM or treated with integrin-activating monoclonal antibodies, integrin activation is manifested by modulations of affinity and avidity [172, 173]. Tzima et al. have shown an increased immunostaining of integrins in sheared ECs, indicating a modulation of integrin affinity by shear stress [136]. Using multiple monoclonal antibodies to assess ligand-bound form of β1 and β3, Jalali et al. have demonstrated that shear stress leads to an increase in integrin avidity in the endothelium [174]. Because integrins lack enzymatic activity, activation of signaling factors requires interaction with cellular proteins that have kinase activity. In ECs, the cytoplasmic tail of the β-subunit has been shown to directly bind to several cytoskeletal proteins that associate with signaling molecules [175]. In other cell types, β1 integrin has been shown to be important for coupling mechanical stretch to activation of the well-known mechanosensitive signaling pathway members mitogen activated protein kinases (MAPKs), as well as focal adhesion kinase (FAK) and Rho GTPases [176, 177]. EC motility is a critical function that is regulated by integrins in response to hemodynamic forces. The effect of shear stress on EC migration and the contribution of the integrins and integrin-dependent signaling pathways have been studied in vitro using scratch-wound assays. LS-induced endothelial migration was significantly reduced by integrin-receptor blocking with RGD peptides or with neutralizing antibodies against integrin subunits αv and β1, whereas antibodies against αvβ3 or α2β1 had no effect. Cell-surface levels of the integrin α5β1 were specifically upregulated in migrating ECs at the wound edges. Consistent with these previous results, blockade of the integrin-associated adapter protein Shc by overexpression of dominant negative construct inhibited shear stress-stimulated endothelial migration [178, 179]. Collectively, these studies imply that integrins are important endothelial mechanosensors and mechanotransducers.
Mechanical forces can also be directly sensed by the actin cortex and, in turn, be transmitted as signaling events. However, the mechanisms are poorly understood. Using an integrative approach of combining molecular and mechanical experimental perturbations with theoretical multiscale modeling, the process of cortical mechanosensing from molecular to cellular scales has been elucidated. Early evidence that the nucleus is under tension came from Ingber and coworkers in 1992, who showed that perturbing actomyosin forces altered cell and nuclear shape [180]. In a landmark paper in 1997, they showed that tugging on integrin receptors in the cell membrane causes nuclear distortion and motion [181]. This established the concept that forces applied externally to the cell are propagated to the nuclear surface, which was consistent with mechanical models of the cell cytoskeleton that are ‘hardwired’ to the nuclear envelope [182–187]. These external forces have now been shown to induce clearly-detectable nuclear deformation [181, 188–195]. The F-actin cytoskeleton plays a major role in propagating the mechanical forces from the integrin receptors to the nuclear surface, although the molecules which connect the nucleus to the cytoskeleton have only recently been identified. In recent years, members of the so-called LINC complex ( Linker of Nucleoskeleton to the Cytoskeleton) have been discovered in the nuclear envelope [196–201]. The LINC complex is comprised of two protein families that span the nuclear envelope, and physically connect the cytoskeleton to the nucleoskeleton. The SUN (Sad1p, UNC-84) domain proteins span the inner nuclear membrane and translumenally bind the KASH (Klarsicht/ANC-1/Syne Homology) domain proteins that span the outer nuclear membrane. In this way the KASH and SUN domain proteins create a mechanical tether that connects both membranes of the nuclear envelope. The KASH domain proteins bind to various cytoskeletal constituents, whereas the SUN domain proteins associate with the nuclear lamina [199–201]. Thus, the mechanical connections created by the LINC complex can integrate the forces of the cytoskeleton and the nucleus.
In the past decade, numerous studies have indicated that the nucleus itself may act as a cellular mechanosensor to directly modulate the expression of mechanosensitive genes [202]. The nucleus is tightly integrated into the structural network of the cell through LINC, which transmits forces between the nucleus and the cytoskeleton [200]. Lamins, which are type V nuclear intermediate filaments, contribute to the nuclear lamina as an extended part of the LINC complex. Through this interaction, lamins play a central role in the nuclear mechanosensory process. Lamins can be separated into A-type and B-type, with lamins A and C as the major A-type isoforms, and lamins B1 and B2 as the major B-type isoforms in somatic cells [203]. Lamins A and C provide structural support to the nucleus [190] and play a major role in physically connecting the nucleus to the cytoskeleton, thereby enabling forces to be transmitted from the cytoskeleton and extracellular matrix to the nuclear interior. Lamins A and C are important contributors to the mechanical stiffness of nuclei, whereas lamin B1 contributes to nuclear integrity instead. Cells lacking lamins A and C have reduced nuclear stiffness and increased nuclear fragility, which leads to increased cell death under mechanical strain. Mutations in lamins A and C result in a variety of severe diseases, including dilated cardiomyopathy, which further indicates the critical role of these nuclear envelope proteins in maintaining normal cellular function. Recent studies have provided some insight into how the nuclear lamina responds to force-induced nuclear deformation and couples to biochemical responses in ECs in response to shear stress [188, 204–207]. However, it remains to be determined whether these changes reflect the role of lamins as mechanosensors or if transcriptional regulation of lamins is downstream of other mechanosensing pathways. Several other key questions remain to be addressed regarding the role of the nuclear envelope proteins in endothelial mechanotransduction. Issues that remain to be addressed include whether lamins are mechanosensors or merely serve as processing hubs, how the nuclear mechanosensing system is integrated with signaling originating from the plasma membrane, and the role of this system during adaptive and maladaptive stages of vessel remodeling.
Finally, G-protein coupled receptor (GPCRs), such as AT1R, can also serve as mechanosensors/transducers. One such, AT1R, binds angiotensin II (Ang II) in the canonical pathway. However, recently it was found that there are biased ligands of AT1R which can be activated by shear stress through an AngII-independent mechanism [208–212]. It is well established that AT1R, the first mechanosensitive GPCR discovered, mediates transformation of mechanical stimuli into biochemical information and gives rise to a variety of mechanosensor-induced cellular responses (such as inflammation, cell growth, and differentiation etc.) [212, 213]. Recent studies suggest that d-flow induces β-arrestin-based signaling downstream of AT1R in the absence of ligand or G protein activation [214]. Mechanical stimulus triggered AT1R receptors mediate conformational changes in β-arrestin, similar to that induced by a β-arrestin-biased ligand, to selectively stimulate receptor signaling in the absence of detectable G protein activation [211, 212, 215]. Yatabe et al., demonstrated that mechanical stress caused an increase in the phosphorylation levels of ERK in rat mesangial cells through the AngII-independent AT1R activation [210]. Taken together, this suggests that our knowledge of the mechanosensory networks in ECs is still not complete, even in well-studied pathways such as the AT1R pathway. This will be an interesting area for future research in endothelial mechanobiology.
4.2. Mechanosensitive Signaling Pathways
Shear stress is able to be translated from the cell surface through a variety of mechanosensors [216]. Once the shear stress stimulus is applied, various intracellular pathways are triggered. Interestingly, many of these pathways converge on common signaling pathways, such as the MAPK pathway and the PI3K/Akt pathway [154]. The MAPK pathway in particular can be activated through integrins (as previously discussed), among others. Briefly, integrins activated by mechanical stimuli phosphorylate and activate a complex of kinases, adaptor proteins, and guanine nucleotide exchange factors, which ultimately lead to the activation of Ras (Figure 5A). When Ras becomes activated, this leads to the activation of MAPKs. ERK1/2, members of the MAPK family, then activate transcription factors (such as c-myc, c-jun, and c-fos) and/or eNOS [217]. Furthermore, mechanosensitive membrane proteins activate the MAPK pathway through protein kinase C (PKC) [217], whereas NADPH oxidase activates the MAPK pathway through ROS [218]. An additional example of such is the activation of PECAM1, as discussed previously. Furthermore, the PI3K/Akt pathway can converge with the same integrins that the MAPKs interact with and can lead to activation of eNOS [219, 220].
These shear-responsive pathways often activate the MAPK and PI3K/Akt pathways differentially in response to LS vs. OS (Figure 5B) [131]. In LS, atheroprotective genes become upregulated [221–223]. Particularly, eNOS becomes phosphorylated and activated by Akt via a PI3K-dependent pathway [224, 225] and leads to an anti-atherogenic phenotype in ECs [226].
Krüppel-Like Factor 2 (KLF2) is a mechanosensitive transcription factor which is intimately involved in the aforementioned mechanosensitive pathways. Klf2 is critical for vascular homeostasis and is a potent anti-atherogenic transcription factor [227]. Klf2 is a highly expressed in straight sections of the human aorta that experience LS, but is lower in the regions experiencing OS, such as the bifurcation in the iliac and carotid arteries [228]. Consequently, KLF2 downregulates a host of pro-inflammatory genes in ECs exposed to LS, such as vascular cell adhesion molecule (VCAM-1) and E-selectin, which mediate monocyte and T-cell adhesion to the endothelium [229]. Furthermore, KLF2 can inhibit the NFκB pathway by recruiting transcriptional coactivators to inhibit interleukin 1 (IL-1β) and tumor necrosis factor (TNFα)-mediated stimulation. KLF2 also inhibits thrombin-mediated induction of inflammatory factors, such as monocyte chemoattractant protein (MCP-1), IL-6, and IL-8, by inhibiting expression of its principal receptor protease-activated receptor 1 [230, 231]. Additionally, KLF2 prevents endothelial inflammation and thrombosis by preventing nuclear localization of phosphorylated activating transcription factor 2 (ATF2), which mediates the expression of proinflammatory and procoagulant genes [232]. To further solidify the role of KLF2 as a potent anti-thrombotic agent, it was found that KLF2 overexpression strongly increases TM, which is a cell surface factor that inhibits coagulation, and KLF2 inhibits tissue factor and plasminogen activator inhibitor 1 production. However, not only does KLF2 downregulate inflammation and prevent thrombosis, KLF2 is also responsible for the shear-induced alignment of ECs. Horrevoets et al first linked KLF2 expression to the morphological changes seen in endothelial cells exposed to shear. In this study, they found that Jun NH2-terminal kinase and MAPK signaling lead to inhibition of the phosphorylation of actin cytoskeleton-associated proteins [233]. KLF2 promotes vasodilation through the direct transcription of eNOS [234–237] and by inhibiting caveolin-1, which regulates eNOS by inducing arginosuccinate synthase, a limiting enzyme in eNOS substrate bioavailability [234–237]. In the long term, sustained expression of KLF2 inhibits expression of endothelin-1, adrenomedullin, and angiotensin converting enzyme, all ofwhich increase vascular contractile tone [238].
Furthermore, KLF2 inhibits angiogenesis and EC proliferation via inhibition of VEGF-A[239] and VEGFR2 expression by competing with its transcription factor Sp1 at the promoter. In addition to these mechanisms, KLF2 can inhibit angiogenesis through semaphorins, have been shown to inhibit EC migration. Klf2 can be upregulated by LS indirectly through the LS effect on the MAPK pathway (namely MEK5 and ERK5) [240]. KLF2 is regulated through a variety of mechanosensitive pathways. These include LS induction of MAPK pathway, which causes histone deacetylase 5 (HDAC5) dissociation from MEF2, thus allowing MEF2 transcription of KLF2 mRNA [241], PI3K inhibition by prolonged flow via Tie2, AMP-activated protein kinase activation by LS activating ERK5-MEF2. KLF2 mRNA is also stabilized via PI3K pathway [242]. Furthermore, OS causes prolonged suppression of KLF2 by the src signaling pathway [128]. Additionally, the OS sensitive gene regulator HuR plays a role in KLF2 regulation. Specifically, downregulation of HuR leads to KLF2 mRNA stabilization [243]. Pro-inflammatory cytokines such as TNFα and IL1β repress KLF2 expression via HDAC4 and p65 (component of NFκB) cooperatively inhibiting MEF2 [244]. Finally, oxidative stress can also decrease KLF2. SUMOylation of ERK5 by hydrogen peroxide and advanced glycation end products inhibits KLF2 expression by decreasing MEF2 [245] and the Shc oxidative stress protein p66shc [246] reduces MEF2A expression as well.
Another transcription factor, nuclear factor (erythroid-derived 2)-like 2 (Nrf2) is a target of KLF2, is also mechanosensitive, and in conjunction with KLF2, controls 70% of the genes induced by LS [237]. Nrf2 becomes translocated to the nucleus in cultured endothelial cells exposed to LS [247]. Nrf2 which exerts its anti-atherogenic effects by modulating ROS and reactive nitrogen species (RNS) [223]. Shear stress plays a critical role in the production of ROS and RNS in ECs. Shear stress activates NADPH oxidase, resulting in production of superoxide (O2-) [248–251]. Xanthine oxidoreductase also contributes to superoxide production in response to OS [252]. In addition, NO is generated (via eNOS) [156, 253–256] Although NO plays an important role in vasodilation [257–259] and inflammation [260, 261]. NO may react with superoxide, thereby forming peroxynitrite (ONOO−), which is highly reactive. Peroxynitrite can modify proteins and lipids and induce oxidative damage [262, 263]. Although both OS and LS induce superoxide and NO, the balance between the species determines the overall effect. In the case of LS, NO production is significantly higher than that of OS [264, 265]). Whereas in OS, superoxide production is much higher [252, 266–268] but eNOS is upregulated to a much lesser extent [269–271], which in turn leads to the reaction of NO with superoxide to form peroxynitrite, resulting in less bioavailable NO. In contrast, LS upregulates the expression of eNOS strongly, as well as dismutases that can convert superoxide into other species, thus preventing peroxynitrite formation. These include copper/zinc superoxide dismutase (CuZnSOD) and manganese (MnSOD) [272, 273]. Nrf2 is essential for upregulating cytoprotective genes under LS [223, 274, 275]. Nrf2 binds to the antioxidant response element (ARE) in its target genes, which include phase II detoxification enzymes and antioxidant proteins, such as glutathione-S-transferase, HO-1, peroxiredoxin 1, NQO1, GCLM, and GCLC [263, 276, 277]. These enzymes are crucial for protecting cells from electrophile toxicity and oxidative stress.
Nrf2 is negatively regulated by Kelch-like ECH-associated protein 1 (Keap1), which facilitates the degradation of Nrf2 through the proteasome [278]. In response to oxidative stimuli, Nrf2 becomes activated by its dissociation from Keap1, which undergoes electrophilic attack that causes it to undergo the conformational change to dissociate from Nrf2 [245, 246, 279, 280]. In turn, Nrf2 upregulates its anti-oxidant target proteins [281, 282]. The upregulation of Nrf2 in LS is ROS dependent, as Nrf2 activation in LS was blocked by ROS scavengers [275, 283–287] and NADPH oxidase inhibitors. The subsequent ROS/RNS modifies Keap1 under LS conditions so that Nrf2 is no longer suppressed.
As opposed to LS, in OS, ECs express pro-inflammatory cytokines such as MCP-1 [219] and inflammatory cell adhesion molecules such as VCAM1 and ICAM1 [288]. The chemokine MCP-1, contains a phorbol ester (TPA)-responsive element (TRE) in its promoter region, which was also found to be shear-sensitive and regulated through MAPKs [219]. Similarly, VCAM1 and ICAM1 also contain these shear-responsive elements in their promoters [288]. Furthermore, ECs express other pro-inflammatory, shear-sensitive proteins such as NADPH oxidase [223], and bone morphogenetic protein (BMP4) [289]. Two of the main transcription factors that are responsible for the upregulation of many of these pro-inflammatory genes such as ICAM1, VCAM1, and E-selectin are the activator protein complex (AP-1) and nuclear factor (NFκB) complex [233, 290–295].
NFκB is comprised of p65 (RelA) and p50, and its activity is regulated by its intracellular location. Under basal conditions, NFκB is located in the cytosol bound to IκBα. Under stimulation, IκBα becomes phosphorylated by IκB kinase, and thus NFκB becomes free to pass into the nucleus and bind to target genes for transcription. Whereas AP-1 is a heterodimer composed of different proteins from either the c-Fos, c-Jun, ATF, or JDP families depending on the target gene. ROS produced by NADPH oxidase or other oxidases can directly activate NFκB and AP-1 [296, 297] in MAPK, ERK, and c-Jun N-terminal kinase (JNK) dependent pathways [221, 298–302].
Ultimately, these findings have demonstrated that LS upregulates “atheroprotective” genes and downregulates “pro-atherogenic” genes while OS results in the opposite phenomenon. However, although the PI3K/Akt and the MAPK pathways have been well-documented, other pathways still remain to be discovered.
4.3. Mechanosensitive Endothelial-Derived Factors and Their Role in Atherosclerosis
Mechanical forces generated at the endothelium due to altered blood flow conditions are important in order to ensure a continuous release of vasoactive factors, including endothelium-derived growth factor, miRNAs, autacoids, etc. Although the mechanism by which endothelial cells are able to detect and convert these physical stimuli into chemical signals is unclear, this process involves the activation of integrins, G proteins, miRNAs, and cascades of protein kinases. Importantly, eNOS can be activated by LS as described above. Likewise, these hemodynamic forces are also able to elicit the synthesis of ROS and endothelium-derived hyperpolarizing factors, all of which play a role in modulating arterial compliance in particular vascular beds. Fluid shear stress can modulate EC and vascular SMC gene expression, cellular function, and pathophysiology of atherosclerosis. Studies assessing eNOS gene expression, proliferation, angiogenesis, migration during atherogenesis further highlight the importance of EC-SMC interactions, suggesting a complex eNOS regulation by shear stress-induced epigenetic modification at the transcriptional and post-transcriptional levels [303].
Vascular smooth muscle cells (SMCs) are important targets for endothelium-derived nitric oxide (NO) and the vasodilator action of the endothelium is mediated by the release of substances–including NO, prostacyclin, and eicosatrienoic acid–that hyperpolarize smooth muscle by activating calcium-dependent potassium channels and are collectively referred to as endothelium-derived hyperpolarizing factors [304]. eNOS catalyses the production of nitric oxide from the cationic amino acid L-arginine. The enzyme is activated via changes in intracellular calcium in response to changes in shear forces or via a receptor-mediated process. Released nitric oxide activates soluble guanylate cyclase in smooth muscle cells, converting GTP to cGMP, activating protein kinases which lead to the inhibition of calcium influx into the smooth muscle cell thereby reducing calcium-calmodulin stimulation of myosin light chain kinase. This in turn downregulates the phosphorylation of myosin light chains thus reducing smooth muscle constriction and causing vasodilatation [305]. These molecules are released from the endothelium by many of the same agonists (e.g., acetylcholine and bradykinin) that stimulate nitric oxide synthesis after receptor-activated increases in endothelial cytosolic calcium concentration. Additionally, 15(S)-Hydroxy-11,12-epoxyeicosatrienoic acid (15-H-11,12-EETA) and 11(R),12(S),15(S)-trihydroxyeicosatrienoic acid (11,12,15-THETA) are endothelial metabolites of the 15-lipoxygenase (15-LO) pathway of arachidonic acid metabolism and are also hyperpolarizing factors. 11,12,15-THETA activates small conductance, calcium-activated potassium channels on smooth muscle cells causing membrane hyperpolarization, and relaxation. Regulation of its expression is by transcriptional, translational, and epigenetic mechanisms. Hypoxia, hypercholesterolemia, atherosclerosis, anemia, estrogen, interleukins, and possibly other hormones increase 15-LO expression. In addition to 15-LO metabolites, a number of other biochemicals have been identified as endothelial-derived factors and their contributions to vascular tone vary with species and vascular bed [306–308].
As mechanistic studies about endothelium-derived factors and their transport to adjacent cells are still emerging, it has become clear thus far that part from the direct transfer of these factors to smooth muscle cells, circulating extracellular vesicles (EVs) comprised of exosomes, microvesicles, and apoptotic bodies can serve as transporters and can be easily differentiated in their size, formation, and release mechanisms. EVs were shown to act as a messengers that serves a long-distance delivery of complex cellular messages. The cargo of EVs consists of a variety of biomolecules including proteins, DNA, mRNA, miRNAs and long non-coding RNAs. In normal or pathological conditions, EVs deliver various molecules to the recipient cells. Those molecules greatly vary depending on the microenvironmental stimuli. During atherogenesis, EVs derived from vascular endothelial cells, vascular smooth muscle cells, macrophages, and other circulating immune cells mainly possess proinflammatory properties. However, the capacity of circulating EVs to stably maintain and deliver a variety of biomolecules makes these microparticles to be a promising therapeutic tool for treatment of cardiovascular pathology. To date, circulating EVs were evaluated to be as a source of valuable diagnostic and prognostic biomarkers such as microRNA. Circulating EVs have the potential to serve as natural vehicles for targeted therapy of cardiovascular diseases [309].
Recent studies have shown that miRNAs can be rapidly transported from the endothelium to other cell types and can perform athero-relevant functions. Zhou et al showed that miR-126 is secreted by endothelial cells in protein complexes by OS conditions. This miR is then transported to smooth muscle cells, where it inhibits the expression of proteins that normally keep the cells in a contractile and quiescent state [310]. There has been conflicting reports on the role of miR-126 in pathophysiology of atherosclerosis. Initially, miR-126 was implicated in atheroprotection especially in the endothelial cells [311]; however, its role in smooth muscle cells is proatherogenic [310]. It is not clear how these miR-containing protein complexes are secreted by endothelial cells and how they enter smooth muscle cells, but answering these questions require additional studies.
From our previous studies, we found that miR-712 and miR-205 expression is also increased in circulation and were also increased in the medial SMCs and circulating immune cells [79]. Although, their mechanism of transport is not clear, it is clear that these miRNAs play an important role in smooth muscle migration and leukocyte-endothelial interactions, respectively, which are key steps in atherogenesis. On the same lines, various shear stress-modulated miRNAs, including miR-126, miR-19a, miR-23b, miR-92a, miR-10a, miR-21 and miR-663, play a crucial role in EC angiogenesis, proliferation, and atherosclerosis [312–314]. The role of shear stress-induced expression of these miRNAs and their impact on the vascular physiology and pathophysiology can be demonstrated by their regulatory roles in differentially regulating various cellular function in ECs vs. SMCs during atherosclerosis [310, 313, 315–320].
Finally, the mechanosensitive transcription factor KLF2 (discussed previously) binds to the promoter of the miR-143/145 cluster in ECs and thus induces the expression of miRNAs contained within the cluster and their subsequent transport to SMCs via EVs. These EVs derived from KLF2-expressing ECs also reduced atherosclerotic lesion formation in the aortas of ApoE-deficient mice, suggesting that atheroprotective stimuli induce communication between ECs and SMCs through miRNAs [321].
5. Identification of Mechanosensitive Genes by ‘Omics’
The greatest challenge of cardiovascular research is to understand the causes and consequences of vascular pathology. In recent years, many resources have been devoted to unraveling the basic mechanisms of atherosclerosis. Because of the complex and multifactorial pathophysiology, different research techniques have increasingly been combined to disentangle various aspects, molecular pathways, and cellular functions involved in atherogenesis and endothelial inflammation so as to get a holistic picture. Research in this area has made great strides due to a rapid evolution of high-throughput technologies to identify molecular changes at DNA, RNA, and protein levels. With the help of high-tech computational tools, these data sets are integrated to enhance information extraction and are being increasingly used in a systems biology approach to model biological processes as interconnected and regulated networks. Here, we briefly review how a high-throughput “omics” approach, such as epigenomics, transcriptomics, miRnomics, proteomics, and metabolomics can be used to explore the mechanisms of disturbed flow-induced endothelial inflammation and atherosclerosis.
5.1. Methylomics Approach
From “omics” data, it has become increasingly clear that key regulatory elements control the mechanosensitivity of vast networks of genes. Therefore, in order to use “omics” data such as the methylome to find mechanosensitive genes, it is critical to understand epigenetic regulation. Epigenetics is defined as the modification of genetic information without direct alteration of the DNA sequence. One such mechanism is through direct DNA structural modifications such as DNA methylation, histone modifications, and chromatin remodeling complexes. Histone modifications and chromatin remodeling complexes alter the structure of genomic DNA interchangeably between euchromatin and heterochromatin in order to control accessibility of DNA sequences [322]. Genomic DNA in an open, relaxed conformation is known as euchromatin and is associated with acetylated histones, whereas condensed genomic DNA is defined as heterochromatin and is associated with methylated histones (reviewed elsewhere [323, 324]). Whereas DNA methylation directly alters the bases and the physical interaction (or lack thereof) with transcription factors impact transcription of genes. In this mechanism, transcription factors are recruited to specific regions of a gene promoter to form a transcriptional complex that assists RNA polymerase to bind and transcribe the gene to mRNA reviewed elsewhere [324].
One of the first indications of aberrant DNA methylation in atherosclerosis was reported by Lund et al. [325]. In this study, they found that atherosclerosis-prone ApoE-deficient mice develop specific changes in DNA methylation in peripheral blood leukocytes and the aorta, both pre and post-lesion formation. Specifically, there is both hypomethylation and hypermethylation in the aortas and leukocytes of ApoE knockout mice. Furthermore, they found that atherogenic lipoproteins promote global DNA hypermethylation in a human monocyte cell line. Review of additional studies indicate there is global hypomethylation in advanced lesions due to the increased proliferation of smooth muscle cells in humans, ApoE knockout mice, and New Zealand White rabbits [326, 327]. Later, Zaina et al. reported that in donor-matched healthy and atherosclerotic human aorta samples, the atherosclerotic portion of the aorta was hypermethylated across many genomic loci in comparison with the healthy counterpart [328]. Furthermore, they identified several differentially methylated genes associated with atherosclerosis onset that are involved in endothelial and smooth muscle functions including HOXA6, HOXA9, MIR23b, PDGFA, PLAT, PRRX1, and PXDN. Taken together, these early studies indicate that there is hypomethylation of pro-atherosclerotic genes and hypermethylation of anti-atherosclerotic genes.
Although the previous studies greatly contributed to our understanding of epigenetics in atherosclerosis, more recent studies focus more in depth on the role of methylation in specific cell types. Several groups independently found that DNA methyltransferases (DNMTs) are shear responsive proteins that regulate flow-mediated endothelial gene expression programs [329–331]. Jiang et al. first discovered that in the endothelium of pig aortas and in cultured human aortic ECs, the promoter of Klf4 is hypermethylated [329]. Klf4 is a key mediator of endothelial function and has been well documented to maintain an anti-inflammatory, quiescent endothelial state in unidirectional flow conditions [329, 332–334]. In this study, both DNMT3A expression and DNMT3A binding to the Klf4 promoter were found to increase due to d-flow. This led to DNA hypermethylation and decreased MEF2 binding. MEF2 is a key transcription factor that controls Klf4 upregulation in ECs [335]. DNMT inhibition by the chemical inhibitors 5Aza and RG108 rescued Klf4 expression and reversed the d-flow-induced suppression of the downstream targets eNOS and thrombomodulin. Additionally, the d-flow-induced overexpression of MCP-1 was blunted. Thus, the methylation status of key genes such as Klf4 and eNOS can be a crucial indicator of endothelial phenotype.
Genome-wide studies of DNA methylation and gene expression, recently reported by our lab and others, provided a link between mechanosensitive DNA methylation changes and atherosclerosis development in vivo. Our DNA methylome and transcriptome studies, using reduced representation bisulfite sequencing (RRBS) and microarray, respectively, revealed that d-flow regulates global DNA methylation patterns in a DNMT-dependent manner. Using the PCL model, we found a dramatic increase in DNMT1 expression in the LCA. Further, DNMT1 expression was induced by OS in cultured ECs, and inhibition of DNMT with either 5Aza or DNMT1 siRNA markedly reduced OS-induced endothelial inflammation. Additionally, 5Aza reduced lesion formation in both acute and chronic mouse models of atherosclerosis. Genome-wide RRBS and microarray studies from animals treated with 5Aza demonstrated that DNMT1 inhibition in d-flow regions reverts DNA methylation and gene expression back to the healthy conditions [330]. These DNA methylome and transcriptome results were used to identify 11 mechanosensitive genes that were hypermethylated in the promoter regions, downregulated by d-flow, and 5Aza-reversible (Figure 6). They were HoxA5, Tmem184b, Adamtsl5, Klf3, Cmklr1, Pkp4, Acvrl1, Dok4, Spry2, and Zfp46. Of these 11 genes, 5 (HoxA5, Klf3, Cmklr1, Acvrl1, and Spry2) contain CRE (cyclic AMP response element) in their promoters. The relevance of these genes to atherosclerosis and endothelial biology is currently unknown, but is under investigation. [330].
Figure 6. Methylation study in PCL model identifies 11 mechanosensitive genes regulated by promoter methylation.

Following 5Aza treatment of partially ligated animals, endothelial gDNA was collected and the methylation status was determined. On comparison between the genes identified as having hypermethylated promoters and a standard gene microarray using mRNA, 11 genes were found that hypermethylated promoters, were downregulated in the LCA, and were rescued by 5Aza treatment. (data are shown as the mean ± SEM. *P < 0.05; **P < 0.05). Gene names containing hypermethylated promoter CRE sites in are indicated with a single asterisk (*). Adapted from Dunn et al. (2014).
5.2 Transcriptomics Approach
Transcriptomics approach covers the genome-wide study of RNA (mRNA, microRNA and long noncoding RNA) expression that is primarily identified using microarray analyses. It is a widely applied technique for studying the underlying mechanism(s) of diseases, and it has also extensively been used to study expression profiles of atherosclerotic-prone or atherosclerotic vessels, [336] predominantly in human [337–339] and other experimental animals [340–344]. Advanced techniques like laser-capture microdissection have furthermore enabled scientists to isolate and analyze specific subregions within atherosclerotic vessels or lesions, such as, e.g., plaque area versus media and adventitia, etc. [345]. Furthermore innovative methods have been developed to allow enriched isolation of particular cell types, such as endothelium [43, 312, 346] or macrophages [347] for subsequent genome-wide expression profiling, enhancing our understanding of cell type-specific functions in atherogenesis. Since the use of transcriptomic approach in atherosclerosis has been reviewed else [345, 348–351], we will focus on how this approach has been used to identify mechanosensitive genes in the endothelium that play a critical role in endothelial dysfunction and atherosclerosis.
McCormick et al. studied changes in gene expression of sheared human umbilical vein ECs using microarray and identified that genes responsible for cell proliferation and differentiation, vascular tone, ECM, RNA degradation, thrombosis, chemotaxis, and inflammation were differentially regulated [352, 353]. Chen et al. investigated the effects of 24 h shear stress on gene expression profiles of HAECs by microarrays and identified that genes related to inflammatory cytokines, cell proliferation, ECM/cytoskeleton remodeling, and signal transduction were altered by long-term LS that kept the ECs quiescent under laminar flow [354]. Using a custom-designed microarray, Dekker et al. showed that the majority of flow-regulated endothelial genes are also influenced by increased cytokine levels, which results in cross-talk between flow and inflammatory-mediated downstream signaling mechanisms. They also identified a flow-sensitive endothelial-specific transcription factor LKLF [228]. Ohura et al. showed that LS, but not OS, decreased DNA synthesis and cell cycle regulators in ECs and that OS affects genes responsible for vascular remodeling, such as endothelin-1, TGFβ, collagen type IV, and ephrin A1 [355]. Viemann et al. reviewed the knowledge from these array datasets and identified that endothelial subtype heterogeneity and limited quantity of RNA samples were the important limitations for studying the global gene expression using arrays. Moreover, these datasets were obtained from in vitro ECs, which do not necessary show the same genotypic/phenotypic correlation as the ECs experiencing LS or OS in in vivo conditions. This was further complicated by a lack of robust and reproducible animal models to produce d-flow and endothelial dysfunction. To overcome this, we recently developed the PCL model (see above) [43]. We also developed a novel method to collect endothelial-enriched RNAs from the carotids of these animals, which enabled us to perform genome-wide expression analyses of mRNAs in the arterial endothelium exposed to either d-flow or laminar flow [356] (Figure 7).
Figure 7. Microarray and miRNA array from endothelial RNA in PCL model identifies novel mechanosensitive miRNAs and genes.

Following partial carotid ligation, endothelial RNA was collected either 12 or 48 hours after ligation and subject to a microarray. Both scatter plots of the normalized intensities of each probe and heat maps of single samples pooled from 3 different LCAs or RCAs show the number of genes affected by flow increase from 12 to 48 hours. The Venn diagrams also show the temporal effects of d-flow on the number of up- or down-regulated mechanosensitive genes Adapted from Ni et al. (2010) (A). Endothelial RNA collected 48 hours post-ligation (pooled from three mice) was also analyzed by miRNA array. The heat map shows several miRNAs which are differentially regulated by flow, several of which were validated by qPCR (data shown as mean±s.e.m; *P<0.05 as determined by paired t-test). Adapted from Son et al. (2013) (B).
Recently small non-coding RNAs, microRNAs (miRNAs), have emerged as important regulatory RNAs that have been implicated in gene expression regulation [357]. miRNAs interact with the 3′untranslated region (UTR) of specific target mRNAs in a sequence-specific manner, resulting in mRNA degradation or translational inhibition [357]. With the advancements in the field and availability of miRNA microarray platforms, many research groups used this “omics” approach to identify flow-sensitive regulators of endothelial transcriptome. It has been previously demonstrated that LS and OS differentially regulate the expression of miRNAs in ECs. Initially, the majority of flow-sensitive miRNAs has been identified and characterized using cultured ECs that were subjected to LS or OS conditions. Using an omics approach on cultured ECs Weber et al. showed that miR-21 is induced in ECs by shear stress and modulates apoptosis and eNOS activity [358]. Likewise, Wang et al. showed that miR-19a suppresses the expression of cyclin D1 under LS [313]. Our lab showed that miR-663 is one of the most flow sensitive miRNAs this is upregulated by OS [359]. Of these, a select few were subsequently validated in vivo. Therefore, only a limited number of direct linkages to atherogenesis have been established. Below is a summary of the current knowledge of flow-sensitive microRNAs and their role in endothelial dysfunction and atherosclerosis. Shu Chien and colleagues were the first to report flow-sensitive miRNAs (miR-19a and 23b) in cultured ECs while Peter Davies and colleagues reported miR-10a as the first flow-sensitive microRNA identified directly from the porcine endothelium in vivo [312, 319]. Although the majority of flow-sensitive microRNA have been identified using ECs in vitro, it is important to validate them in vivo since numerous mechanosensitive genes identified in vivo are known to be either dysregulated or lost during endothelial cell culture [79, 356]. Our lab identified two important and novel d-flow induced miRNAs, miR-712 and miR-205, by using the PCL model described above [79, 81] (Figure 7). These miRNAs play a critical role in regulation of the MMPs by regulating their upstream inhibitors (TIMP3 and RECK). Using a systems biology approach to integrate the knowledge from these two datasets, we identified key hub genes and important gene networks that are crucial in the pathophysiological process of atherosclerosis. Further mining of these two important datasets (d-flow-altered miRNAs and d-flow altered genes) could provide additional insight into the underlying mechanisms of d-flow-induced atherosclerosis.
5.3 Proteomics Approach
Although proteomics has been extensively used in other diseases such as cancer, there are only a few reports on proteome analysis of arterial ECs. In this section, we will summarize the proteome studies that utilized cultured ECs to identify molecular mediators of shear stress and the roles they play in the regulation of endothelial function and atherosclerosis.
Traditional techniques to understand changes at the protein level are mainly based on immunological detection methods such as Western blots and ELISAs. However, these approaches have identified some of the shear stress-responsive proteins [189, 190, 200, 288, 360–370]. But these techniques can identify only one or a few proteins at a time and heavily depend on the abundance of protein of interest in the sample and the availability of specific antibodies, thus limiting their feasibility for comprehensive analyses [371]. Using mass spectrometry-based strategies, however, the up- or downregulation of a large numbers of proteins in response to shear stress can be examined pursue the changes at the protein level in a greater details. Proteomic studies on cultured ECs involving the analysis of ECs from human and other animal sources is summarized below.
Previous studies identified that a point mutation in the 5′-flanking region of the eNOS gene, −786T→C, renders it insensitive to laminar flow, thereby suppressing its transcription. Consequently, human ECs homozygous for the eNOS mutant variant do not respond to shear stress and lead to endothelial dysfunction [372–374]. Severely reduced NO production, however, could indirectly affect the expression of other proteins in CC genotype cells. A total of 14 proteins were identified to be differentially expressed and primarily affected the NO-dependent endoplasmic reticulum stress response. Antioxidant gene manganese-containing superoxide dismutase (SOD-2) expression increased in the CC genotype ECs compared to those carrying the TT genotype and possibly contributed to an anti-atherosclerotic phenotype [375]. Shear stress not only affects eNOS expression, but also indirectly enhances eNOS activity via phosphorylation [376]. Chen et al. used a nano-LC–MS/MS-based proteomics approach and identified that AMP-activated protein kinase-dependent phosphorylation of eNOS Ser-633 in response to LS acts as a functional signaling event for NO production [376]. Similarly, Gallis et al. reported increased phosphorylation of eNOS in response to LS in bovine aortic ECs. Furthermore, using metal affinity chromatography followed by solid phase extraction capillary electrophoresis, two key phosphorylation sites on eNOS, Ser-116 and Ser-1179, were identified and activation of PI3K and Akt was found to be involved in shear stress-dependent eNOS phosphorylation and stimulation [377].
The stimulation of eNOS results in an increased production of NO, which, in turn, can interact with susceptible cysteine residues, resulting in S-nitrosylation of proteins. S-nitrosylation is an important posttranslational modification that plays a role in the modulation of cardiovascular function via the regulation of mitochondrial metabolism, intracellular Ca2+ handling, protein trafficking, and cellular defense against apoptosis and oxidative stress [378–380]. Huang et al. analyzed the changes in S-nitrosylation of reactive cysteine residues present in endothelial proteins post LS [381]. Using a similar approach, Huang et al. showed that increased S-nitrosylation on cytoskeletal proteins is critical for adaptation and remodeling of endothelium in response to laminar flow conditions [382]. Wang et al. used a proteomics approach on bovine arterial ECs to identify the effect of shear stress at multiple time points and confirmed that many previously-identified proteins change as a result of LS within a few minutes to a few hours [371]. Using a labeled quantitative proteomics approach, Freed et al. analyzed the role of phosphatidylserine in preventing apoptosis under LS [383]. Additionally, phosphatidylserine was shown to be essential for induction of the shear stress-mediated activation of the PI3K/AKT cell proliferation and survival pathway [384, 385]. In some recent studies, experimental set-ups were modified to create pulsatile shear stress by utilizing dynamic flow systems [272, 386, 387]. In ECs, exposure to shear stress for 18 h conferred protection from TNF-α-induced apoptosis through an NO-independent mechanism that relied on de novo protein synthesis [387]. Pulsatile shear stress also provides antioxidative and anti-inflammatory benefits on ECs, at least in part, by the induction of sirtuin 1 (SIRT1). Wen et al. identified phosphorylation sites based on nano-LC–MS/MS. Their study revealed that pulsatile shear stress induces an upregulation of SIRT1 and Ca2+/calmodulin-dependent protein kinase kinase β (CaMKK-β) phosphorylation of SIRT1 at Ser-27 and Ser-47. The role of CaMKK-β in SIRT1 activation was validated in mice lacking CaMKK-β or endothelial SIRT1, which shows a remarkable increase in atherosclerosis [386]. Taken together, these data from following proteomics approach suggest that both laminar as well as pulsatile shear stress provide protection to ECs via NO production as well as other mechanisms.
In contrast to laminar or pulsatile shear, Ai et al. analyzed the pathophysiological significance of SOD-2 in response to OS using LDL particles to assess protein nitration via peroxynitrite (ONOO−). The analysis of apolipoprotein B-100 (apoB-100), the protein component of LDL, by LC–MS/MS revealed that OS increased the extent of LDL protein nitration in comparison to static controls. OS also induces oxidative stress as it enhances ONOO− formation through alteration of the O2− to NO ratio, leading to protein nitrotyrosination that further induces atherosclerosis [271, 388]. Burghoff and Schrader analyzed the secretome of ECs under static and shear stress (both LS and OS) conditions using a quantitative proteomics approach and found that out of a total of 240 secreted proteins, 101 were differentially regulated under shear stress. This finding highlights the impact of shear stress on the contribution of ECs to the regulation of vascular homeostasis [389].
Studying one protein at a time, our lab also identified that LS upregulates endothelial Ca2+-activated K+ channels KCa2.3 and KCa3.1 via a Ca2+/calmodulin-dependent protein kinase kinase/Akt/p300 cascade. Laminar shear also upregulates peroxiredoxins in ECs, which serve as mechanosensitive antioxidants. Laminar shear also inhibits the activity of the protease cathepsin L in ECs. While, OS stimulates endothelial production of BMP4 and loss of BMPR2, thereby contributing to endothelial dysfunction and atherosclerosis [82, 91, 266, 390–396].
In summary, the proteomic studies represent a cutting edge tool which can be used to understand the underlying mechanisms of atherosclerosis and identify novel disease-associated biomarkers that will provide specificity and sensitivity to diagnostics and improvement in prognostics. Investigations of endothelial dysfunction and atherosclerosis using direct proteomic studies have been very challenging in particular due to the heterogeneity of the vascular tissue, hemodynamic forces, as well as that of the plaque composition. Presently, there is a limited insight into the endothelial proteome under shear stress. These studies have already identified some key shear stress-responsive proteins that were not previously known and may pave the way for future investigations to understand the mechanisms linking cause and effect in atherosclerosis. Further research is needed in order to complete understand the specific role of these proteins in activation or inhibition of specific signaling pathways with regard to the response to shear stress.
5.4 Metabolomics Approach
Metabolites are small molecule intermediates and products of cellular metabolism. Over the past several years, researchers have gleaned valuable information from analyzing the metabolite profile in human plasma. With the inventions of new platforms to analyze hundreds of biomolecules, a wealth of metabolite information is within easy grasp. The most common method for metabolic profiling uses mass spectrometry [397]. Metabolite information can be integrated with biological phenotypes and can improve our understanding of the metabolic basis of disease [397]. One of the first studies to identify differential expression of biomarkers in atherosclerosis was by Mayr et al. They found there was a decreased ratio of alanine to pyruvate in ApoE knockout mice and there was a difference in the metabolism of trimethylamine oxide (TMAO), a byproduct of cholate metabolism, in female vs. male aortas [398]. Later studies by Chen et al. found that the fatty acid palmitate significantly contributed to atherogenesis through its effects on apoptosis and inflammation [399]. Ultimately, their study concluded that the development of atherosclerosis is linked to dysfunctional fatty acid metabolism. Another study by Cheng et al. found that ApoE and LDL receptor knockout mice fed high fat diet led to significant differences in tricarboxylic acid cycle and fatty acid metabolism, and in choline metabolism, notably the choline oxidation pathway [400]. Specifically, the loss in the LDLR caused a marked reduction in the urinary excretion of betaine and dimethylglycine. Later, Wang et al. discovered that three metabolites of the dietary lipid phosphatidylcholine, choline, TMAO and betaine, were enriched in a cardiovascular disease clinical cohort. Dietary supplementation of mice with choline, TMAO, or betaine promoted upregulation of multiple macrophage scavenger receptors linked to atherosclerosis, and supplementation with choline or TMAO promoted atherosclerosis. Furthermore, they found that the interaction of dietary choline with gut flora was playing a critical role in this process [401].
Although these studies were pioneering work in the field of metabolomics and atherosclerosis, our more recent study investigated the role of d-flow on the metabolite profile in order to elucidate specific mechanisms of d-flow-induced atherosclerosis. In this study, we used blood plasma samples from ApoE−/− mice collected one week after undergoing a partial carotid ligation to induce d-flow. Mice receiving sham ligation were used as a control. A metabolome-wide association study showed that 128 metabolites were significantly altered in the ligated mice compared to the sham group. Of these, sphingomyelin (SM), a common mammalian cell membrane sphingolipid, was the most significantly increased in the ligated mice. 171 metabolites were associated with SM. 18 and 41 of the 128 discriminatory metabolites were positively and negatively correlated with SM, respectively. Furthermore, Metabolic network analysis of these 59 metabolites significantly altered by d-flow and correlated with sphingomyelin was performed for pathway maps using MetaCore. From the analysis, 13 significant metabolic networks were discovered as being altered [402] (Figure 8). These results suggest that local signaling from d-flow can induce systemic metabolic changes associated with atherosclerosis.
Figure 8. Mechanosensitive metabolites identified by the PCL model.

Metabolic network analysis on the 59 metabolites significantly altered by d-flow and correlated with sphingomyelin was performed for pathway maps using MetaCore pathway analysis software. 13 significant metabolic networks are shown with associated FDR adjusted p-value. The 59 ions significantly altered by d-flow and correlated with sphingomyelin were classified by KEGG Compound Brite mapping and % of each category is shown in a pie chart. Adapted from Go et al. (2014).
Summary and Future Perspectives
The endothelium is essential to maintaining homeostasis in the vasculature, and in turn, the effect of forces on the endothelium is critical to maintaining a healthy endothelial phenotype. In particular, fluid shear stress from blood flow is one of the stimuli that is paramount to the regulation of vessel physiology. The effect of WSS on the endothelium has far-reaching consequences in the cardiovascular system, namely the endpoints of atherosclerosis, yet the initiation can be traced back to the intracellular level. A variety of mechanosensors identified in the endothelium have been shown to be directly responsible for the change in endothelial phenotype, which in turn leads to atherosclerosis. These include PECAM1, glycocalyx, caveolins, cytoskeletal structures, integrins, AT1R, stretch-activated channels, TRPCs, and the apelin receptor. Through their effects on the levels of NO, ROS, MAPK, and Akt pathways, WSS is transduced from a physiological phenomenon to a disease in regions where d-flow arises, namely where there is low magnitude WSS in oscillating, complex flow pattern. This is due to conversion of ECs into a pro-inflammatory phenotype.
A variety of models of shear stress and atherosclerosis are currently in use. These include the cone-and-plate viscometer, the parallel plate chamber, the partial carotid ligation, and the carotid cuff model. These models directly study the effect of disturbances in WSS. However, despite the progress made in these models, we still lack answers to major questions. Current and future work is needed to identify all endothelial shear sensors, elucidate the translation of these sensors’ responses to biological responses, and how these signals synergize with systemic risk factors to promote atherosclerosis.
Furthermore, the budding field of bioinformatics allows us access to glean useful information about the pathogenesis of atherosclerosis from large datasets generated in the above models. Each large dataset, including the methylome, the transcriptome, the miRnome, the proteome, and the metabolome uncovered a variety of important genes and small molecules. Although we have not found the clear link between all of the genes and small molecules that were uncovered in these studies, in the future, pathway analyses will certainly provide connections.
Using an integrative “omics” approach, we can identify novel biomarkers and design therapeutic strategies to treat atherosclerosis. In particular, locked nucleic acids (LNAs) and siRNAs have already been developed against miRNAs and genes respectively, which have been successfully used in animal models. Likewise, methylomics studies provide novel insight into the mechanism by which flow regulates gene expression in a methylation-dependent manner and will aid in the discovery of novel anti-atherogenic therapeutic targets. In order to optimize the delivery of therapeutics, better strategies should be developed in the future to deliver these inhibitors either specifically to cells of interest or to design more specific inhibitors that can specifically block a unique but desired mRNA–miRNA interaction or gene.
Atherosclerosis occurs in arterial regions exposed to oscillating blood flow
Blood flow is sensed by endothelial cells and leads to mechanotransduction
Integrative omics approach can simplify the complex mechanotransduction signaling
Studying endothelial mechanosensitive pathways can provide clues for therapies
Acknowledgments
This work was supported by funding from National Institutes of Health grants HL119798, HL113451, HL095070 and HL124879 to HJ. HJ is John and Jan Portman Professor. RS is an American Heart Association pre-doctoral fellow.
Abbreviations
- WSS
Wall shear stress
- GC
Greater Curvature
- LC
Lesser Curvature
- PCL
Partial Carotid Ligation
- LCA
Left Carotid Artery
- RCA
Right Carotid Artery
- LDL
Low density lipoprotein
- d-flow
disturbed flow
- OS
Oscillatory Shear Stress
- LS
Laminar Shear Stress
- EC
Endothelial Cell
- NO
Nitric Oxide
- ECM
Extracellular Matrix
- MMPs
Matrix metalloproteinases
- eNOS
endothelial Nitric Oxide Synthase
- ApoE
Apolipoprotein E
- SSRE
Shear Stress Responsive Element
- PECAM1
Platelet Endothelial Cell Adhesion Molecule-1
- VEGF
vascular endothelial growth factor
- AT1R
Angiotensin Type 1 (AT1) Receptor
- TRPCs
Transient Receptor Potential Channels
- ERK
Extracellular Signal-Regulated Kinase
- GAGs
glycosaminoglycans
- PKCα
Protein Kinase C-α
- CASK
Calcium/Calmodulin-Dependent Serine Protein Kinase
- LINC
Linker of Nucleoskeleton to the Cytoskeleton
- SUN
Sad1p, UNC-84
- KASH
Klarsicht/ANC-1/Syne Homology
- GPCR
G-protein coupled receptor
- AngII
Angiotensin II
- MAPK
Mitogen Activated Protein Kinases
- FAK
Focal Adhesion Kinase
- PI3K
phosphatidylinositol-3-OH kinase
- NADPH
Nicotinamide Adenine Dinucleotide Phosphate
- ROS
Reactive Oxygen Species
- IL
interleukin
- HDAC
Histone deacetylase
- TNFα
Tumor Necrosis Factor α
- Nrf2
Nuclear factor (erythroid-derived 2)-like 2
- Keap1
Kelch-like ECH-associated protein 1
- AP-1
Activator Protein Complex
- NFkB
Nuclear Factor kappa B
- Klf
Krüppel-like factor
- BMP4
bone morphogenetic protein
- MCP-1
Monocyte Chemottractant Protein
- TRE
TPA (phorbol ester)-responsive element
- 15-H-11, 12-EETA
15(S)-Hydroxy-11,12-epoxyeicosatrienoic acid
- 11,12,15-THETA
11(R),12(S),15(S)-trihydroxyeicosatrienoic acid
- 15-LO
15-lipoxygenase
- EV
extracellular vesicles
- DNMT
DNA MethylTransferases
- RRBS
Reduced Representation Bisulfite Sequencing
- CRE
cyclic AMP Response Element
- TGFβ
Transforming Growth Factor
- miRNA
microRNA
- UTR
untranslated region
- SOD
Superoxide Dismutase
- SIRT1
sirtuin 1
- TMAO
trimethylamine oxide
- SM
Sphingomyelin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Malek AM, Alper SL, Izumo S. JAMA. 1999;282:2035–2042. doi: 10.1001/jama.282.21.2035. [DOI] [PubMed] [Google Scholar]
- 2.LaBarbera M. Science. 1990;249:992–1000. doi: 10.1126/science.2396104. [DOI] [PubMed] [Google Scholar]
- 3.Girerd X, London G, Boutouyrie P, Mourad JJ, Safar M, Laurent S. Hypertension. 1996;27:799–803. doi: 10.1161/01.hyp.27.3.799. [DOI] [PubMed] [Google Scholar]
- 4.Kamiya A, Bukhari R, Togawa T. Bull Math Biol. 1984;46:127–137. doi: 10.1007/BF02463726. [DOI] [PubMed] [Google Scholar]
- 5.Rossitti S, Lofgren J. Stroke. 1993;24:371–377. doi: 10.1161/01.str.24.3.371. [DOI] [PubMed] [Google Scholar]
- 6.Gnasso A, Carallo C, Irace C, De Franceschi MS, Mattioli PL, Motti C, Cortese C. Atherosclerosis. 2001;156:171–176. doi: 10.1016/s0021-9150(00)00617-1. [DOI] [PubMed] [Google Scholar]
- 7.Jiang Y, Kohara K, Hiwada K. Hypertens Res. 1999;22:203–207. doi: 10.1291/hypres.22.203. [DOI] [PubMed] [Google Scholar]
- 8.Joannides R, Bizet-Nafeh C, Costentin A, Iacob M, Derumeaux G, Cribier A, Thuillez C. Hypertension. 2001;38:1446–1450. doi: 10.1161/hy1201.096529. [DOI] [PubMed] [Google Scholar]
- 9.Taber LA, Ng S, Quesnel AM, Whatman J, Carmen CJ. J Biomech. 2001;34:121–124. doi: 10.1016/s0021-9290(00)00173-1. [DOI] [PubMed] [Google Scholar]
- 10.Taber LA. Biophys J. 1998;74:109–114. doi: 10.1016/S0006-3495(98)77772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zamir M. J Gen Physiol. 1977;69:449–461. doi: 10.1085/jgp.69.4.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murray CD. Proc Natl Acad Sci U S A. 1926;12:207–214. doi: 10.1073/pnas.12.3.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murray CD. J Gen Physiol. 1926;9:835–841. doi: 10.1085/jgp.9.6.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu SP, Ringgaard S, Oyre S, Hansen MS, Rasmus S, Pedersen EM. J Magn Reson Imaging. 2004;19:188–193. doi: 10.1002/jmri.10441. [DOI] [PubMed] [Google Scholar]
- 15.Kornet L, Hoeks AP, Lambregts J, Reneman RS. J Vasc Res. 2000;37:112–122. doi: 10.1159/000025722. [DOI] [PubMed] [Google Scholar]
- 16.Cheng CP, Herfkens RJ, Taylor CA. Atherosclerosis. 2003;168:323–331. doi: 10.1016/s0021-9150(03)00099-6. [DOI] [PubMed] [Google Scholar]
- 17.Gnasso A, Carallo C, Irace C, Spagnuolo V, De Novara G, Mattioli PL, Pujia A. Circulation. 1996;94:3257–3262. doi: 10.1161/01.cir.94.12.3257. [DOI] [PubMed] [Google Scholar]
- 18.Silber HA, Ouyang P, Bluemke DA, Gupta SN, Foo TK, Lima JA. Am J Physiol Heart Circ Physiol. 2005;288:H822–828. doi: 10.1152/ajpheart.00612.2004. [DOI] [PubMed] [Google Scholar]
- 19.Gaenzer H, Neumayr G, Marschang P, Sturm W, Kirchmair R, Patsch JR. J Am Coll Cardiol. 2001;38:1313–1319. doi: 10.1016/s0735-1097(01)01575-3. [DOI] [PubMed] [Google Scholar]
- 20.Tang BT, Cheng CP, Draney MT, Wilson NM, Tsao PS, Herfkens RJ, Taylor CA. Am J Physiol Heart Circ Physiol. 2006;291:H668–676. doi: 10.1152/ajpheart.01301.2005. [DOI] [PubMed] [Google Scholar]
- 21.Pedersen EM, Oyre S, Agerbaek M, Kristensen IB, Ringgaard S, Boesiger P, Paaske WP. Eur J Vasc Endovasc Surg. 1999;18:328–333. doi: 10.1053/ejvs.1999.0913. [DOI] [PubMed] [Google Scholar]
- 22.Oyre S, Pedersen EM, Ringgaard S, Boesiger P, Paaske WP. Eur J Vasc Endovasc Surg. 1997;13:263–271. doi: 10.1016/s1078-5884(97)80097-4. [DOI] [PubMed] [Google Scholar]
- 23.Oshinski JN, Ku DN, Mukundan S, Jr, Loth F, Pettigrew RI. J Magn Reson Imaging. 1995;5:640–647. doi: 10.1002/jmri.1880050605. [DOI] [PubMed] [Google Scholar]
- 24.Samijo SK, Willigers JM, Barkhuysen R, Kitslaar PJ, Reneman RS, Brands PJ, Hoeks AP. Cardiovasc Res. 1998;39:515–522. doi: 10.1016/s0008-6363(98)00074-1. [DOI] [PubMed] [Google Scholar]
- 25.Oyre S, Ringgaard S, Kozerke S, Paaske WP, Erlandsen M, Boesiger P, Pedersen EM. J Am Coll Cardiol. 1998;32:128–134. doi: 10.1016/s0735-1097(98)00207-1. [DOI] [PubMed] [Google Scholar]
- 26.Dammers R, Stifft F, Tordoir JH, Hameleers JM, Hoeks AP, Kitslaar PJ. J Appl Physiol (1985) 2003;94:485–489. doi: 10.1152/japplphysiol.00823.2002. [DOI] [PubMed] [Google Scholar]
- 27.Li YH, Reddy AK, Taffet GE, Michael LH, Entman ML, Hartley CJ. Ultrasound Med Biol. 2003;29:1281–1289. doi: 10.1016/s0301-5629(03)00986-4. [DOI] [PubMed] [Google Scholar]
- 28.Ross G, White FN, Brown AW, Kolin A. J Appl Physiol. 1966;21:1273–1275. doi: 10.1152/jappl.1966.21.4.1273. [DOI] [PubMed] [Google Scholar]
- 29.Marano G, Palazzesi S, Vergari A, Ferrari AU. Arterioscler Thromb Vasc Biol. 1999;19:2609–2614. doi: 10.1161/01.atv.19.11.2609. [DOI] [PubMed] [Google Scholar]
- 30.Kamiya A, Togawa T. Am J Physiol. 1980;239:H14–21. doi: 10.1152/ajpheart.1980.239.1.H14. [DOI] [PubMed] [Google Scholar]
- 31.Wu SP, Ringgaard S, Pedersen EM. Magn Reson Imaging. 2004;22:345–351. doi: 10.1016/j.mri.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 32.Cheng C, Helderman F, Tempel D, Segers D, Hierck B, Poelmann R, van Tol A, Duncker DJ, Robbers-Visser D, Ursem NT, van Haperen R, Wentzel JJ, Gijsen F, van der Steen AF, de Crom R, Krams R. Atherosclerosis. 2007;195:225–235. doi: 10.1016/j.atherosclerosis.2006.11.019. [DOI] [PubMed] [Google Scholar]
- 33.de Crom R, Cheng C, Helderman F, Krams R. Atherosclerosis. 2009;204:16–17. doi: 10.1016/j.atherosclerosis.2008.08.010. author reply 18–19. [DOI] [PubMed] [Google Scholar]
- 34.Masuda H, Zhuang YJ, Singh TM, Kawamura K, Murakami M, Zarins CK, Glagov S. Arterioscler Thromb Vasc Biol. 1999;19:2298–2307. doi: 10.1161/01.atv.19.10.2298. [DOI] [PubMed] [Google Scholar]
- 35.Miyashiro JK, Poppa V, Berk BC. Circ Res. 1997;81:311–319. doi: 10.1161/01.res.81.3.311. [DOI] [PubMed] [Google Scholar]
- 36.Ibrahim J, Miyashiro JK, Berk BC. Circ Res. 2003;92:1001–1009. doi: 10.1161/01.RES.0000069687.54486.B1. [DOI] [PubMed] [Google Scholar]
- 37.Lee K, Choi M, Yoon J, Jung J. Vet Radiol Ultrasound. 2004;45:166–171. doi: 10.1111/j.1740-8261.2004.04027.x. [DOI] [PubMed] [Google Scholar]
- 38.Lie M, Sejersted OM, Kiil F. Circ Res. 1970;27:727–737. doi: 10.1161/01.res.27.5.727. [DOI] [PubMed] [Google Scholar]
- 39.Serhatlioglu S, Kiris A, Kocakoc E, Canpolat I, Bozgeyik Z, Han MC. Urol Int. 2003;71:103–107. doi: 10.1159/000071105. [DOI] [PubMed] [Google Scholar]
- 40.Pohl U, Holtz J, Busse R, Bassenge E. Hypertension. 1986;8:37–44. doi: 10.1161/01.hyp.8.1.37. [DOI] [PubMed] [Google Scholar]
- 41.Samady H, Eshtehardi P, McDaniel MC, Suo J, Dhawan SS, Maynard C, Timmins LH, Quyyumi AA, Giddens DP. Circulation. 2011;124:779–788. doi: 10.1161/CIRCULATIONAHA.111.021824. [DOI] [PubMed] [Google Scholar]
- 42.Suo J, Ferrara DE, Sorescu D, Guldberg RE, Taylor WR, Giddens DP. Arterioscler Thromb Vasc Biol. 2007;27:346–351. doi: 10.1161/01.ATV.0000253492.45717.46. [DOI] [PubMed] [Google Scholar]
- 43.Nam D, Ni CW, Rezvan A, Suo J, Budzyn K, Llanos A, Harrison D, Giddens D, Jo H. American journal of physiology. Heart and circulatory physiology. 2009;297:H1535–1543. doi: 10.1152/ajpheart.00510.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Domanski M, Lloyd-Jones D, Fuster V, Grundy S. Nat Rev Cardiol. 2011;8:721–725. doi: 10.1038/nrcardio.2011.158. [DOI] [PubMed] [Google Scholar]
- 45.Glaser R, Selzer F, Faxon DP, Laskey WK, Cohen HA, Slater J, Detre KM, Wilensky RL. Circulation. 2005;111:143–149. doi: 10.1161/01.CIR.0000150335.01285.12. [DOI] [PubMed] [Google Scholar]
- 46.Finn AV, Nakano M, Narula J, Kolodgie FD, Virmani R. Arterioscler Thromb Vasc Biol. 2010;30:1282–1292. doi: 10.1161/ATVBAHA.108.179739. [DOI] [PubMed] [Google Scholar]
- 47.Finn AV, Jain RK. JACC Cardiovasc Imaging. 2010;3:41–44. doi: 10.1016/j.jcmg.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maldonado N, Kelly-Arnold A, Vengrenyuk Y, Laudier D, Fallon JT, Virmani R, Cardoso L, Weinbaum S. Am J Physiol Heart Circ Physiol. 2012;303:H619–628. doi: 10.1152/ajpheart.00036.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spain DM. Sci Am. 1966;215:48–56. doi: 10.1038/scientificamerican0866-48. [DOI] [PubMed] [Google Scholar]
- 50.Suo J, Oshinski JN, Giddens DP. Mol Cell Biomech. 2008;5:9–18. [PubMed] [Google Scholar]
- 51.Steinman DA, Taylor CA. Ann Biomed Eng. 2005;33:1704–1709. doi: 10.1007/s10439-005-8772-2. [DOI] [PubMed] [Google Scholar]
- 52.Davies PF, Polacek DC, Handen JS, Helmke BP, DePaola N. Trends Biotechnol. 1999;17:347–351. doi: 10.1016/s0167-7799(99)01348-7. [DOI] [PubMed] [Google Scholar]
- 53.Libby P. Am J Cardiol. 2000;86:3J–8J. doi: 10.1016/s0002-9149(00)01339-4. discussion 8J–9J. [DOI] [PubMed] [Google Scholar]
- 54.Caro CG, Fitz-Gerald JM, Schroter RC. Nature. 1969;223:1159–1160. doi: 10.1038/2231159a0. [DOI] [PubMed] [Google Scholar]
- 55.Caro CG. Arterioscler Thromb Vasc Biol. 2009;29:158–161. doi: 10.1161/ATVBAHA.108.166736. [DOI] [PubMed] [Google Scholar]
- 56.Caro CG, Nerem RM. Circ Res. 1973;32:187–205. doi: 10.1161/01.res.32.2.187. [DOI] [PubMed] [Google Scholar]
- 57.Friedman MH, Hutchins GM, Bargeron CB, Deters OJ, Mark FF. J Biomech Eng. 1981;103:204–207. doi: 10.1115/1.3138279. [DOI] [PubMed] [Google Scholar]
- 58.Friedman MH, Hutchins GM, Bargeron CB, Deters OJ, Mark FF. Atherosclerosis. 1981;39:425–436. doi: 10.1016/0021-9150(81)90027-7. [DOI] [PubMed] [Google Scholar]
- 59.Zarins CK, Giddens DP, Bharadvaj BK, Sottiurai VS, Mabon RF, Glagov S. Circ Res. 1983;53:502–514. doi: 10.1161/01.res.53.4.502. [DOI] [PubMed] [Google Scholar]
- 60.Ku DN, Giddens DP, Zarins CK, Glagov S. Arteriosclerosis. 1985;5:293–302. doi: 10.1161/01.atv.5.3.293. [DOI] [PubMed] [Google Scholar]
- 61.Tada S, Tarbell JM. Ann Biomed Eng. 2005;33:1202–1212. doi: 10.1007/s10439-005-5630-1. [DOI] [PubMed] [Google Scholar]
- 62.Davies PF. Nat Clin Pract Cardiovasc Med. 2009;6:16–26. doi: 10.1038/ncpcardio1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Janiczek RL, Blackman BR, Roy RJ, Meyer CH, Acton ST, Epstein FH. Magn Reson Med. 2011;66:1382–1390. doi: 10.1002/mrm.22937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nerem RM, Levesque MJ, Cornhill JF. J Biomech Eng. 1981;103:172–176. doi: 10.1115/1.3138275. [DOI] [PubMed] [Google Scholar]
- 65.Dewey CF, Jr, Bussolari SR, Gimbrone MA, Jr, Davies PF. J Biomech Eng. 1981;103:177–185. doi: 10.1115/1.3138276. [DOI] [PubMed] [Google Scholar]
- 66.Frangos JA, Eskin SG, McIntire LV, Ives CL. Science. 1985;227:1477–1479. doi: 10.1126/science.3883488. [DOI] [PubMed] [Google Scholar]
- 67.Grabowski EF, Jaffe EA, Weksler BB. J Lab Clin Med. 1985;105:36–43. [PubMed] [Google Scholar]
- 68.Mo M, Eskin SG, Schilling WP. Am J Physiol. 1991;260:H1698–1707. doi: 10.1152/ajpheart.1991.260.5.H1698. [DOI] [PubMed] [Google Scholar]
- 69.Shen J. Zhonghua Yi Xue Za Zhi. 1991;71:488–491. 434. [PubMed] [Google Scholar]
- 70.Kuchan MJ, Jo H, Frangos JA. Am J Physiol. 1994;267:C753–758. doi: 10.1152/ajpcell.1994.267.3.C753. [DOI] [PubMed] [Google Scholar]
- 71.Kuchan MJ, Frangos JA. Am J Physiol. 1994;266:C628–636. doi: 10.1152/ajpcell.1994.266.3.C628. [DOI] [PubMed] [Google Scholar]
- 72.Korenaga R, Ando J, Tsuboi H, Yang W, Sakuma I, Toyo-oka T, Kamiya A. Biochem Biophys Res Commun. 1994;198:213–219. doi: 10.1006/bbrc.1994.1030. [DOI] [PubMed] [Google Scholar]
- 73.Jo H, Dull RO, Hollis TM, Tarbell JM. Am J Physiol. 1991;260:H1992–1996. doi: 10.1152/ajpheart.1991.260.6.H1992. [DOI] [PubMed] [Google Scholar]
- 74.Warboys CM, Eric Berson R, Mann GE, Pearson JD, Weinberg PD. Acute and chronic exposure to shear stress have opposite effects on endothelial permeability to macromolecules. 2010 doi: 10.1152/ajpheart.00114.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kurose I, Miura S, Fukumura D, Tsuchiya M. Eur J Pharmacol. 1993;250:85–94. doi: 10.1016/0014-2999(93)90624-q. [DOI] [PubMed] [Google Scholar]
- 76.Baldwin AL, Thurston G, al Naemi H. Am J Physiol. 1998;274:H1776–1784. doi: 10.1152/ajpheart.1998.274.5.H1776. [DOI] [PubMed] [Google Scholar]
- 77.Hillsley MV, Tarbell JM. Biochem Biophys Res Commun. 2002;293:1466–1471. doi: 10.1016/S0006-291X(02)00410-2. [DOI] [PubMed] [Google Scholar]
- 78.Tarbell JM. Cardiovasc Res. 2010;87:320–330. doi: 10.1093/cvr/cvq146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Son DJ, Kumar S, Takabe W, Kim CW, Ni CW, Alberts-Grill N, Jang IH, Kim S, Kim W, Won Kang S, Baker AH, Woong Seo J, Ferrara KW, Jo H. Nat Commun. 2013;4:3000. doi: 10.1038/ncomms4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Huynh J, Nishimura N, Rana K, Peloquin JM, Califano JP, Montague CR, King MR, Schaffer CB, Reinhart-King CA. Sci Transl Med. 2011;3:112ra122. doi: 10.1126/scitranslmed.3002761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim CW, Kumar S, Son DJ, Jang IH, Griendling KK, Jo H. Arterioscler Thromb Vasc Biol. 2014;34:1412–1421. doi: 10.1161/ATVBAHA.113.303134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tressel SL, Huang RP, Tomsen N, Jo H. Arterioscler Thromb Vasc Biol. 2007;27:2150–2156. doi: 10.1161/ATVBAHA.107.150920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Harrison M, Smith E, Ross E, Krams R, Segers D, Buckley CD, Nash GB, Rainger GE. Arterioscler Thromb Vasc Biol. 2013;33:694–701. doi: 10.1161/ATVBAHA.112.300379. [DOI] [PubMed] [Google Scholar]
- 84.Chien S, Chiu JJ, Li YS. Am J Physiol Cell Physiol. 2009;297:C800–801. doi: 10.1152/ajpcell.00364.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chiu JJ, Usami S, Chien S. Ann Med. 2009;41:19–28. doi: 10.1080/07853890802186921. [DOI] [PubMed] [Google Scholar]
- 86.Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI. Proc Natl Acad Sci U S A. 2000;97:9052–9057. doi: 10.1073/pnas.97.16.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Passerini AG, Polacek DC, Shi C, Francesco NM, Manduchi E, Grant GR, Pritchard WF, Powell S, Chang GY, Stoeckert CJ, Jr, Davies PF. Proc Natl Acad Sci U S A. 2004;101:2482–2487. doi: 10.1073/pnas.0305938101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bussolari SR, Dewey CF, Jr, Gimbrone MA., Jr The Review of scientific instruments. 1982;53:1851–1854. doi: 10.1063/1.1136909. [DOI] [PubMed] [Google Scholar]
- 89.Franke RP, Grafe M, Schnittler H, Seiffge D, Mittermayer C, Drenckhahn D. Nature. 1984;307:648–649. doi: 10.1038/307648a0. [DOI] [PubMed] [Google Scholar]
- 90.Go YM, Boo YC, Park H, Maland MC, Patel R, Pritchard KA, Jr, Fujio Y, Walsh K, Darley-Usmar V, Jo H. J Appl Physiol (1985) 2001;91:1574–1581. doi: 10.1152/jappl.2001.91.4.1574. [DOI] [PubMed] [Google Scholar]
- 91.Jo H, Song H, Mowbray A. Antioxid Redox Signal. 2006;8:1609–1619. doi: 10.1089/ars.2006.8.1609. [DOI] [PubMed] [Google Scholar]
- 92.Frangos JA, McIntire LV, Eskin SG. Biotechnol Bioeng. 1988;32:1053–1060. doi: 10.1002/bit.260320812. [DOI] [PubMed] [Google Scholar]
- 93.Schaff UY, Xing MM, Lin KK, Pan N, Jeon NL, Simon SI. Lab Chip. 2007;7:448–456. doi: 10.1039/b617915k. [DOI] [PubMed] [Google Scholar]
- 94.Ashpole NE, Overby DR, Ethier CR, Stamer WD. Invest Ophthalmol Vis Sci. 2014;55:8067–8076. doi: 10.1167/iovs.14-14722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ganguly A, Zhang H, Sharma R, Parsons S, Patel KD. J Vis Exp. 2012:e4032. doi: 10.3791/4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rezvan A, Ni CW, Alberts-Grill N, Jo H. Antioxid Redox Signal. 2011;15:1433–1448. doi: 10.1089/ars.2010.3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bardy N, Karillon GJ, Merval R, Samuel JL, Tedgui A. Circ Res. 1995;77:684–694. doi: 10.1161/01.res.77.4.684. [DOI] [PubMed] [Google Scholar]
- 98.Gambillara V, Chambaz C, Montorzi G, Roy S, Stergiopulos N, Silacci P. Am J Physiol Heart Circ Physiol. 2006;290:H2320–2328. doi: 10.1152/ajpheart.00486.2005. [DOI] [PubMed] [Google Scholar]
- 99.Lu X, Kassab GS. J Physiol. 2004;561:575–582. doi: 10.1113/jphysiol.2004.075218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gleason RL, Gray SP, Wilson E, Humphrey JD. J Biomech Eng. 2004;126:787–795. doi: 10.1115/1.1824130. [DOI] [PubMed] [Google Scholar]
- 101.Paigen B, Morrow A, Holmes PA, Mitchell D, Williams RA. Atherosclerosis. 1987;68:231–240. doi: 10.1016/0021-9150(87)90202-4. [DOI] [PubMed] [Google Scholar]
- 102.Paigen B, Holmes PA, Mitchell D, Albee D. Atherosclerosis. 1987;64:215–221. doi: 10.1016/0021-9150(87)90249-8. [DOI] [PubMed] [Google Scholar]
- 103.Paigen B, Morrow A, Brandon C, Mitchell D, Holmes P. Atherosclerosis. 1985;57:65–73. doi: 10.1016/0021-9150(85)90138-8. [DOI] [PubMed] [Google Scholar]
- 104.Paigen B, Havens MB, Morrow A. Cancer Res. 1985;45:3850–3855. [PubMed] [Google Scholar]
- 105.Piedrahita JA, Zhang SH, Hagaman JR, Oliver PM, Maeda N. Proc Natl Acad Sci U S A. 1992;89:4471–4475. doi: 10.1073/pnas.89.10.4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Plump AS, Smith JD, Hayek T, Aalto-Setala K, Walsh A, Verstuyft JG, Rubin EM, Breslow JL. Cell. 1992;71:343–353. doi: 10.1016/0092-8674(92)90362-g. [DOI] [PubMed] [Google Scholar]
- 107.Zhang SH, Reddick RL, Piedrahita JA, Maeda N. Science. 1992;258:468–471. doi: 10.1126/science.1411543. [DOI] [PubMed] [Google Scholar]
- 108.Ishibashi S, Herz J, Maeda N, Goldstein JL, Brown MS. Proc Natl Acad Sci U S A. 1994;91:4431–4435. doi: 10.1073/pnas.91.10.4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lichtman AH, Clinton SK, Iiyama K, Connelly PW, Libby P, Cybulsky MI. Arterioscler Thromb Vasc Biol. 1999;19:1938–1944. doi: 10.1161/01.atv.19.8.1938. [DOI] [PubMed] [Google Scholar]
- 110.Hardin NJ, Minick CR, Murphy GE. Am J Pathol. 1973;73:301–326. [PMC free article] [PubMed] [Google Scholar]
- 111.Melkumyants AM, Balashov SA, Khayutin VM. Cardiovasc Res. 1989;23:741–747. doi: 10.1093/cvr/23.9.741. [DOI] [PubMed] [Google Scholar]
- 112.Clowes AW, Ryan GB, Breslow JL, Karnovsky MJ. Lab Invest. 1976;35:6–17. [PubMed] [Google Scholar]
- 113.Lindner V, Fingerle J, Reidy MA. Circ Res. 1993;73:792–796. doi: 10.1161/01.res.73.5.792. [DOI] [PubMed] [Google Scholar]
- 114.Feuerstein GZ, Ruffolo RR., Jr Eur Heart J 17 Suppl B. 1996:24–29. doi: 10.1093/eurheartj/17.suppl_b.24. [DOI] [PubMed] [Google Scholar]
- 115.Morishita T, Tsutsui M, Shimokawa H, Horiuchi M, Tanimoto A, Suda O, Tasaki H, Huang PL, Sasaguri Y, Yanagihara N, Nakashima Y. FASEB J. 2002;16:1994–1996. doi: 10.1096/fj.02-0155fje. [DOI] [PubMed] [Google Scholar]
- 116.Hollestelle SC, De Vries MR, Van Keulen JK, Schoneveld AH, Vink A, Strijder CF, Van Middelaar BJ, Pasterkamp G, Quax PH, De Kleijn DP. Circulation. 2004;109:393–398. doi: 10.1161/01.CIR.0000109140.51366.72. [DOI] [PubMed] [Google Scholar]
- 117.Lardenoye JH, Delsing DJ, de Vries MR, Deckers MM, Princen HM, Havekes LM, van Hinsbergh VW, van Bockel JH, Quax PH. Circ Res. 2000;87:248–253. doi: 10.1161/01.res.87.3.248. [DOI] [PubMed] [Google Scholar]
- 118.Sasaguri Y, Wang KY, Tanimoto A, Tsutsui M, Ueno H, Murata Y, Kohno Y, Yamada S, Ohtsu H. Circ Res. 2005;96:974–981. doi: 10.1161/01.RES.0000166325.00383.ed. [DOI] [PubMed] [Google Scholar]
- 119.da Cunha V, Martin-McNulty B, Vincelette J, Choy DF, Li WW, Schroeder M, Mahmoudi M, Halks-Miller M, Wilson DW, Vergona R, Sullivan ME, Wang YX. J Vasc Surg. 2006;44:364–371. doi: 10.1016/j.jvs.2006.04.033. [DOI] [PubMed] [Google Scholar]
- 120.Ivan E, Khatri JJ, Johnson C, Magid R, Godin D, Nandi S, Lessner S, Galis ZS. Circulation. 2002;105:2686–2691. doi: 10.1161/01.cir.0000016825.17448.11. [DOI] [PubMed] [Google Scholar]
- 121.Jonsson-Rylander AC, Nilsson T, Fritsche-Danielson R, Hammarstrom A, Behrendt M, Andersson JO, Lindgren K, Andersson AK, Wallbrandt P, Rosengren B, Brodin P, Thelin A, Westin A, Hurt-Camejo E, Lee-Sogaard CH. Arterioscler Thromb Vasc Biol. 2005;25:180–185. doi: 10.1161/01.ATV.0000150045.27127.37. [DOI] [PubMed] [Google Scholar]
- 122.Leidenfrost JE, Khan MF, Boc KP, Villa BR, Collins ET, Parks WC, Abendschein DR, Choi ET. Am J Pathol. 2003;163:773–778. doi: 10.1016/S0002-9440(10)63704-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Liu SL, Li YH, Shi GY, Tang SH, Jiang SJ, Huang CW, Liu PY, Hong JS, Wu HL. Cardiovasc Res. 2009;82:161–169. doi: 10.1093/cvr/cvp043. [DOI] [PubMed] [Google Scholar]
- 124.Nakamura K, Sasaki T, Cheng XW, Iguchi A, Sato K, Kuzuya M. Atherosclerosis. 2009;206:355–361. doi: 10.1016/j.atherosclerosis.2009.02.014. [DOI] [PubMed] [Google Scholar]
- 125.Rekhter M, Staschke K, Estridge T, Rutherford P, Jackson N, Gifford-Moore D, Foxworthy P, Reidy C, Huang XD, Kalbfleisch M, Hui K, Kuo MS, Gilmour R, Vlahos CJ. Biochem Biophys Res Commun. 2008;367:642–648. doi: 10.1016/j.bbrc.2007.12.186. [DOI] [PubMed] [Google Scholar]
- 126.Tsutsui M. J Atheroscler Thromb. 2004;11:41–48. doi: 10.5551/jat.11.41. [DOI] [PubMed] [Google Scholar]
- 127.Yamada S, Wang KY, Tanimoto A, Fan J, Shimajiri S, Kitajima S, Morimoto M, Tsutsui M, Watanabe T, Yasumoto K, Sasaguri Y. Am J Pathol. 2008;172:1419–1429. doi: 10.2353/ajpath.2008.070604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang LN, Wilson DW, da Cunha V, Sullivan ME, Vergona R, Rutledge JC, Wang YX. Arterioscler Thromb Vasc Biol. 2006;26:765–772. doi: 10.1161/01.ATV.0000207319.28254.8c. [DOI] [PubMed] [Google Scholar]
- 129.Sawchuk AP, Unthank JL, Davis TE, Dalsing MC. J Vasc Surg. 1994;19:58–63. doi: 10.1016/s0741-5214(94)70120-2. discussion 63–54. [DOI] [PubMed] [Google Scholar]
- 130.Tarbell JM, Shi ZD, Dunn J, Jo H. Annual review of fluid mechanics. 2014;46:591–614. doi: 10.1146/annurev-fluid-010313-141309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chien S. Am J Physiol Heart Circ Physiol. 2007;292:H1209–1224. doi: 10.1152/ajpheart.01047.2006. [DOI] [PubMed] [Google Scholar]
- 132.Davies PF. Arteriosclerosis Thrombosis and Vascular Biology. 2002;22:1755–1757. doi: 10.1161/01.atv.0000034391.00347.71. [DOI] [PubMed] [Google Scholar]
- 133.Chrétien ML, Zhang M, Jackson MR, Kapus A, Langille BL. Journal of Cellular Physiology. 2010;224:352–361. doi: 10.1002/jcp.22125. [DOI] [PubMed] [Google Scholar]
- 134.Osawa M, Masuda M, Harada N, Lopes RB, Fujiwara K. Eur J Cell Biol. 1997;72:229–237. [PubMed] [Google Scholar]
- 135.Osawa M, Masuda M, Kusano K, Fujiwara K. J Cell Biol. 2002;158:773–785. doi: 10.1083/jcb.200205049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tzima E, del Pozo MA, Shattil SJ, Chien S, Schwartz MA. Activation of integrins in endothelial cells by fluid shear stress mediates Rho-dependent cytoskeletal alignment. 2001 doi: 10.1093/emboj/20.17.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, Cao G, DeLisser H, Schwartz MA. Nature. 2005;437:426–431. doi: 10.1038/nature03952. [DOI] [PubMed] [Google Scholar]
- 138.Pries AR, Secomb TW, Gaehtgens P. Pflugers Arch. 2000;440:653–666. doi: 10.1007/s004240000307. [DOI] [PubMed] [Google Scholar]
- 139.Adamson RH, Clough G. J Physiol. 1992;445:473–486. doi: 10.1113/jphysiol.1992.sp018934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Koo A, Dewey CF, Jr, Garcia-Cardena G. Am J Physiol Cell Physiol. 2013;304:C137–146. doi: 10.1152/ajpcell.00187.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ihrcke NS, Platt JL. J Cell Physiol. 1996;168:625–637. doi: 10.1002/(SICI)1097-4652(199609)168:3<625::AID-JCP15>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 142.Fuster MM, Wang L. Prog Mol Biol Transl Sci. 2010;93:179–212. doi: 10.1016/S1877-1173(10)93009-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Coombe DR, Kett WC. Cell Mol Life Sci. 2005;62:410–424. doi: 10.1007/s00018-004-4293-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hileman RE, Fromm JR, Weiler JM, Linhardt RJ. Bioessays. 1998;20:156–167. doi: 10.1002/(SICI)1521-1878(199802)20:2<156::AID-BIES8>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 145.Munoz EM, Linhardt RJ. Arterioscler Thromb Vasc Biol. 2004;24:1549–1557. doi: 10.1161/01.ATV.0000137189.22999.3f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jackson RL, Busch SJ, Cardin AD. Physiol Rev. 1991;71:481–539. doi: 10.1152/physrev.1991.71.2.481. [DOI] [PubMed] [Google Scholar]
- 147.Florian JA, Kosky JR, Ainslie K, Pang Z, Dull RO, Tarbell JM. Circ Res. 2003;93:e136–142. doi: 10.1161/01.RES.0000101744.47866.D5. [DOI] [PubMed] [Google Scholar]
- 148.Hecker M, Mulsch A, Bassenge E, Busse R. Am J Physiol. 1993;265:H828–833. doi: 10.1152/ajpheart.1993.265.3.H828. [DOI] [PubMed] [Google Scholar]
- 149.Mochizuki S, Vink H, Hiramatsu O, Kajita T, Shigeto F, Spaan JA, Kajiya F. Am J Physiol Heart Circ Physiol. 2003;285:H722–726. doi: 10.1152/ajpheart.00691.2002. [DOI] [PubMed] [Google Scholar]
- 150.Moon JJ, Matsumoto M, Patel S, Lee L, Guan JL, Li S. J Cell Physiol. 2005;203:166–176. doi: 10.1002/jcp.20220. [DOI] [PubMed] [Google Scholar]
- 151.Thi MM, Tarbell JM, Weinbaum S, Spray DC. Proc Natl Acad Sci U S A. 2004;101:16483–16488. doi: 10.1073/pnas.0407474101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M. Annu Rev Biochem. 1999;68:729–777. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- 153.Rizzo V, Morton C, DePaola N, Schnitzer JE, Davies PF. Am J Physiol Heart Circ Physiol. 2003;285:H1720–1729. doi: 10.1152/ajpheart.00344.2002. [DOI] [PubMed] [Google Scholar]
- 154.Li Y, Ouyang J, Zheng H, Yu Z, Wang B. Sheng Wu Yi Xue Gong Cheng Xue Za Zhi. 2005;22:1020–1023. [PubMed] [Google Scholar]
- 155.Balligand JL, Feron O, Dessy C. Physiol Rev. 2009;89:481–534. doi: 10.1152/physrev.00042.2007. [DOI] [PubMed] [Google Scholar]
- 156.Arnal JF, Dinh-Xuan AT, Pueyo M, Darblade B, Rami J. Cell Mol Life Sci. 1999;55:1078–1087. doi: 10.1007/s000180050358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Yang B, Rizzo V. Cell Mol Bioeng. 2013;6:346–354. doi: 10.1007/s12195-013-0276-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Tkachenko E, Rhodes JM, Simons M. Circ Res. 2005;96:488–500. doi: 10.1161/01.RES.0000159708.71142.c8. [DOI] [PubMed] [Google Scholar]
- 159.Yoneda A, Couchman JR. Matrix Biol. 2003;22:25–33. doi: 10.1016/s0945-053x(03)00010-6. [DOI] [PubMed] [Google Scholar]
- 160.Yoo J, Jeong MJ, Cho HJ, Oh ES, Han MY. Biochem Biophys Res Commun. 2005;328:424–431. doi: 10.1016/j.bbrc.2004.12.179. [DOI] [PubMed] [Google Scholar]
- 161.Essner JJ, Chen E, Ekker SC. Int J Biochem Cell Biol. 2006;38:152–156. doi: 10.1016/j.biocel.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 162.Simons M, Horowitz A. Cell Signal. 2001;13:855–862. doi: 10.1016/s0898-6568(01)00190-5. [DOI] [PubMed] [Google Scholar]
- 163.Zimmermann P, David G. FASEB J. 1999;13(Suppl):S91–S100. doi: 10.1096/fasebj.13.9001.s91. [DOI] [PubMed] [Google Scholar]
- 164.Park PW, Reizes O, Bernfield M. J Biol Chem. 2000;275:29923–29926. doi: 10.1074/jbc.R000008200. [DOI] [PubMed] [Google Scholar]
- 165.Rost B. J Struct Biol. 2001;134:204–218. doi: 10.1006/jsbi.2001.4336. [DOI] [PubMed] [Google Scholar]
- 166.Lin K, Simossis VA, Taylor WR, Heringa J. Bioinformatics. 2005;21:152–159. doi: 10.1093/bioinformatics/bth487. [DOI] [PubMed] [Google Scholar]
- 167.Juliano RL, Haskill S. J Cell Biol. 1993;120:577–585. doi: 10.1083/jcb.120.3.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Schwartz MA, Schaller MD, Ginsberg MH. Annu Rev Cell Dev Biol. 1995;11:549–599. doi: 10.1146/annurev.cb.11.110195.003001. [DOI] [PubMed] [Google Scholar]
- 169.Hynes RO. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 170.Hynes RO. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 171.Hynes RO. Nat Med. 2002;8:918–921. doi: 10.1038/nm0902-918. [DOI] [PubMed] [Google Scholar]
- 172.Loftus JC, Liddington RC. J Clin Invest. 1997;99:2302–2306. doi: 10.1172/JCI119408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Humphries MJ, Newham P. Trends Cell Biol. 1998;8:78–83. [PubMed] [Google Scholar]
- 174.Jalali S, del Pozo MA, Chen K-D, Miao H, Li Y-S, Schwartz MA, Shyy JY-J, Chien S. Proceedings of the National Academy of Sciences. 2001;98:1042–1046. doi: 10.1073/pnas.031562998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Liu S, Calderwood DA, Ginsberg MH. J Cell Sci. 2000;113(Pt 20):3563–3571. doi: 10.1242/jcs.113.20.3563. [DOI] [PubMed] [Google Scholar]
- 176.Verma SK, Lal H, Golden HB, Gerilechaogetu F, Smith M, Guleria RS, Foster DM, Lu G, Dostal DE. Cardiovasc Res. 2011;90:88–96. doi: 10.1093/cvr/cvq385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lal H, Verma SK, Smith M, Guleria RS, Lu G, Foster DM, Dostal DE. J Mol Cell Cardiol. 2007;43:137–147. doi: 10.1016/j.yjmcc.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Urbich C, Dernbach E, Reissner A, Vasa M, Zeiher AM, Dimmeler S. Arterioscler Thromb Vasc Biol. 2002;22:69–75. doi: 10.1161/hq0102.101518. [DOI] [PubMed] [Google Scholar]
- 179.Chavakis E, Aicher A, Heeschen C, Sasaki K, Kaiser R, El Makhfi N, Urbich C, Peters T, Scharffetter-Kochanek K, Zeiher AM, Chavakis T, Dimmeler S. J Exp Med. 2005;201:63–72. doi: 10.1084/jem.20041402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Sims JR, Karp S, Ingber DE. J Cell Sci. 1992;103(Pt 4):1215–1222. doi: 10.1242/jcs.103.4.1215. [DOI] [PubMed] [Google Scholar]
- 181.Maniotis AJ, Chen CS, Ingber DE. Proc Natl Acad Sci U S A. 1997;94:849–854. doi: 10.1073/pnas.94.3.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wang N, Tolic-Norrelykke IM, Chen J, Mijailovich SM, Butler JP, Fredberg JJ, Stamenovic D. Am J Physiol Cell Physiol. 2002;282:C606–616. doi: 10.1152/ajpcell.00269.2001. [DOI] [PubMed] [Google Scholar]
- 183.Stamenovic D, Mijailovich SM, Tolic-Norrelykke IM, Chen J, Wang N. Am J Physiol Cell Physiol. 2002;282:C617–624. doi: 10.1152/ajpcell.00271.2001. [DOI] [PubMed] [Google Scholar]
- 184.Wang N, Naruse K, Stamenovic D, Fredberg JJ, Mijailovich SM, Tolic-Norrelykke IM, Polte T, Mannix R, Ingber DE. Proc Natl Acad Sci U S A. 2001;98:7765–7770. doi: 10.1073/pnas.141199598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Ingber DE. J Cell Sci. 2003;116:1157–1173. doi: 10.1242/jcs.00359. [DOI] [PubMed] [Google Scholar]
- 186.Chen CS, Ingber DE. Osteoarthritis Cartilage. 1999;7:81–94. doi: 10.1053/joca.1998.0164. [DOI] [PubMed] [Google Scholar]
- 187.Ingber DE. Annu Rev Physiol. 1997;59:575–599. doi: 10.1146/annurev.physiol.59.1.575. [DOI] [PubMed] [Google Scholar]
- 188.Poh YC, Shevtsov SP, Chowdhury F, Wu DC, Na S, Dundr M, Wang N. Nat Commun. 2012;3:866. doi: 10.1038/ncomms1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Pajerowski JD, Dahl KN, Zhong FL, Sammak PJ, Discher DE. Proc Natl Acad Sci U S A. 2007;104:15619–15624. doi: 10.1073/pnas.0702576104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, Stewart CL, Lee RT. J Clin Invest. 2004;113:370–378. doi: 10.1172/JCI19670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Lammerding J, Fong LG, Ji JY, Reue K, Stewart CL, Young SG, Lee RT. J Biol Chem. 2006;281:25768–25780. doi: 10.1074/jbc.M513511200. [DOI] [PubMed] [Google Scholar]
- 192.Lammerding J, Lee RT. Methods Mol Biol. 2009;464:13–26. doi: 10.1007/978-1-60327-461-6_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Lombardi ML, Jaalouk DE, Shanahan CM, Burke B, Roux KJ, Lammerding J. J Biol Chem. 2011;286:26743–26753. doi: 10.1074/jbc.M111.233700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Guilak F. J Biomech. 1995;28:1529–1541. doi: 10.1016/0021-9290(95)00100-x. [DOI] [PubMed] [Google Scholar]
- 195.Guilak F, Tedrow JR, Burgkart R. Biochem Biophys Res Commun. 2000;269:781–786. doi: 10.1006/bbrc.2000.2360. [DOI] [PubMed] [Google Scholar]
- 196.Horn HF, Brownstein Z, Lenz DR, Shivatzki S, Dror AA, Dagan-Rosenfeld O, Friedman LM, Roux KJ, Kozlov S, Jeang KT, Frydman M, Burke B, Stewart CL, Avraham KB. J Clin Invest. 2013;123:740–750. doi: 10.1172/JCI66911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Wang W, Shi Z, Jiao S, Chen C, Wang H, Liu G, Wang Q, Zhao Y, Greene MI, Zhou Z. Cell Res. 2012;22:1440–1452. doi: 10.1038/cr.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Simon DN, Wilson KL. Nat Rev Mol Cell Biol. 2011;12:695–708. doi: 10.1038/nrm3207. [DOI] [PubMed] [Google Scholar]
- 199.Starr DA, Fridolfsson HN. Annu Rev Cell Dev Biol. 2010;26:421–444. doi: 10.1146/annurev-cellbio-100109-104037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Crisp M, Liu Q, Roux K, Rattner JB, Shanahan C, Burke B, Stahl PD, Hodzic D. J Cell Biol. 2006;172:41–53. doi: 10.1083/jcb.200509124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Burke B, Roux KJ. Dev Cell. 2009;17:587–597. doi: 10.1016/j.devcel.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 202.Wang N, Tytell JD, Ingber DE. Nat Rev Mol Cell Biol. 2009;10:75–82. doi: 10.1038/nrm2594. [DOI] [PubMed] [Google Scholar]
- 203.Ho CY, Lammerding J. J Cell Sci. 2012;125:2087–2093. doi: 10.1242/jcs.087288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Tkachenko E, Gutierrez E, Saikin SK, Fogelstrand P, Kim C, Groisman A, Ginsberg MH. Biol Open. 2013;2:1007–1012. doi: 10.1242/bio.20134622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Morgan JT, Wood JA, Shah NM, Hughbanks ML, Russell P, Barakat AI, Murphy CJ. Biomaterials. 2012;33:4126–4135. doi: 10.1016/j.biomaterials.2012.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Dragt BS, van Agtmaal EL, de Laat B, Voorberg J. Thromb Res. 2012;130:741–745. doi: 10.1016/j.thromres.2012.08.301. [DOI] [PubMed] [Google Scholar]
- 207.Fedorchak GR, Kaminski A, Lammerding J. Prog Biophys Mol Biol. 2014;115:76–92. doi: 10.1016/j.pbiomolbio.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Zimmerman B, Beautrait A, Aguila B, Charles R, Escher E, Claing A, Bouvier M, Laporte SA. Sci Signal. 2012;5:ra33. doi: 10.1126/scisignal.2002522. [DOI] [PubMed] [Google Scholar]
- 209.Mehta PK, Griendling KK. Am J Physiol Cell Physiol. 2007;292:C82–97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 210.Yatabe J, Sanada H, Yatabe MS, Hashimoto S, Yoneda M, Felder RA, Jose PA, Watanabe T. Am J Physiol Renal Physiol. 2009;296:F1052–1060. doi: 10.1152/ajprenal.00580.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Zou Y, Akazawa H, Qin Y, Sano M, Takano H, Minamino T, Makita N, Iwanaga K, Zhu W, Kudoh S, Toko H, Tamura K, Kihara M, Nagai T, Fukamizu A, Umemura S, Iiri T, Fujita T, Komuro I. Nat Cell Biol. 2004;6:499–506. doi: 10.1038/ncb1137. [DOI] [PubMed] [Google Scholar]
- 212.Yasuda N, Akazawa H, Qin Y, Zou Y, Komuro I. Naunyn Schmiedebergs Arch Pharmacol. 2008;377:393–399. doi: 10.1007/s00210-007-0215-1. [DOI] [PubMed] [Google Scholar]
- 213.Storch U, Mederos y Schnitzler M, Gudermann T. Am J Physiol Heart Circ Physiol. 2012;302:H1241–1249. doi: 10.1152/ajpheart.00818.2011. [DOI] [PubMed] [Google Scholar]
- 214.Rakesh K, Yoo B, Kim IM, Salazar N, Kim KS, Rockman HA. Sci Signal. 2010;3:ra46. doi: 10.1126/scisignal.2000769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Mederos y Schnitzler M, Storch U, Gudermann T. Curr Opin Pharmacol. 2011;11:112–116. doi: 10.1016/j.coph.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 216.Chatzizisis YS, Coskun AU, Jonas M, Edelman ER, Feldman CL, Stone PH. Journal of the American College of Cardiology. 2007;49:2379–2393. doi: 10.1016/j.jacc.2007.02.059. [DOI] [PubMed] [Google Scholar]
- 217.Traub O, Hertlein B, Kasper M, Eckert R, Krisciukaitis A, Hulser D, Willecke K. Eur J Cell Biol. 1998;77:313–322. doi: 10.1016/S0171-9335(98)80090-3. [DOI] [PubMed] [Google Scholar]
- 218.Hwang J, Ing MH, Salazar A, Lassegue B, Griendling K, Navab M, Sevanian A, Hsiai TK. Circ Res. 2003;93:1225–1232. doi: 10.1161/01.RES.0000104087.29395.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Shyy JY, Chien S. Circ Res. 2002;91:769–775. doi: 10.1161/01.res.0000038487.19924.18. [DOI] [PubMed] [Google Scholar]
- 220.Liu Y, Chen BP, Lu M, Zhu Y, Stemerman MB, Chien S, Shyy JY. Arterioscler Thromb Vasc Biol. 2002;22:76–81. doi: 10.1161/hq0102.101822. [DOI] [PubMed] [Google Scholar]
- 221.Topper JN, Cai J, Falb D, Gimbrone MA., Jr Proc Natl Acad Sci U S A. 1996;93:10417–10422. doi: 10.1073/pnas.93.19.10417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Wang N, Miao H, Li YS, Zhang P, Haga JH, Hu Y, Young A, Yuan S, Nguyen P, Wu CC, Chien S. Biochem Biophys Res Commun. 2006;341:1244–1251. doi: 10.1016/j.bbrc.2006.01.089. [DOI] [PubMed] [Google Scholar]
- 223.Takabe W, Warabi E, Noguchi N. Antioxid Redox Signal. 2011;15:1415–1426. doi: 10.1089/ars.2010.3433. [DOI] [PubMed] [Google Scholar]
- 224.Dimmeler S, Zeiher AM. Cell Death Differ. 1999;6:964–968. doi: 10.1038/sj.cdd.4400581. [DOI] [PubMed] [Google Scholar]
- 225.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 226.Hecker M, Fleming I, Busse R. Agents Actions Suppl. 1995;45:119–127. doi: 10.1007/978-3-0348-7346-8_17. [DOI] [PubMed] [Google Scholar]
- 227.Nayak L, Lin Z, Jain MK. Antioxid Redox Signal. 2011;15:1449–1461. doi: 10.1089/ars.2010.3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Dekker RJ, van Soest S, Fontijn RD, Salamanca S, de Groot PG, VanBavel E, Pannekoek H, Horrevoets AJG. Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Krüppel-like factor (KLF2) 2002 doi: 10.1182/blood-2002-01-0046. [DOI] [PubMed] [Google Scholar]
- 229.Das H, Kumar A, Lin Z, Patino WD, Hwang PM, Feinberg MW, Majumder PK, Jain MK. Proc Natl Acad Sci U S A. 2006;103:6653–6658. doi: 10.1073/pnas.0508235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Lin Z, Hamik A, Jain R, Kumar A, Jain MK. Arterioscler Thromb Vasc Biol. 2006;26:1185–1189. doi: 10.1161/01.ATV.0000215638.53414.99. [DOI] [PubMed] [Google Scholar]
- 231.SenBanerjee S, Lin Z, Atkins GB, Greif DM, Rao RM, Kumar A, Feinberg MW, Chen Z, Simon DI, Luscinskas FW, Michel TM, Gimbrone MA, Jr, Garcia-Cardena G, Jain MK. J Exp Med. 2004;199:1305–1315. doi: 10.1084/jem.20031132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Fledderus JO, Boon RA, Volger OL, Hurttila H, Yla-Herttuala S, Pannekoek H, Levonen AL, Horrevoets AJ. Arterioscler Thromb Vasc Biol. 2008;28:1339–1346. doi: 10.1161/ATVBAHA.108.165811. [DOI] [PubMed] [Google Scholar]
- 233.Boon RA, Leyen TA, Fontijn RD, Fledderus JO, Baggen JM, Volger OL, van Nieuw Amerongen GP, Horrevoets AJ. Blood. 2010;115:2533–2542. doi: 10.1182/blood-2009-06-228726. [DOI] [PubMed] [Google Scholar]
- 234.Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, Di Vizio D, Hou H, Jr, Kneitz B, Lagaud G, Christ GJ, Edelmann W, Lisanti MP. J Biol Chem. 2001;276:38121–38138. doi: 10.1074/jbc.M105408200. [DOI] [PubMed] [Google Scholar]
- 235.Goodwin BL, Solomonson LP, Eichler DC. J Biol Chem. 2004;279:18353–18360. doi: 10.1074/jbc.M308160200. [DOI] [PubMed] [Google Scholar]
- 236.Dekker RJ, van Thienen JV, Rohlena J, de Jager SC, Elderkamp YW, Seppen J, de Vries CJ, Biessen EA, van Berkel TJ, Pannekoek H, Horrevoets AJ. Am J Pathol. 2005;167:609–618. doi: 10.1016/S0002-9440(10)63002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Dekker RJ, Boon RA, Rondaij MG, Kragt A, Volger OL, Elderkamp YW, Meijers JC, Voorberg J, Pannekoek H, Horrevoets AJ. Blood. 2006;107:4354–4363. doi: 10.1182/blood-2005-08-3465. [DOI] [PubMed] [Google Scholar]
- 238.Boon RA, Fledderus JO, Volger OL, van Wanrooij EJ, Pardali E, Weesie F, Kuiper J, Pannekoek H, ten Dijke P, Horrevoets AJ. Arterioscler Thromb Vasc Biol. 2007;27:532–539. doi: 10.1161/01.ATV.0000256466.65450.ce. [DOI] [PubMed] [Google Scholar]
- 239.Bhattacharya R, Senbanerjee S, Lin Z, Mir S, Hamik A, Wang P, Mukherjee P, Mukhopadhyay D, Jain MK. J Biol Chem. 2005;280:28848–28851. doi: 10.1074/jbc.C500200200. [DOI] [PubMed] [Google Scholar]
- 240.Parmar KM, Larman HB, Dai G, Zhang Y, Wang ET, Moorthy SN, Kratz JR, Lin Z, Jain MK, Gimbrone MA, Jr, Garcia-Cardena G. J Clin Invest. 2006;116:49–58. doi: 10.1172/JCI24787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Wang W, Ha CH, Jhun BS, Wong C, Jain MK, Jin ZG. Blood. 2010;115:2971–2979. doi: 10.1182/blood-2009-05-224824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.van Thienen JV, Fledderus JO, Dekker RJ, Rohlena J, van Ijzendoorn GA, Kootstra NA, Pannekoek H, Horrevoets AJ. Cardiovasc Res. 2006;72:231–240. doi: 10.1016/j.cardiores.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 243.Rhee WJ, Ni CW, Zheng Z, Chang K, Jo H, Bao G. Proc Natl Acad Sci U S A. 2010;107:6858–6863. doi: 10.1073/pnas.1000444107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Kumar A, Lin Z, SenBanerjee S, Jain MK. Molecular and cellular biology. 2005;25:5893–5903. doi: 10.1128/MCB.25.14.5893-5903.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Woo CH, Shishido T, McClain C, Lim JH, Li JD, Yang J, Yan C, Abe J. Circ Res. 2008;102:538–545. doi: 10.1161/CIRCRESAHA.107.156877. [DOI] [PubMed] [Google Scholar]
- 246.Kumar A, Hoffman TA, Dericco J, Naqvi A, Jain MK, Irani K. Faseb j. 2009;23:4344–4352. doi: 10.1096/fj.09-138743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Yoshida T, Kaestner KH, Owens GK. Circ Res. 2008;102:1548–1557. doi: 10.1161/CIRCRESAHA.108.176974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Duerrschmidt N, Stielow C, Muller G, Pagano PJ, Morawietz H. J Physiol. 2006;576:557–567. doi: 10.1113/jphysiol.2006.111070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Doehner W, Schoene N, Rauchhaus M, Leyva-Leon F, Pavitt DV, Reaveley DA, Schuler G, Coats AJ, Anker SD, Hambrecht R. Circulation. 2002;105:2619–2624. doi: 10.1161/01.cir.0000017502.58595.ed. [DOI] [PubMed] [Google Scholar]
- 250.Kumar S, Sud N, Fonseca FV, Hou Y, Black SM. American journal of physiology. Lung cellular and molecular physiology. 2010;298:L105–116. doi: 10.1152/ajplung.00290.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Kang-Decker N, Cao S, Chatterjee S, Yao J, Egan LJ, Semela D, Mukhopadhyay D, Shah V. J Cell Sci. 2007;120:492–501. doi: 10.1242/jcs.03361. [DOI] [PubMed] [Google Scholar]
- 252.McNally JS, Davis ME, Giddens DP, Saha A, Hwang J, Dikalov S, Jo H, Harrison DG. Am J Physiol Heart Circ Physiol. 2003;285:H2290–2297. doi: 10.1152/ajpheart.00515.2003. [DOI] [PubMed] [Google Scholar]
- 253.Ziegler T, Silacci P, Harrison VJ, Hayoz D. Hypertension. 1998;32:351–355. doi: 10.1161/01.hyp.32.2.351. [DOI] [PubMed] [Google Scholar]
- 254.Xiao Z, Zhang Z, Ranjan V, Diamond SL. J Cell Physiol. 1997;171:205–211. doi: 10.1002/(SICI)1097-4652(199705)171:2<205::AID-JCP11>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 255.Redmond EM, Cahill PA, Sitzmann JV. Arterioscler Thromb Vasc Biol. 1998;18:75–83. doi: 10.1161/01.atv.18.1.75. [DOI] [PubMed] [Google Scholar]
- 256.Hori N, Wiest R, Groszmann RJ. Hepatology (Baltimore Md) 1998;28:1467–1473. doi: 10.1002/hep.510280604. [DOI] [PubMed] [Google Scholar]
- 257.Villa LM, Salas E, Darley-Usmar VM, Radomski MW, Moncada S. Proc Natl Acad Sci U S A. 1994;91:12383–12387. doi: 10.1073/pnas.91.26.12383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Hartsfield CL, Alam J, Cook JL, Choi AM. Am J Physiol. 1997;273:L980–988. doi: 10.1152/ajplung.1997.273.5.L980. [DOI] [PubMed] [Google Scholar]
- 259.Bouloumie A, Bauersachs J, Linz W, Scholkens BA, Wiemer G, Fleming I, Busse R. Hypertension. 1997;30:934–941. doi: 10.1161/01.hyp.30.4.934. [DOI] [PubMed] [Google Scholar]
- 260.Zingarelli B, Hasko G, Salzman AL, Szabo C. Critical care medicine. 1999;27:1701–1707. doi: 10.1097/00003246-199909000-00001. [DOI] [PubMed] [Google Scholar]
- 261.Li JM, Shah AM. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1014–1030. doi: 10.1152/ajpregu.00124.2004. [DOI] [PubMed] [Google Scholar]
- 262.Kalyanaraman B, Darley-Usmar V, Davies KJ, Dennery PA, Forman HJ, Grisham MB, Mann GE, Moore K, Roberts LJ, 2nd, Ischiropoulos H. Free Radic Biol Med. 2012;52:1–6. doi: 10.1016/j.freeradbiomed.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Alfieri A, Srivastava S, Siow RC, Cash D, Modo M, Duchen MR, Fraser PA, Williams SC, Mann GE. Free Radic Biol Med. 2013;65:1012–1022. doi: 10.1016/j.freeradbiomed.2013.08.190. [DOI] [PubMed] [Google Scholar]
- 264.Tsao PS, Lewis NP, Alpert S, Cooke JP. Circulation. 1995;92:3513–3519. doi: 10.1161/01.cir.92.12.3513. [DOI] [PubMed] [Google Scholar]
- 265.Noris M, Morigi M, Donadelli R, Aiello S, Foppolo M, Todeschini M, Orisio S, Remuzzi G, Remuzzi A. Circ Res. 1995;76:536–543. doi: 10.1161/01.res.76.4.536. [DOI] [PubMed] [Google Scholar]
- 266.Hwang J, Saha A, Boo YC, Sorescu GP, McNally JS, Holland SM, Dikalov S, Giddens DP, Griendling KK, Harrison DG, Jo H. J Biol Chem. 2003;278:47291–47298. doi: 10.1074/jbc.M305150200. [DOI] [PubMed] [Google Scholar]
- 267.Dickhout JG, Hossain GS, Pozza LM, Zhou J, Lhotak S, Austin RC. Arterioscler Thromb Vasc Biol. 2005;25:2623–2629. doi: 10.1161/01.ATV.0000189159.96900.d9. [DOI] [PubMed] [Google Scholar]
- 268.Chappell DC, Varner SE, Nerem RM, Medford RM, Alexander RW. Circ Res. 1998;82:532–539. doi: 10.1161/01.res.82.5.532. [DOI] [PubMed] [Google Scholar]
- 269.Brooks AR, Lelkes PI, Rubanyi GM. Physiol Genomics. 2002;9:27–41. doi: 10.1152/physiolgenomics.00075.2001. [DOI] [PubMed] [Google Scholar]
- 270.Silacci P, Formentin K, Bouzourene K, Daniel F, Brunner HR, Hayoz D. Nitric oxide : biology and chemistry/official journal of the Nitric Oxide Society. 2000;4:47–56. doi: 10.1006/niox.2000.0271. [DOI] [PubMed] [Google Scholar]
- 271.Hsiai TK, Hwang J, Barr ML, Correa A, Hamilton R, Alavi M, Rouhanizadeh M, Cadenas E, Hazen SL. Free Radic Biol Med. 2007;42:519–529. doi: 10.1016/j.freeradbiomed.2006.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Ai L, Rouhanizadeh M, Wu JC, Takabe W, Yu H, Alavi M, Li R, Chu Y, Miller J, Heistad DD, Hsiai TK. Am J Physiol Cell Physiol. 2008;294:C1576–1585. doi: 10.1152/ajpcell.00518.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Cunningham KS, Gotlieb AI. Lab Invest. 2005;85:9–23. doi: 10.1038/labinvest.3700215. [DOI] [PubMed] [Google Scholar]
- 274.Warabi E, Takabe W, Minami T, Inoue K, Itoh K, Yamamoto M, Ishii T, Kodama T, Noguchi N. Free Radic Biol Med. 2007;42:260–269. doi: 10.1016/j.freeradbiomed.2006.10.043. [DOI] [PubMed] [Google Scholar]
- 275.Hsieh CY, Hsiao HY, Wu WY, Liu CA, Tsai YC, Chao YJ, Wang DL, Hsieh HJ. Journal of biomedical science. 2009;16:12. doi: 10.1186/1423-0127-16-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Okouchi M, Okayama N, Alexander JS, Aw TY. Current neurovascular research. 2006;3:249–261. doi: 10.2174/156720206778792876. [DOI] [PubMed] [Google Scholar]
- 277.Acar N, Soylu H, Edizer I, Ozbey O, Er H, Akkoyunlu G, Gemici B, Ustunel I. Acta histochemica. 2014;116:1289–1300. doi: 10.1016/j.acthis.2014.07.012. [DOI] [PubMed] [Google Scholar]
- 278.Zhang Y, Crouch DH, Yamamoto M, Hayes JD. The Biochemical journal. 2006;399:373–385. doi: 10.1042/BJ20060725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Meadows SM, Salanga MC, Krieg PA. Development (Cambridge England) 2009;136:1115–1125. doi: 10.1242/dev.029538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Wu J, Bohanan CS, Neumann JC, Lingrel JB. J Biol Chem. 2008;283:3942–3950. doi: 10.1074/jbc.M707882200. [DOI] [PubMed] [Google Scholar]
- 281.Knorr-Wittmann C, Hengstermann A, Gebel S, Alam J, Muller T. Free Radic Biol Med. 2005;39:1438–1448. doi: 10.1016/j.freeradbiomed.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 282.Kataoka K, Handa H, Nishizawa M. J Biol Chem. 2001;276:34074–34081. doi: 10.1074/jbc.M105383200. [DOI] [PubMed] [Google Scholar]
- 283.Chen XL, Varner SE, Rao AS, Grey JY, Thomas S, Cook CK, Wasserman MA, Medford RM, Jaiswal AK, Kunsch C. J Biol Chem. 2003;278:703–711. doi: 10.1074/jbc.M203161200. [DOI] [PubMed] [Google Scholar]
- 284.Dai G, Vaughn S, Zhang Y, Wang ET, Garcia-Cardena G, Gimbrone MA., Jr Circ Res. 2007;101:723–733. doi: 10.1161/CIRCRESAHA.107.152942. [DOI] [PubMed] [Google Scholar]
- 285.Han Z, Chen YR, Jones CI, 3rd, Meenakshisundaram G, Zweier JL, Alevriadou BR. Am J Physiol Cell Physiol. 2007;292:C1103–1112. doi: 10.1152/ajpcell.00389.2006. [DOI] [PubMed] [Google Scholar]
- 286.Hosoya T, Maruyama A, Kang MI, Kawatani Y, Shibata T, Uchida K, Warabi E, Noguchi N, Itoh K, Yamamoto M. J Biol Chem. 2005;280:27244–27250. doi: 10.1074/jbc.M502551200. [DOI] [PubMed] [Google Scholar]
- 287.Jones CI, 3rd, Zhu H, Martin SF, Han Z, Li Y, Alevriadou BR. Ann Biomed Eng. 2007;35:683–693. doi: 10.1007/s10439-007-9279-9. [DOI] [PubMed] [Google Scholar]
- 288.Nagel T, Resnick N, Atkinson WJ, Dewey CF, Jr, Gimbrone MA., Jr J Clin Invest. 1994;94:885–891. doi: 10.1172/JCI117410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Rhee WJ, Santangelo PJ, Jo H, Bao G. Nucleic Acids Res. 2008;36:e30. doi: 10.1093/nar/gkn039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Zhou Z, Liu Y, Miao AD, Wang SQ. Eur J Pharmacol. 2005;513:1–8. doi: 10.1016/j.ejphar.2005.01.059. [DOI] [PubMed] [Google Scholar]
- 291.Ueno H, Pradhan S, Schlessel D, Hirasawa H, Sumpio BE. Cardiovasc Toxicol. 2006;6:39–50. doi: 10.1385/ct:6:1:39. [DOI] [PubMed] [Google Scholar]
- 292.Lin SJ, Shyue SK, Hung YY, Chen YH, Ku HH, Chen JW, Tam KB, Chen YL. Arterioscler Thromb Vasc Biol. 2005;25:334–340. doi: 10.1161/01.ATV.0000152114.00114.d8. [DOI] [PubMed] [Google Scholar]
- 293.Ishii H, Takada K. Toxicol Appl Pharmacol. 2002;184:88–97. [PubMed] [Google Scholar]
- 294.Hubbard AK, Rothlein R. Free Radic Biol Med. 2000;28:1379–1386. doi: 10.1016/s0891-5849(00)00223-9. [DOI] [PubMed] [Google Scholar]
- 295.Fan H, Sun B, Gu Q, Lafond-Walker A, Cao S, Becker LC. Am J Physiol Heart Circ Physiol. 2002;282:H1778–1786. doi: 10.1152/ajpheart.00796.2000. [DOI] [PubMed] [Google Scholar]
- 296.Suzuki Y, Wang W, Vu TH, Raffin TA. Biochem Biophys Res Commun. 1992;184:1339–1343. doi: 10.1016/s0006-291x(05)80029-4. [DOI] [PubMed] [Google Scholar]
- 297.Weber C, Erl W, Pietsch A, Strobel M, Ziegler-Heitbrock HW, Weber PC. Arterioscler Thromb. 1994;14:1665–1673. doi: 10.1161/01.atv.14.10.1665. [DOI] [PubMed] [Google Scholar]
- 298.Holland JA, Meyer JW, Schmitt ME, Sauro MD, Johnson DK, Abdul-Karim RW, Patel V, Ziegler LM, Schillinger KJ, Small RF, Lemanski LF. Endothelium. 1997;5:191–207. doi: 10.3109/10623329709053398. [DOI] [PubMed] [Google Scholar]
- 299.Khachigian LM, Resnick N, Gimbrone MA, Jr, Collins T. J Clin Invest. 1995;96:1169–1175. doi: 10.1172/JCI118106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Lan Q, Mercurius KO, Davies PF. Biochem Biophys Res Commun. 1994;201:950–956. doi: 10.1006/bbrc.1994.1794. [DOI] [PubMed] [Google Scholar]
- 301.Sorescu D, Weiss D, Lassegue B, Clempus RE, Szocs K, Sorescu GP, Valppu L, Quinn MT, Lambeth JD, Vega JD, Taylor WR, Griendling KK. Circulation. 2002;105:1429–1435. doi: 10.1161/01.cir.0000012917.74432.66. [DOI] [PubMed] [Google Scholar]
- 302.Woodman CR, Muller JM, Rush JW, Laughlin MH, Price EM. Am J Physiol. 1999;276:H1058–1063. doi: 10.1152/ajpheart.1999.276.3.H1058. [DOI] [PubMed] [Google Scholar]
- 303.Nazari-Jahantigh M, Wei Y, Schober A. Thromb Haemost. 2012;107:611–618. doi: 10.1160/TH11-12-0826. [DOI] [PubMed] [Google Scholar]
- 304.Cohen RA, Vanhoutte PM. Circulation. 1995;92:3337–3349. doi: 10.1161/01.cir.92.11.3337. [DOI] [PubMed] [Google Scholar]
- 305.Lee TJ, Yu JG. Annals of the New York Academy of Sciences. 2002;962:73–80. doi: 10.1111/j.1749-6632.2002.tb04057.x. [DOI] [PubMed] [Google Scholar]
- 306.Archer SL, Gragasin FS, Wu X, Wang S, McMurtry S, Kim DH, Platonov M, Koshal A, Hashimoto K, Campbell WB, Falck JR, Michelakis ED. Circulation. 2003;107:769–776. doi: 10.1161/01.cir.0000047278.28407.c2. [DOI] [PubMed] [Google Scholar]
- 307.Bolz SS, de Wit C, Pohl U. Br J Pharmacol. 1999;128:124–134. doi: 10.1038/sj.bjp.0702775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Hayabuchi Y, Nakaya Y, Matsuoka S, Kuroda Y. J Cardiovasc Pharmacol. 1998;32:642–649. doi: 10.1097/00005344-199810000-00018. [DOI] [PubMed] [Google Scholar]
- 309.Chistiakov DA, Orekhov AN, Bobryshev YV. Cell Mol Life Sci. 2015;72:2697–2708. doi: 10.1007/s00018-015-1906-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Zhou J, Li YS, Nguyen P, Wang KC, Weiss A, Kuo YC, Chiu JJ, Shyy JY, Chien S. Circ Res. 2013;113:40–51. doi: 10.1161/CIRCRESAHA.113.280883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA, Richardson JA, Bassel-Duby R, Olson EN. Dev Cell. 2008;15:261–271. doi: 10.1016/j.devcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Fang Y, Shi C, Manduchi E, Civelek M, Davies PF. Proc Natl Acad Sci U S A. 2010;107:13450–13455. doi: 10.1073/pnas.1002120107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Qin X, Wang X, Wang Y, Tang Z, Cui Q, Xi J, Li YS, Chien S, Wang N. Proc Natl Acad Sci U S A. 2010;107:3240–3244. doi: 10.1073/pnas.0914882107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Fang Y, Davies PF. Arterioscler Thromb Vasc Biol. 2012;32:979–987. doi: 10.1161/ATVBAHA.111.244053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Chen LJ, Chuang L, Huang YH, Zhou J, Lim SH, Lee CI, Lin WW, Lin TE, Wang WL, Chen L, Chien S, Chiu JJ. Circ Res. 2015;116:1157–1169. doi: 10.1161/CIRCRESAHA.116.305987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Chen Z, Wen L, Martin M, Hsu CY, Fang L, Lin FM, Lin TY, Geary MJ, Geary GG, Zhao Y, Johnson DA, Chen JW, Lin SJ, Chien S, Huang HD, Miller YI, Huang PH, Shyy JY. Circulation. 2015;131:805–814. doi: 10.1161/CIRCULATIONAHA.114.013675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Wang KC, Nguyen P, Weiss A, Yeh YT, Chien HS, Lee A, Teng D, Subramaniam S, Li YS, Chien S. Arterioscler Thromb Vasc Biol. 2014;34:1437–1445. doi: 10.1161/ATVBAHA.114.303473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Wu W, Xiao H, Laguna-Fernandez A, Villarreal G, Jr, Wang KC, Geary GG, Zhang Y, Wang WC, Huang HD, Zhou J, Li YS, Chien S, Garcia-Cardena G, Shyy JY. Circulation. 2011;124:633–641. doi: 10.1161/CIRCULATIONAHA.110.005108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Zhou J, Wang KC, Wu W, Subramaniam S, Shyy JY, Chiu JJ, Li JY, Chien S. Proc Natl Acad Sci U S A. 2011;108:10355–10360. doi: 10.1073/pnas.1107052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Wang KC, Garmire LX, Young A, Nguyen P, Trinh A, Subramaniam S, Wang N, Shyy JY, Li YS, Chien S. Proc Natl Acad Sci U S A. 2010;107:3234–3239. doi: 10.1073/pnas.0914825107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Hergenreider E, Heydt S, Treguer K, Boettger T, Horrevoets AJ, Zeiher AM, Scheffer MP, Frangakis AS, Yin X, Mayr M, Braun T, Urbich C, Boon RA, Dimmeler S. Nat Cell Biol. 2012;14:249–256. doi: 10.1038/ncb2441. [DOI] [PubMed] [Google Scholar]
- 322.Robertson KD. Oncogene. 2002;21:5361–5379. doi: 10.1038/sj.onc.1205609. [DOI] [PubMed] [Google Scholar]
- 323.Mason K, Liu Z, Aguirre-Lavin T, Beaujean N. Animal reproduction science. 2012;134:45–55. doi: 10.1016/j.anireprosci.2012.08.010. [DOI] [PubMed] [Google Scholar]
- 324.Dunn J, Simmons R, Thabet S, Jo H. Int J Biochem Cell Biol. 2015 doi: 10.1016/j.biocel.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Lund G, Andersson L, Lauria M, Lindholm M, Fraga MF, Villar-Garea A, Ballestar E, Esteller M, Zaina S. J Biol Chem. 2004;279:29147–29154. doi: 10.1074/jbc.M403618200. [DOI] [PubMed] [Google Scholar]
- 326.Baccarelli A, Rienstra M, Benjamin EJ. Circ Cardiovasc Genet. 2010;3:567–573. doi: 10.1161/CIRCGENETICS.110.958744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Turunen MP, Aavik E, Yla-Herttuala S. Biochim Biophys Acta. 2009;1790:886–891. doi: 10.1016/j.bbagen.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 328.Zaina S, Heyn H, Carmona FJ, Varol N, Sayols S, Condom E, Ramirez-Ruz J, Gomez A, Goncalves I, Moran S, Esteller M. Circ Cardiovasc Genet. 2014;7:692–700. doi: 10.1161/CIRCGENETICS.113.000441. [DOI] [PubMed] [Google Scholar]
- 329.Jiang YZ, Jimenez JM, Ou K, McCormick ME, Zhang LD, Davies PF. Circ Res. 2014 doi: 10.1161/CIRCRESAHA.115.303883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Dunn J, Qiu H, Kim S, Jjingo D, Hoffman R, Kim CW, Jang I, Son DJ, Kim D, Pan C, Fan Y, Jordan IK, Jo H. J Clin Invest. 2014;124:3187–3199. doi: 10.1172/JCI74792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Zhou J, Li YS, Wang KC, Chien S. Cell Mol Bioeng. 2014;7:218–224. doi: 10.1007/s12195-014-0325-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Hamik A, Lin Z, Kumar A, Balcells M, Sinha S, Katz J, Feinberg MW, Gerzsten RE, Edelman ER, Jain MK. J Biol Chem. 2007;282:13769–13779. doi: 10.1074/jbc.M700078200. [DOI] [PubMed] [Google Scholar]
- 333.Hamik A, Jain MK. Arteriosclerosis thrombosis and vascular biology. 2012;32:839–840. doi: 10.1161/ATVBAHA.112.245563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Huddleson JP, Ahmad N, Srinivasan S, Lingrel JB. J Biol Chem. 2005;280:23371–23379. doi: 10.1074/jbc.M413839200. [DOI] [PubMed] [Google Scholar]
- 335.Villarreal G, Jr, Zhang Y, Larman HB, Gracia-Sancho J, Koo A, Garcia-Cardena G. Biochem Biophys Res Commun. 2010;391:984–989. doi: 10.1016/j.bbrc.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Bijnens APJJ, Lutgens E, Ayoubi T, Kuiper J, Horrevoets AJ, Daemen MJAP. Arteriosclerosis Thrombosis and Vascular Biology. 2006;26:1226–1235. doi: 10.1161/01.ATV.0000219289.06529.f1. [DOI] [PubMed] [Google Scholar]
- 337.Cagnin S, Biscuola M, Patuzzo C, Trabetti E, Pasquali A, Laveder P, Faggian G, Iafrancesco M, Mazzucco A, Pignatti PF, Lanfranchi G. BMC Genomics. 2009;10:13. doi: 10.1186/1471-2164-10-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Sluimer JC, Kisters N, Cleutjens KB, Volger OL, Horrevoets AJ, van den Akker LH, Bijnens AP, Daemen MJ. Physiol Genomics. 2007;30:335–341. doi: 10.1152/physiolgenomics.00076.2007. [DOI] [PubMed] [Google Scholar]
- 339.Ashley EA, Ferrara R, King JY, Vailaya A, Kuchinsky A, He X, Byers B, Gerckens U, Oblin S, Tsalenko A, Soito A, Spin JM, Tabibiazar R, Connolly AJ, Simpson JB, Grube E, Quertermous T. Circulation. 2006;114:2644–2654. doi: 10.1161/CIRCULATIONAHA.106.637025. [DOI] [PubMed] [Google Scholar]
- 340.Tabibiazar R, Wagner RA, Ashley EA, King JY, Ferrara R, Spin JM, Sanan DA, Narasimhan B, Tibshirani R, Tsao PS, Efron B, Quertermous T. Physiol Genomics. 2005;22:213–226. doi: 10.1152/physiolgenomics.00001.2005. [DOI] [PubMed] [Google Scholar]
- 341.Skogsberg J, Lundstrom J, Kovacs A, Nilsson R, Noori P, Maleki S, Kohler M, Hamsten A, Tegner J, Bjorkegren J. PLoS Genet. 2008;4:e1000036. doi: 10.1371/journal.pgen.1000036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Herrera VM, Didishvili T, Lopez LV, Ruiz-Opazo N. Mol Med. 2002;8:367–375. [PMC free article] [PubMed] [Google Scholar]
- 343.Geary RL, Wong JM, Rossini A, Schwartz SM, Adams LD. Arterioscler Thromb Vasc Biol. 2002;22:2010–2016. doi: 10.1161/01.atv.0000038147.93527.35. [DOI] [PubMed] [Google Scholar]
- 344.Civelek M, Manduchi E, Riley RJ, Stoeckert CJ, Jr, Davies PF. Circ Cardiovasc Genet. 2011;4:243–252. doi: 10.1161/CIRCGENETICS.110.958926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Doring Y, Noels H, Weber C. Arterioscler Thromb Vasc Biol. 2012;32:182–195. doi: 10.1161/ATVBAHA.111.232686. [DOI] [PubMed] [Google Scholar]
- 346.Volger OL, Fledderus JO, Kisters N, Fontijn RD, Moerland PD, Kuiper J, van Berkel TJ, Bijnens AP, Daemen MJ, Pannekoek H, Horrevoets AJ. Am J Pathol. 2007;171:326–337. doi: 10.2353/ajpath.2007.061196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Trogan E, Choudhury RP, Dansky HM, Rong JX, Breslow JL, Fisher EA. Proc Natl Acad Sci U S A. 2002;99:2234–2239. doi: 10.1073/pnas.042683999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Shalhoub J, Sikkel MB, Davies KJ, Vorkas PA, Want EJ, Davies AH. Vasc Endovascular Surg. 2014;48:5–17. doi: 10.1177/1538574413510628. [DOI] [PubMed] [Google Scholar]
- 349.Ramsey SA, Gold ES, Aderem A. EMBO Mol Med. 2010;2:79–89. doi: 10.1002/emmm.201000063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Ghazalpour A, Doss S, Yang X, Aten J, Toomey EM, Van Nas A, Wang S, Drake TA, Lusis AJ. J Lipid Res. 2004;45:1793–1805. doi: 10.1194/jlr.R400006-JLR200. [DOI] [PubMed] [Google Scholar]
- 351.Bijnens AP, Lutgens E, Ayoubi T, Kuiper J, Horrevoets AJ, Daemen MJ. Arterioscler Thromb Vasc Biol. 2006;26:1226–1235. doi: 10.1161/01.ATV.0000219289.06529.f1. [DOI] [PubMed] [Google Scholar]
- 352.McCormick SM, Eskin SG, McIntire LV, Teng CL, Lu CM, Russell CG, Chittur KK. Proc Natl Acad Sci U S A. 2001;98:8955–8960. doi: 10.1073/pnas.171259298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.McCormick SM, Frye SR, Eskin SG, Teng CL, Lu CM, Russell CG, Chittur KK, McIntire LV. Biorheology. 2003;40:5–11. [PubMed] [Google Scholar]
- 354.Chen BP, Li YS, Zhao Y, Chen KD, Li S, Lao J, Yuan S, Shyy JY, Chien S. Physiol Genomics. 2001;7:55–63. doi: 10.1152/physiolgenomics.2001.7.1.55. [DOI] [PubMed] [Google Scholar]
- 355.Ohura N, Yamamoto K, Ichioka S, Sokabe T, Nakatsuka H, Baba A, Shibata M, Nakatsuka T, Harii K, Wada Y, Kohro T, Kodama T, Ando J. J Atheroscler Thromb. 2003;10:304–313. doi: 10.5551/jat.10.304. [DOI] [PubMed] [Google Scholar]
- 356.Ni CW, Qiu H, Rezvan A, Kwon K, Nam D, Son DJ, Visvader JE, Jo H. Blood. 2010;116:e66–73. doi: 10.1182/blood-2010-04-278192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Bartel DP. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 358.Weber M, Baker MB, Moore JP, Searles CD. Biochem Biophys Res Commun. 2010;393:643–648. doi: 10.1016/j.bbrc.2010.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Ni CW, Qiu H, Jo H. Am J Physiol Heart Circ Physiol. 2011;300:H1762–1769. doi: 10.1152/ajpheart.00829.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Wang X, Fang X, Zhou J, Chen Z, Zhao B, Xiao L, Liu A, Li YS, Shyy JY, Guan Y, Chien S, Wang N. Proc Natl Acad Sci U S A. 2013;110:13174–13179. doi: 10.1073/pnas.1312065110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Shepherd RD, Kos SM, Rinker KD. Am J Physiol Heart Circ Physiol. 2011;301:H98–H107. doi: 10.1152/ajpheart.00668.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Potter CM, Lundberg MH, Harrington LS, Warboys CM, Warner TD, Berson RE, Moshkov AV, Gorelik J, Weinberg PD, Mitchell JA. Arterioscler Thromb Vasc Biol. 2011;31:384–391. doi: 10.1161/ATVBAHA.110.214031. [DOI] [PubMed] [Google Scholar]
- 363.Prigent-Tessier A, Quirie A, Maguin-Gate K, Szostak J, Mossiat C, Nappey M, Devaux S, Marie C, Demougeot C. Cardiovasc Res. 2013;100:374–382. doi: 10.1093/cvr/cvt219. [DOI] [PubMed] [Google Scholar]
- 364.Barauna VG, Campos LC, Miyakawa AA, Krieger JE. PLoS One. 2011;6:e22803. doi: 10.1371/journal.pone.0022803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Xue L, Yu C, Liu X, Lai Y, Zeng Y, Zhang Y. Sheng Wu Yi Xue Gong Cheng Xue Za Zhi. 2007;24:1054–1057. [PubMed] [Google Scholar]
- 366.Urbich C, Walter DH, Zeiher AM, Dimmeler S. Circ Res. 2000;87:683–689. doi: 10.1161/01.res.87.8.683. [DOI] [PubMed] [Google Scholar]
- 367.Soghomonians A, Thirkill TL, Mariano NF, Barakat AI, Douglas GC. Toxicol Sci. 2004;81:408–418. doi: 10.1093/toxsci/kfh210. [DOI] [PubMed] [Google Scholar]
- 368.Ozawa N, Shichiri M, Iwashina M, Fukai N, Yoshimoto T, Hirata Y. Hypertens Res. 2004;27:93–99. doi: 10.1291/hypres.27.93. [DOI] [PubMed] [Google Scholar]
- 369.Eng E, Ballermann BJ. Microvasc Res. 2003;65:137–144. doi: 10.1016/s0026-2862(03)00004-9. [DOI] [PubMed] [Google Scholar]
- 370.Kudo FA, Warycha B, Juran PJ, Asada H, Teso D, Aziz F, Frattini J, Sumpio BE, Nishibe T, Cha C, Dardik A. Am J Surg. 2005;190:763–769. doi: 10.1016/j.amjsurg.2005.07.017. [DOI] [PubMed] [Google Scholar]
- 371.Wang XL, Fu A, Raghavakaimal S, Lee HC. Proteomics. 2007;7:588–596. doi: 10.1002/pmic.200600568. [DOI] [PubMed] [Google Scholar]
- 372.Cattaruzza M, Guzik TJ, Slodowski W, Pelvan A, Becker J, Halle M, Buchwald AB, Channon KM, Hecker M. Circ Res. 2004;95:841–847. doi: 10.1161/01.RES.0000145359.47708.2f. [DOI] [PubMed] [Google Scholar]
- 373.Rossi GP, Cesari M, Zanchetta M, Colonna S, Maiolino G, Pedon L, Cavallin M, Maiolino P, Pessina AC. J Am Coll Cardiol. 2003;41:930–937. doi: 10.1016/s0735-1097(02)03012-7. [DOI] [PubMed] [Google Scholar]
- 374.Colombo MG, Paradossi U, Andreassi MG, Botto N, Manfredi S, Masetti S, Biagini A, Clerico A. Clin Chem. 2003;49:389–395. doi: 10.1373/49.3.389. [DOI] [PubMed] [Google Scholar]
- 375.Asif AR, Hecker M, Cattaruzza M. Arterioscler Thromb Vasc Biol. 2009;29:1890–1893. doi: 10.1161/ATVBAHA.109.190678. [DOI] [PubMed] [Google Scholar]
- 376.Chen Z, Peng IC, Sun W, Su MI, Hsu PH, Fu Y, Zhu Y, DeFea K, Pan S, Tsai MD, Shyy JY. Circ Res. 2009;104:496–505. doi: 10.1161/CIRCRESAHA.108.187567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Gallis B, Corthals GL, Goodlett DR, Ueba H, Kim F, Presnell SR, Figeys D, Harrison DG, Berk BC, Aebersold R, Corson MA. J Biol Chem. 1999;274:30101–30108. doi: 10.1074/jbc.274.42.30101. [DOI] [PubMed] [Google Scholar]
- 378.Hare JM, Stamler JS. Nat Med. 1999;5:273–274. doi: 10.1038/6486. [DOI] [PubMed] [Google Scholar]
- 379.Sun J, Murphy E. Circ Res. 2010;106:285–296. doi: 10.1161/CIRCRESAHA.109.209452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Hare JM, Stamler JS. J Clin Invest. 2005;115:509–517. doi: 10.1172/JCI200524459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Kettenhofen NJ, Wang X, Gladwin MT, Hogg N. Methods Enzymol. 2008;441:53–71. doi: 10.1016/S0076-6879(08)01204-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Huang B, Chen SC, Wang DL. Cardiovasc Res. 2009;83:536–546. doi: 10.1093/cvr/cvp154. [DOI] [PubMed] [Google Scholar]
- 383.Freed JK, Shortreed MR, Kleefisch CJ, Smith LM, Greene AS. Endothelium. 2008;15:225–230. doi: 10.1080/10623320802228849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Thakker GD, Hajjar DP, Muller WA, Rosengart TK. J Biol Chem. 1999;274:10002–10007. doi: 10.1074/jbc.274.15.10002. [DOI] [PubMed] [Google Scholar]
- 385.Bell RM, Burns DJ. J Biol Chem. 1991;266:4661–4664. [PubMed] [Google Scholar]
- 386.Wen L, Chen Z, Zhang F, Cui X, Sun W, Geary GG, Wang Y, Johnson DA, Zhu Y, Chien S, Shyy JY. Proc Natl Acad Sci U S A. 2013;110:E2420–2427. doi: 10.1073/pnas.1309354110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Freed JK, Greene AS. Microcirculation. 2010;17:259–270. doi: 10.1111/j.1549-8719.2010.00031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Irani K. Circ Res. 2000;87:179–183. doi: 10.1161/01.res.87.3.179. [DOI] [PubMed] [Google Scholar]
- 389.Burghoff S, Schrader J. J Proteome Res. 2011;10:1160–1169. doi: 10.1021/pr100937a. [DOI] [PubMed] [Google Scholar]
- 390.Platt MO, Ankeny RF, Shi GP, Weiss D, Vega JD, Taylor WR, Jo H. Am J Physiol Heart Circ Physiol. 2007;292:H1479–1486. doi: 10.1152/ajpheart.00954.2006. [DOI] [PubMed] [Google Scholar]
- 391.Platt MO, Ankeny RF, Jo H. Arterioscler Thromb Vasc Biol. 2006;26:1784–1790. doi: 10.1161/01.ATV.0000227470.72109.2b. [DOI] [PubMed] [Google Scholar]
- 392.Mowbray AL, Kang DH, Rhee SG, Kang SW, Jo H. J Biol Chem. 2008;283:1622–1627. doi: 10.1074/jbc.M707985200. [DOI] [PubMed] [Google Scholar]
- 393.Csiszar A, Labinskyy N, Smith KE, Rivera A, Bakker EN, Jo H, Gardner J, Orosz Z, Ungvari Z. Arterioscler Thromb Vasc Biol. 2007;27:776–782. doi: 10.1161/01.ATV.0000259355.77388.13. [DOI] [PubMed] [Google Scholar]
- 394.Chang K, Weiss D, Suo J, Vega JD, Giddens D, Taylor WR, Jo H. Circulation. 2007;116:1258–1266. doi: 10.1161/CIRCULATIONAHA.106.683227. [DOI] [PubMed] [Google Scholar]
- 395.Boyd NL, Park H, Yi H, Boo YC, Sorescu GP, Sykes M, Jo H. Am J Physiol Heart Circ Physiol. 2003;285:H1113–1122. doi: 10.1152/ajpheart.00302.2003. [DOI] [PubMed] [Google Scholar]
- 396.Takai J, Santu A, Zheng H, Koh SD, Ohta M, Filimban LM, Lemaitre V, Teraoka R, Jo H, Miura H. Am J Physiol Heart Circ Physiol. 2013;305:H484–493. doi: 10.1152/ajpheart.00642.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Johnson JM, Yu T, Strobel FH, Jones DP. Analyst. 2010;135:2864–2870. doi: 10.1039/c0an00333f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Mayr M, Chung YL, Mayr U, Yin X, Ly L, Troy H, Fredericks S, Hu Y, Griffiths JR, Xu Q. Arterioscler Thromb Vasc Biol. 2005;25:2135–2142. doi: 10.1161/01.ATV.0000183928.25844.f6. [DOI] [PubMed] [Google Scholar]
- 399.Chen X, Liu L, Palacios G, Gao J, Zhang N, Li G, Lu J, Song T, Zhang Y, Lv H. J Sep Sci. 2010;33:2776–2783. doi: 10.1002/jssc.201000395. [DOI] [PubMed] [Google Scholar]
- 400.Cheng KK, Benson GM, Grimsditch DC, Reid DG, Connor SC, Griffin JL. Physiol Genomics. 2010;41:224–231. doi: 10.1152/physiolgenomics.00188.2009. [DOI] [PubMed] [Google Scholar]
- 401.Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, Wu Y, Schauer P, Smith JD, Allayee H, Tang WH, DiDonato JA, Lusis AJ, Hazen SL. Nature. 2011;472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Go YM, Kim CW, Walker DI, Kang DW, Kumar S, Orr M, Uppal K, Quyyumi AA, Jo H, Jones DP. Am J Physiol Regul Integr Comp Physiol. 2015;308:R62–72. doi: 10.1152/ajpregu.00278.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]