

Significance
The increasing number of patients presenting with severe asthma throughout the world present a clear unmet medical need. This study identified putative transcriptional regulators in T-helper 2 (TH2) cells with the aim of identifying previously unidentified targets to inhibit TH2-mediated allergy. Genetic deletion of Trim24 (tripartite motif-containing 24) in T cells showed that Trim24 was essential for TH2-mediated allergy. Transcriptional analysis showed that Trim24 was required for many of the pathogenic properties of TH2 cells and that IL-1–regulated signaling is compromised in Trim24−/− cells. In vivo, in vitro, and in silico approaches identified a previously overlooked role for Trim24 in TH2-mediated allergy and validate a combined approach to interrogate transcriptional datasets to identify new therapeutic targets to prevent allergy and asthma.
Keywords: Trim24, Th2, allergy, inflammation, asthma
Abstract
There is a paucity of new therapeutic targets to control allergic reactions and forestall the rising trend of allergic diseases. Although a variety of immune cells contribute to allergy, cytokine-secreting αβ+CD4+ T-helper 2 (TH2) cells orchestrate the type-2–driven immune response in a large proportion of atopic asthmatics. To identify previously unidentified putative targets in pathogenic TH2 cells, we performed in silico analyses of recently published transcriptional data from a wide variety of pathogenic TH cells [Okoye IS, et al. (2014) Proc Natl Acad Sci USA 111(30):E3081–E3090] and identified that transcription intermediary factor 1 regulator-alpha (Tif1α)/tripartite motif-containing 24 (Trim24) was predicted to be active in house dust mite (HDM)- and helminth-elicited Il4gfp+αβ+CD4+ TH2 cells but not in TH1, TH17, or Treg cells. Testing this prediction, we restricted Trim24 deficiency to T cells by using a mixed bone marrow chimera system and found that T-cell–intrinsic Trim24 is essential for HDM-mediated airway allergy and antihelminth immunity. Mechanistically, HDM-elicited Trim24−/− T cells have reduced expression of many TH2 cytokines and chemokines and were predicted to have compromised IL-1–regulated signaling. Following this prediction, we found that Trim24−/− T cells have reduced IL-1 receptor (IL-1R) expression, are refractory to IL-1β–mediated activation in vitro and in vivo, and fail to respond to IL-1β–exacerbated airway allergy. Collectively, these data identify a previously unappreciated Trim24-dependent requirement for IL-1R expression on TH2 cells and an important nonredundant role for T-cell–intrinsic Trim24 in TH2-mediated allergy and antihelminth immunity.
Allergic diseases, including allergic asthma, have continued to rise in the past 50 y. With few new drugs available to treat allergic diseases and many patients with severe asthma refractory to currently available drugs (1), there is a growing need for new molecular targets to curb symptoms and prevent exacerbations. Dysregulated T-cell responses underpin the hyperinflammatory allergic reaction leading to asthma. Although many T-cell populations contribute to the spectrum of allergic asthma phenotypes (2), cytokine-secreting T-helper 2 (TH2) cells have the capacity to induce allergen-specific IgE (atopy) and invoke many of the pathophysiological manifestations associated with asthma, including airway eosinophilia, mucus hypersecretion, airway remodeling, and airway hyperreactivity. Targeting specific cytokine-signaling pathways in allergic asthma has had mixed efficacy (3–5), suggesting that additional targets and more focused approaches should be considered (6). Several TH2 cell lineage-promoting transcriptional regulators, including GATA binding protein 3 (GATA-3) (7), STAT-3 (8), STAT-6 (9), and avian musculoaponeurotic fibrosarcoma (cMAF) (10), have been identified in TH2 cells; however, it is unclear whether other transcriptional regulators are required for TH2 cell-mediated responses.
The two-signal model of TH2 cell differentiation, involving T-cell receptor (TCR) and costimulatory engagement coupled with secondary cytokine signaling, is well defined (11). However, the activation of differentiated TH2 cells and the acquisition of cytokine-secreting effector function in the tissues is poorly understood. Tertiary cytokine signals by tissue-associated inflammatory cytokines, including members of the IL-1 family (12, 13), and the “alarmins” (IL-25 and TSLP) (14) have been proposed to activate TH2 cells; however, the regulation and pathways involved are unclear. An IL-1R (IL-1 receptor)/ubiquitin C/Trim24 (tripartite motif-containing 24) axis has been identified previously (15–19), but the involvement of Trim24 in TH2 biology has not been reported.
The tripartite motif (Trim) family of more than 60 proteins is highly conserved throughout metazoans and has been widely studied in innate antiviral immunity (20). However, Trim proteins have a variety of functions, including regulation of transcription and chromatin (21–23), tumor suppression (24), and cytokine signaling and secretion, in both innate and adaptive immune cells (25, 26). Specifically, the transcription intermediary factor 1 regulator-alpha, Tif1α (Trim24), which is structurally related to Tif1β (Trim28) and Tif1γ (Trim33) (22), has important roles in cancer (27, 28), gene regulation (29), and cytokine signaling (30), in part through the interaction of Trim24 with nuclear hormone receptors, vitamin D receptors, estrogen receptors, and retinoic acid receptors (30, 31). Unlike the closely related Trim28 (26), which regulates TH17-mediated immunity, a role for Trim24 in T-cell biology, type-2 immunity, or allergic asthma has not been reported. In this study, we found that deletion of Trim24 in T cells did not lead to any overt autoimmune phenotype. In contrast, Trim24 is essential for TH2 cell-mediated airway allergy and TH2-dependent expulsion of intestinal helminths. Mechanistically, Trim24−/− T cells isolated from the lungs of allergic mice had a dampened IL-1–regulated transcriptome, suggesting that Trim24 is required for IL-1–mediated TH2 cell activation. Indeed, Trim24−/− TH2 cells had reduced IL-1R expression. Furthermore, deletion of T-cell–intrinsic Trim24, similar to the effect of IL-1R deletion on T cells, rendered T cells refractory to the stimulatory effects of IL-1β and curtailed the manifestation of house dust mite (HDM)-induced airway allergy, identifying a critical and nonredundant role for Trim24 in IL-1–mediated TH2 cell-mediated inflammation in vivo.
Results
Tif1α/Trim24 Is a Predicted Transcriptional Regulator in TH2 Cells but Not in TH1, TH17, or Foxp3+ Treg Cells.
Dysregulated TH2-cell responses following allergen exposure contribute directly to allergic disease (32). We recently reported that HDM- or helminth-elicited TH2 cells have distinct transcriptional profiles, compared with ex vivo TH1, TH17, and natural Treg cells (33). Using these transcriptional datasets (more than twofold change relative to naive T cells, P < 0.05) we applied in silico upstream regulator analyses (Ingenuity Pathways Analysis, IPA) (34) and generated a z-score representing the likelihood of activity of putative transcriptional regulators. Briefly, this analysis examines how many known target genes of a transcription regulator are present in the dataset and compares their expression with the level expected from the literature to predict transcriptional regulator activity. If the observed expression of target genes is mostly consistent with a particular activation state of the transcriptional regulator, then a higher prediction (z-score) is made about that activation state. This in silico approach identified GATA-3, STAT-6, and cMAF as putative regulators of TH2 cells (Fig. 1 A and B), in line with previous experimental observations. This analysis also predicted that Tif1α, belonging to the tripartite motif (Trim) family (i.e., Trim24), would be active in TH2 cells but not in TH1, TH17, or Treg cells (Fig. 1 A–C and Table S1). The microarray analysis showed that Trim24, like Gata3, Stat6, and other Trim family members, was not significantly differentially regulated in TH2 cells as compared with naive T cells (Fig. 1A and Fig. S1 A and B). Using Il4gfp-reporter mice, we assessed the expression of Trim24 mRNA by quantitative RT-PCR (qRT-PCR) in freshly isolated Il4gfp+ TH2 cells from the lung tissue or local lymph nodes of HDM-challenged mice and from the local lymph nodes of helminth-infected mice. Il4gfp+ TH2 cells had elevated Il4, Il5, and Il13 mRNA but not Trim24 mRNA (Fig. 1D), in line with the microarray data (Fig. S1A). Western blot analysis of Il4gfp+ TH2 cells generated in vitro identified increased Trim24 protein expression, as compared with naive T cells (Fig. 1E), supporting a role for Trim24 in TH2 cells. Collectively, predictive in silico analyses coupled with the elevated levels of Trim24 protein in TH2 cells suggest that Trim24 may contribute to TH2 cell biology.
Fig. 1.

Trim24 protein is elevated in TH2 cells and is predicted to be functionally active in TH2 cells but not in TH1, TH17, or Treg cells. (A) Transcriptional datasets from TH2 cells purified from helminth (H. polygyrus)-infected or HDM-challenged mice (33) were analyzed using IPA upstream analysis software (34) to generate a z-score of the likelihood of activity. (B and C) Comparative analysis of predicted upstream transcriptional regulators in TH2 cells (B) and TH1, TH17, and Treg cells (C). (D) Expression of Il4, Il5, Il13, and Trim24 in Il4gfp+ TH2 cells isolated from pulmonary tissue [HDM (lung)] and local lymph nodes [HDM (LN)] of mice with HDM-induced airway allergy and from the local lymph nodes of H. polygyrus-infected mice [H.p. (MLN)]. Expression is shown relative to Hprt. (E) Expression of Trim24 protein in naive and in vitro-generated TH2 cells. A.U., arbitrary units.
Table S1.
Trim24-regulated genes, curated from IPA, in Il4gfp+ TH2 cells isolated ex vivo from HDM-challenged or H. polygyrus-infected mice
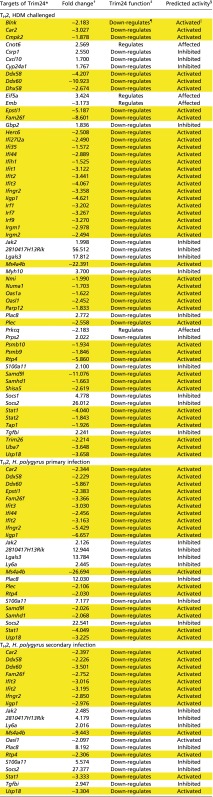 |
Genes highlighted in yellow have an expression profile consistent with active Trim24.
Known Trim24-regulated genes.
Observed fold change in dataset, relative to naive T cells.
Expected direction of regulation by Trim24.
The predicted activity of Trim24 in this dataset is based on the Trim24 function and the observed fold change.
Trim24 is known to down-regulate the target gene.
‖Trim24 is known to down-regulate the target gene, and the target gene is down-regulated.
Fig. S1.

Trim24 and other Trim family members were not significantly transcriptionally regulated in TH and Treg cells. Expression of Trim24 (A) and other Trim family members (B) in ex vivo TH1, TH2, TH17, and Treg cells, as indicated (11). H.p. 1°, H. polygyrus primary infection; H.p. 2°, H. polygyrus secondary infection.
T-Cell–Intrinsic Tif1α/Trim24 Is Required for HDM-Induced Airway Allergy and Antihelminth Immunity.
To test whether the predicted activity of Trim24 (Fig. 1A) and the elevated expression of Trim24 protein in TH2 cells (Fig. 1E) is required for TH2 cell-mediated airway allergy and TH2-dependent antihelminth immunity in vivo, we generated chimeric mice in which Trim24-deficiency was restricted to T cells (Fig. S2A). T-cell–chimeric mice had a comparable number and frequency of naive, memory, and regulatory T cells with no overt immunopathology after 3 mo of bone marrow reconstitution (Fig. S2 B–D). This T-cell–chimera system relied upon on the fact that non-T cells from WT or Trim24−/− bone marrow repopulate at a similar rate in both WT and Trim24−/− chimeric mice and constitute only a minor fraction (20%) of the total bone marrow. To determine whether Trim24−/− cells displayed any potential growth advantage that might compromise the T-cell–chimeric system, we generated separate 50:50 chimeras with 50% WT and 50% Trim24−/− bone marrow. After 8 wk of reconstitution, the 50:50 chimeras retained the 50:50 ratio with comparable ratios of lymphocytes, myeloid cells, and granulocyte populations, indicating that neither the WT nor the Trim24−/− populations had an overt growth advantage (Fig. S2E). Following HDM sensitization and local airway challenge of T-cell–chimeric mice (Fig. S2A), mice with Trim24−/− T cells had significantly reduced TH2-mediated airway allergy, with reduced airway inflammation (Fig. 2A) and reduced airway eosinophilia (Fig. 2B), as compared with mice with WT T cells. Mild perivascular infiltrates were present in mice with Trim24−/− T cells; however, the cells failed to migrate and cause interstitial or peribronchial inflammation or mucus secretion following HDM challenge (Fig. 2C). Reduced airway mucus was supported by reduced Gob5 and Muc5ac expression in pulmonary tissue of mice with Trim24−/− T cells (Fig. 2D). Local lymph node cells restimulated with HDM indicated that Trim24 is required for optimal TH2 responses, because the absence of T-cell–intrinsic Trim24 led to reduced HDM-induced IL-5, IL-13, and IL-10 secretion without any appreciable change in HDM-induced IFNγ (Fig. 2E). Mice with Trim24−/− T cells also had reduced total and HDM-specific IgE (Fig. 2F), confirming the requirement of T-cell–intrinsic Trim24 for TH2-orchestrated type-2 immunity. Using an additional, adjuvant-free HDM-induced airway allergy model, we observed a similar reduction in airway infiltrates, eosinophilia, airway pathology, IgE production, and TH2 cytokine secretions in chimeric mice with Trim24−/− T cells as compared with mice with WT T cells (Fig. S3), further confirming an important requirement for T-cell–intrinsic Trim24.
Fig. S2.

Mice with T-cell–intrinsic Trim24 deficiency had a normal frequency of T cells and did not show any signs of overt pathology. (A) Chimeric mice were generated by reconstituting irradiated Rag2−/− mice with 2–5 × 106 bone marrow cells (20% bone marrow from Trim24−/− or WT donor mice and 80% bone marrow from C57BL/6.Tcra−/− mice) for 6–8 wk. (B) Total number of CD4+ splenocytes in naive chimeric mice. (C) Frequency of CD4+, CD8+, naive (CD4+CD62L+CD25–), and Treg (CD4+CD62L–CD25+) cells in naive chimeric mice. (D) H&E-stained liver, lung, small intestine (SI), and colon sections from naive chimeric mice. (E) The 50:50 chimeric mice were generated by reconstituting irradiated Rag2−/− mice with 5 × 106 bone marrow cells (2.5 × 106 CD45.1+ Trim24+/+ and 2.5 × 106 CD45.2+ Trim24−/− bone marrow cells). After 8 wk of reconstitution, various leukocyte populations were determined, as indicated.
Fig. 2.

T-cell–intrinsic Trim24 is essential for TH2-mediated airway allergy and TH2-dependent antihelminth immunity. Chimeric mice with WT or Trim24−/− T cells were sensitized and challenged with HDM, and airway allergy was assessed 1 d after the last airway challenge. *P ≤ 0.05. One of three representative experiments is shown. (A and B) BAL cells were recovered, counted (A), and used for cytospins for differential analysis (B). (C) H&E- and AB-PAS–stained lung sections from chimeric mice with HDM-induced airway allergy. (D) Gene expression in the pulmonary tissue of mice with HDM-induced airway allergy. Expression is shown relative to PBS-treated mice. (E) Cells from local lymph nodes were restimulated with HDM. Secreted cytokines were measured in supernatants after 4 d. (F) Circulating total and HDM-specific IgE measured in the serum of mice with HDM-induced airway allergy. (G) Chimeric mice with WT or Trim24−/− T cells were infected, drug-treated on days 14 and 15, and given a challenge infection with H. polygyrus on day 21. Adult worms were counted in the small intestine 14 d after the challenge infection (day 35). (H) Gene expression in the small intestine 14 d after the challenge infection. Fold change (FC) is shown relative to uninfected mice. (I) AB-PAS–stained tissue sections from chimeric mice 14 d after the challenge infection.
Fig. S3.

T-cell–intrinsic Trim24 is essential for i.n. HDM-mediated airway allergy. Chimeric mice with WT or Trim24−/− T cells were sensitized (day 0) and challenged i.n. (days 7–11) with HDM. *P ≤ 0.05. One of two representative experiments is shown. (A and B) BAL cells were recovered, counted (A), and used for cytospins for differential analysis (B). (C and D) Gene expression in pulmonary tissue of mice with HDM-induced airway allergy. Expression is shown relative to PBS-treated mice. (E) Local lymph node cells were restimulated with HDM. Secreted cytokines were measured in supernatants after 4 d. (F) Circulating IgE measured in the serum of mice with HDM-induced airway allergy. (G) H&E-stained (Left) and AB-PAS–stained (Right) lung sections from chimeric mice with HDM-induced airway allergy.
Expulsion of a challenge infection with the intestinal helminth Heligmosomoides polygyrus following drug cure of a primary infection requires TH2-dependent orchestration of a type-2 inflammatory cascade around invading larvae (35). To test further whether T-cell–intrinsic Trim24 is required for TH2-dependent antihelminth immunity in the gut, we gave a challenge infection of H. polygyrus to previously infected and drug-cured mice. Mice with Trim24-deficient T cells failed to expel the challenge infection (Fig. 2G), indicating that T-cell–intrinsic Trim24 also is required for proficient antihelminth immunity in the gut. The failure to expel H. polygyrus correlated with reduced expression of type-2 innate immune cell activation, including reduced Relmα and Arg1, as well as reduced Il13 expression in the small intestine (Fig. 2H) and reduced mucus staining in the small intestine (Fig. 2I). Collectively, these data identify an important requirement for T-cell–intrinsic Trim24 for TH2-mediated airway allergy and TH2-dependent antihelminth immunity.
Tif1α/Trim24 Is Required for IL-1R Expression and IL-1β–Mediated Activation of TH2 Cells in Vitro.
To identify how T-cell–intrinsic Trim24 regulated TH2 cell responses and TH2-mediated immunity, we purified activated WT or Trim24−/− T cells (TCRβ+CD4+CD44+) from the lungs of HDM-challenged mice, isolated total RNA, and subjected the RNA to transcriptional profiling (Fig. 3A). Microarray analysis showed that Trim24 regulated many genes in the purified T cells (Fig. 3B), with reduced expression of several chemokines (Ccl1, Ccl4, and Ccl5), chemokine receptors (Ccr5, Ccr6, Ccr9, and Cxcr3), cytokines (Il4, Il10, Il21, Spp1, and Ccl3l3), cytokine receptors (Il1r2, Il1rl1, Il2ra, Il17re, Csf1r, and Csf2rb), and regulators of cytokine secretion (Socs2) (Fig. 3C), compared with WT T cells. These data indicate that Trim24 is required for a spectrum of TH2-associated characteristics and provide a mechanistic explanation for the reduced airway allergy and compromised antihelminth immunity observed in vivo (Fig. 2). To identify how Trim24 regulates such a broad spectrum of pathways, we again used an upstream analysis algorithm [IPA software (34)] similar to that used in Fig. 1 and found that an IL-1–regulated pathway was predicted to be inhibited or absent in Trim24−/− T cells as compared with WT T cells (Fig. 3D and Table S2). These data suggest that Trim24 is required for IL-1–activated pathways in HDM-elicited pulmonary T cells. Several lines of evidence support this Trim24/IL-1 pathway. First, it has been widely reported that IL-1 signaling is required for TH2-mediated immunity and allergy (36–39). More specifically, IL-1 signaling is required to activate TH2 cells directly in vivo (13, 40). Second, a relationship between IL-1R/ubiquitin C and Trim24 has been suggested previously (15–19); however, the requirement of Trim24 for IL-1R expression has not been reported. Thus, based on our in silico prediction (Fig. 3D) and the previous reports mentioned above, we hypothesized that Trim24 regulates IL-1R expression and/or IL-1–mediated TH2 cell activation. To test this hypothesis, we isolated naive (CD4+CD44–CD62Lhi) WT and Trim24−/−T cells and polarized these cells under TH2 conditions. Trim24−/− TH2 cells secreted slightly lower levels of IL-4, IL-5, and IL-13 in vitro, but these differences failed to reach statistical significance (Fig. 3E). However, in vitro Trim24−/− TH2 cells had lower mRNA (Fig. 3F) and protein (Fig. 3G) expression of IL-1R1. Similarly, ex vivo Trim24−/− T cells had lower expression of IL-1R (Fig. S4D), indicating that Trim24 is required for IL-1R expression on TH2 cells. To test whether the reduced IL-1R expression led to a functional reduction of TH2 cell activation, at day 10 in vitro-generated WT or Trim24−/− TH2 cells were washed, counted, and restimulated with anti-CD3 in the presence or absence of IL-1β for 24 h. Trim24−/− TH2 cells proliferated less than WT cells (Fig. 3H) and transcribed (Fig. 3I) and secreted (Fig. 3J) slightly less IL-4 and less IL-13; however, this difference failed to reach statistical significance. In the presence of IL-1β, WT cells increased activation/proliferation (Fig. 3H) with increased Il4 transcription (Fig. 3I) and IL-4 and IL-13 secretion (Fig. 3J). In contrast, Trim24−/− TH2 cells were almost completely refractory to IL-1β–mediated activation/proliferation and cytokine production, suggesting that Trim24 is required for IL-1β–mediated TH2 cell activation in vitro. Similarly, ex vivo Trim24−/− T cells (TCRβ+CD4+CD44+) isolated from the lungs of HDM-challenged mice were refractory to IL-1β–mediated activation in vitro (Fig. S4E). Collectively, these data suggest that Trim24 is required for IL-1R expression on TH2 cells and for IL-1β–mediated activation of TH2 cells in vitro.
Fig. 3.

Transcriptional analysis of activated (CD44+) Trim24−/− T cells from the lungs of mice with identified airway allergy, reduced cytokine and chemokine expression, and reduced IL-1–regulated pathways. Chimeric mice with WT or Trim24−/− T cells were sensitized and challenged with HDM. Activated (CD4+CD44+) WT or Trim24−/− T cells were isolated from the lungs and FACS-purified for transcriptional analysis 1 d after the final airway challenge. *P ≤ 0.05. (A) Heat map of differentially expressed genes in WT and Trim24−/− T cells, relative to naive T cells. (B) Venn diagram showing common and unique genes regulated in WT and Trim24−/− T cells. (C) Noteworthy TH2 effector genes involved in airway allergy. (D) Z-score of the likelihood of activity for IL-1–regulated pathways in WT and Trim24−/− T cells. (E) Naive (CD4+CD44–CD62Lhi) WT and Trim24−/− T cells were FACS-purified and cultured under TH2 conditions for the indicated days. IL-4, IL-5, and IL-13 were measured in supernatant. (F) WT and Trim24−/− T cells cultured under TH2 conditions were harvested at the indicated days. RNA was extracted, and Il1r1 expression was determined by qRT-PCR. (G) IL-1R1 expression on CD4+CD44hi WT and Trim24−/− TH2 cells at day 10. (H) Day 10 WT and Trim24−/− TH2 cells were washed, counted, and replated at 2 × 105 cells per well. Cells were stimulated with anti-CD3 in the presence or absence of IL-1β (10 ng/mL), as indicated. Cell viability/activity was determined by Alamar blue fluorescence. (I) Il4 and Il13 mRNA expression in day 10 WT and Trim24−/− TH2 cells. Expression is shown relative to Hprt. One of three representative experiments is shown. (J) IL-4 and IL-13 protein expression in supernatant from WT or Trim24−/− T cells. Expression is shown relative to Hprt. One of three representative experiments is shown.
Table S2.
IL-1–regulated genes in activated WT and Trim24−/− T cells isolated ex vivo from HDM-challenged mice
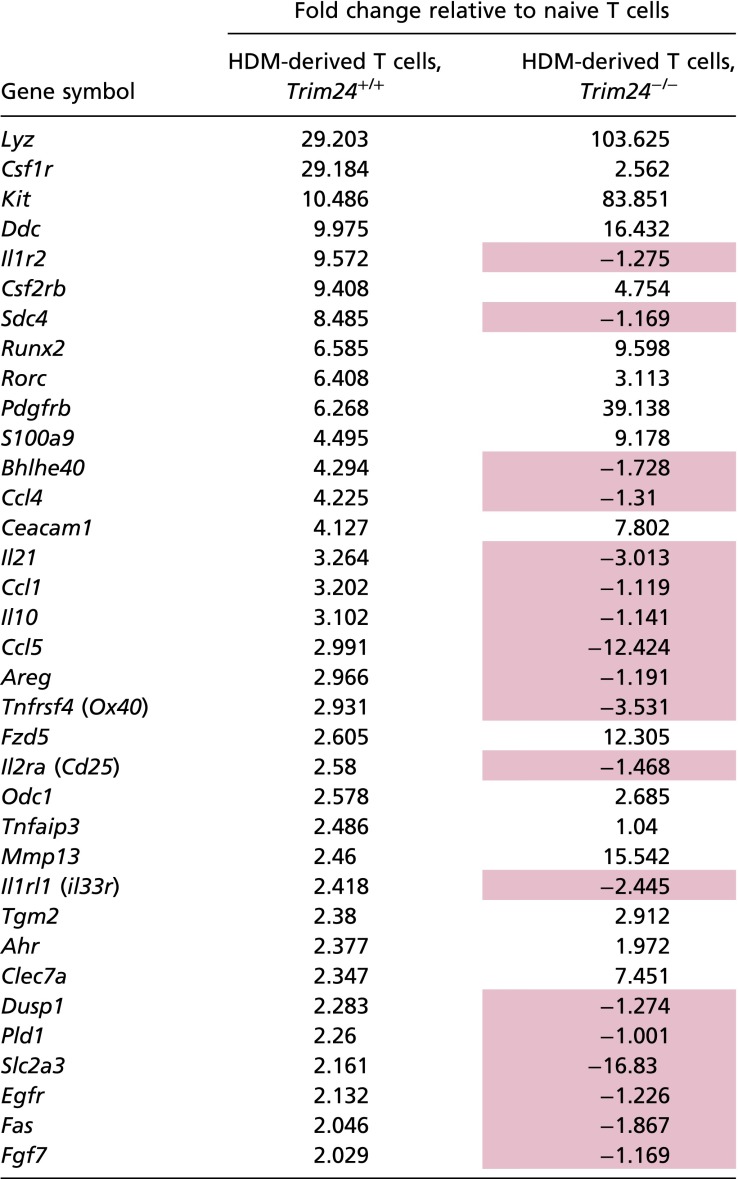 |
IL-1–regulated pathways. Genes highlighted in pink were up-regulated in WT and down-regulated in Trim24−/− T cells.
Fig. S4.

Trim24 does not appear to regulate retinoic acid or estrogen receptor signaling pathways in activated T cells from the allergic lung. Chimeric mice with WT or Trim24−/− T cells were sensitized and challenged with HDM. Activated (CD4+CD44+) WT or Trim24−/− T cells were isolated and FACS-purified from the lungs for transcriptional analysis 1 d after the final airway challenge. (A) Expression of genes in WT or Trim24−/− T cells involved in vitamin D and retinoic acid receptor (VDR/RXR) activation. (B) Expression of genes in WT or Trim24−/− T cells involved in estrogen receptor signaling. (C) Expression of genes in WT or Trim24−/− T cells involved in retinoic acid receptor signaling. (D) Activated (CD4+CD44+) WT or Trim24−/− T cells from the lungs of mice with HDM-induced airway allergy were isolated and FACS-purified 1 d after the final airway challenge. IL-1R expression was determined by FACS. (E) IL-13 secretion from FACS-purified ex vivo WT or Trim24−/− T cells stimulated with anti-CD3 with or without recombinant (r) IL-1β, as indicated.
T-Cell–Intrinsic Trim24 Is Required for IL-1β–Mediated Activation of TH2 Cells in Vivo.
IL-1R signaling is essential for the effector phase of TH2-mediated airway allergy (38, 39), and IL-1 can directly activate TH2 cells in vivo (13, 40). However, it is unclear how this process is regulated and whether Trim24 is required for IL-1–mediated TH2 cell activation in vivo. First, using chimeric mice with an IL-1R deficiency restricted to T cells (Fig. S5A), we confirmed that IL-1R-signaling in T cells is required for a fulminant type-2 allergic airway response. In line with previous reports (38, 39), mice with Il1r−/− T cells had reduced airway inflammation (Fig. S5B), eosinophilia (Fig. S5C), and pulmonary inflammation (Fig. S5D) as compared with chimeric mice with Il1r-sufficient T cells. Furthermore, T-cell expression of IL-1R is required for IL-1β–mediated exacerbation of airway allergy, because intratracheal challenge with HDM and IL-1β had little impact in chimeric mice with Il1r−/− T cells (Fig. S5). These data suggest both that IL-1R signaling in T cells is required for fulminant airway allergy and that T cells are a major target of IL-1β–exacerbated airway allergy.
Fig. S5.

IL1R-expressing T cells are required for IL-1β–exacerbated airway allergy. (A) Chimeric mice were generated by reconstituting irradiated Rag2−/− mice with 2–5 × 106 bone marrow cells (20% bone marrow from Il1r−/− or WT donor mice and 80% bone marrow from C57BL/6.Tcra−/− mice) for 6–8 wk. Chimeric mice with WT or Il1r −/− T cells were sensitized with HDM/alum and were challenged with HDM supplemented with IL-1β or not supplemented as indicated. One of two representative experiments is shown. (B) BAL cells were recovered, counted, and used for cytospins for differential analysis. (C) H&E- and AB-PAS–stained lung sections from chimeric mice with HDM-induced airway allergy. (D) Gene expression in pulmonary tissue of mice with HDM-induced airway allergy. Expression is shown relative to Hprt. *P ≤ 0.05.
To determine whether T-cell–intrinsic Trim24 is required for IL-1β–mediated T-cell activation in vivo, we challenged mice with WT or Trim24−/− T cells with HDM supplemented with IL-1β. After HDM/IL-1β challenge chimeric mice with WT T cells developed significantly enhanced airway inflammation (Fig. 4A) with elevated eosinophilia, lymphocyte recruitment, and, to a lesser extent, increased neutrophilia (Fig. 4 A and B). In contrast, mice with Trim24−/− T cells had only a modest increase in airway inflammation with a small increase in airway neutrophils after HDM/IL-1β challenge (Fig. 4 A and B). HDM/IL-1β challenge also increased systemic IgE in mice with WT cells but not in mice with Trim24−/− T cells (Fig. 4C). Within pulmonary tissue, the expression of mucus-associated genes (Muc5ac and Gob5) and type-2 cytokine genes (Il5 and Il13) also was increased in mice with WT cells but not in those with Trim24−/− T cells (Fig. 4D). Most strikingly, pulmonary inflammation was increased in dramatically HDM/IL-1β–challenged mice with WT T cells and was accompanied by increased mucus staining (Fig. 4E). Supporting the requirement of T-cell–intrinsic Trim24 for TH2-mediated airway allergy and the mechanistic requirement of T-cell–intrinsic Trim24 for IL-1β–mediated exacerbation, mice with Trim24−/− T cells showed very little pulmonary inflammation with only a modest increase in mucus staining following HDM/IL-1β challenge. In summary, we have identified an unappreciated and nonredundant role for T-cell–intrinsic Trim24 in the manifestation of TH2-mediated allergy and immunity and have identified mechanistically that Trim24 is required for IL-1R expression on TH2 cells and for IL-1β–mediated TH2 cell activation in vivo.
Fig. 4.

IL-1β–exacerbated airway allergy is dependent on T-cell–intrinsic Trim24. Chimeric mice with WT or Trim24−/− T cells were sensitized with HDM and challenged with HDM supplemented with IL-1β or unsupplemented as indicated. One of two representative experiments is shown. *P ≤ 0.05. (A and B) BAL cells were recovered, counted (A), and used for cytospins for differential analysis (B). (C) Circulating IgE was measured in the serum of chimeric mice. (D) Gene expression in pulmonary tissue of mice with HDM-induced airway allergy. Expression is shown relative to Hprt. (E) H&E- and AB-PAS–stained lung sections from chimeric mice with HDM-induced airway allergy.
Discussion
In this study we found that T-cell–intrinsic Trim24, which was predicted to be active in TH2 cells but not in TH1, TH17, or Treg cells, is essential for HDM-mediated airway allergy and TH2-dependent expulsion of the intestinal helminth H. polygyrus. Genome-wide transcriptional analysis of activated, tissue-derived Trim24−/− T cells showed that Trim24 is required for cytokine, chemokine, and chemokine receptor expression. In silico upstream analyses of transcriptional data from WT and Trim24−/− T cells suggested that IL-1 signaling is compromised in Trim24−/− T cells. Both in vitro and in vivo experiments supported this mechanistic pathway, with Trim24−/− T cells being refractory to IL-1β–mediated TH2 cell activation. Thus this study has identified a previously unappreciated role for Trim24 in TH2 cells and highlights a previously unidentified therapeutic target to forestall hyperactive TH2-mediated inflammatory diseases.
A large proportion of allergic asthmatic patients mount dysregulated type-2 immune responses to otherwise innocuous allergens. An increasing number of these patients—indeed, the majority of asthmatics—respond poorly to currently available drugs (1), increasing the need to identify new targetable pathways to treat allergic asthma. Cytokine and chemokine-secreting TH2 cells orchestrate many of the pathophysiological features of allergic asthma. We therefore interrogated the transcriptional profile of highly purified TH2 cells that contribute directly to HDM-induced airway allergy, in addition to TH2 cells isolated from helminth-infected mice (33). Rather than focusing on differentially regulated genes in the datasets, we focused on putative upstream pathways that could contribute to the transcriptional profiles. Validating this approach, we identified IL-4 (41) and several transcriptional factors/regulators including Gata3 (7), Stat3 (8), Stat6 (9), and cMaf (10), all of which are necessary for TH2 cell differentiation and/or effector function. This approach also predicted that Trim24 is functionally active in TH2 cells, even though its expression does not change significantly. Trim24 was not transcriptionally regulated in vitro in T cells after 3 d of stimulation with IL-4 and TCR engagement, but the protein levels of Trim24 were increased as compared with naive T cells, supporting the involvement of Trim24 in TH2 cells. The precise mechanisms of posttranscriptional regulation of Trim24, which could explain the disparity in mRNA and protein levels, are currently unclear. There are many miRNAs that are predicted to target Trim24, which, if down-regulated in TH2 cells, could permit the translation of Trim24 without altering the transcription of Trim24. For example, miR-384 and miR-539 are predicted to target Trim24 and are down-regulated in TH2 cells (33). Whether these or other miRNA-mediated pathways regulate Trim24 expression in T cells requires further validation.
Trim24 is expressed early in the developing embryo (42) in the developing nervous system (43), liver (28, 44), intestine (45), lung (46), and microglia (47). Unlike Trim28 (26) and Trim33 (48), which have reported roles in T cells, Trim24 has not been studied within the immune system. However, IL-4 can up-regulate Trim24 expression in microglia as early as 4 h poststimulation (47), and combinatorial transcription factor predictions identified that Trim24 interacts with Stat-6 (49), an IL-4–mediated signal transduction component. Furthermore, elevated Trim24 (28, 44, 45, 50–52) and constitutively active IL-4/STAT6 signaling (53–56) have been observed in many forms of human cancer. However, whether observation describes a causal relationship is currently unclear.
Trim24 can promote glucose metabolism and cellular proliferation (28, 44, 45, 50–52, 57) in a variety of cells, contributing to its involvement in tumorigenesis. TH2 cells are dependent on glucose metabolism (58); thus deletion of Trim24 in T cells also may compromise glucose metabolism, providing an additional explanation for reduced TH2 cell responses in the absence of Trim24. In addition, Trim24 interacts with several nuclear receptors, including the estrogen and retinoic acid receptors (28, 59), regulating a variety of transcriptional programs. Both estrogen receptor (60) and retinoic acid receptor signaling (61) have been widely studied in the context of allergy and T-cell biology; however, our transcriptional analysis of Trim24−/− T cells (Fig. S4) did not indicate that these pathways are dysregulated in the absence of Trim24. Instead, Trim24−/− T cells had dysregulated expression of the genes involved in amino acid transport, lipid metabolism, and glucogenesis (Lat-1, Trib3, Gpt2, Arsb, Chpt1, and Galc). However, the most notable transcriptional changes observed in Trim24−/− T cells included chemokines, cytokines, and their receptors (Fig. 3), many of which have critical functions in airway allergy. For example, Trim24-deficient T cells had reduced expression of Ccl1 and Ccl5, chemokines involved in the recruitment of eosinophils (62) and TH2 cells (63), respectively; reduced expression of Il4, Il21, and Spp1 (Osteopontin), all of which contribute significantly to airway allergy (64–66); and reduced expression of Il1rl1 (Il33r) and il17re (Il25r), both of which contribute to TH2 cell activation and recruitment (67–69). These data highlight the importance of Trim24 for TH2 cell effector function and provide multiple mechanistic explanations for the reduced airway allergy observed in mice with Trim24−/− T cells.
To identify an upstream mechanistic link that could underpin the various deficiencies observed in Trim24−/− T cells, we performed upstream analyses of transcriptional datasets from WT and Trim24−/− T cells. These analyses predicted that IL-1R signaling would be compromised in Trim24−/− T cells. In line with this prediction, IL-1R signaling is required for TH2-dependent immunity (36, 37) and TH2-mediated airway allergy (38, 39), supporting observations presented here with Trim24−/− T cells. Specifically, IL-1 signaling is required to activate TH2 cells in vivo (13, 40), and, as we show here, IL-1R signaling in T cells can exacerbate TH2-driven airway allergy. In support of this upstream mechanistic link, Tim24−/− TH2 cells were refractory to IL-1β–induced activation/proliferation and cytokine production in vitro, and mice with Tim24−/− T cells failed to develop exacerbated disease when activated with allergen and IL-1β. Whether integrated IL-1R and IL-4R signaling pathways converge on Trim24 in T cells is unclear; however, a series of protein interactions involving an IL-1R1–ubiquitin C–Trim24–STAT6 axis (15, 18, 49) has been independently described but as yet not validated. In addition, IL-1 signaling in lymphocytes potently activates NFκB (70), which is regulated by several Trim family members (71). Whether Trim24 regulates IL-1–mediated NFκB also is currently unknown but may explain the requirement of Trim24 for IL-1–mediated TH2 cell activation.
Therapeutically, anakinra, an IL-1R antagonist, has been approved and is used to treat rheumatoid arthritis when there are no signs of infection (72); however, inhibiting IL-1R signaling may compromise continuous immunosurveillance, particularly in the allergic lung. Instead, targeting Trim24 may not disarm the host as much as blocking IL-1R but still may curtail TH2-mediated airway disease. Bromodomain inhibitors targeting Trim24 have been developed (73, 74) and also may prevent dysregulated TH2-mediated diseases. In summary, we have identified an important and previously unappreciated role for T-cell–intrinsic Trim24, which is required for TH2 cell-mediated airway allergy and for IL-1R–mediated exacerbation of allergic airway disease.
Materials and Methods
Animals.
C57BL/6, C57BL/6 Il4gfp, C57BL/6.Tcra−/−, Rag2−/−, and Il1r−/− mice were bred and kept in the specific pathogen-free facility at The Francis Crick Institute. Trim24−/− bone marrow was kindly provided by Gabrielle Mengus and Irwin Davidson (Institut de Génétique et de Biologie Moléculaire et Cellulaire, Illkirch, France). Mice were randomly housed five mice per cage before the experimental procedure. A minimum of five mice per group was used for each experiment, unless indicated.
Generation of Mixed T-Cell Bone Marrow Chimeric Mice.
Rag2−/−mice (6–8 wk old) were irradiated (2 × 450 rad) followed by adoptive transfer of 2–5 × 106 bone marrow cells (20% bone marrow from Trim24−/−, Il1r−/−, or WT donor mice and 80% bone marrow from C57BL/6.Tcra−/− mice) for 6–8 wk before any experimental procedure. Mice were given water ad libitum that was supplemented with Baytril for 3 wk after radiation. For 1:1 chimeric mice, Rag2−/− mice were lethally irradiated (2 × 450 rad) and given 2 × 106 WT and Trim24−/− bone marrow cells for 8 wk before analysis.
CD4 T-Cell Isolation, Flow Cytometry, and TH2 Polarization.
CD4+ T cells were isolated from tissues by mechanical disruption followed by red blood cell lysis and Percoll gradient separation followed by positive or negative cell enrichment using magnetic beads (L3T4; Miltenyi Biotec). Cells then were stained with anti-mouse CD4 (clone RM4-5), CD44 (clone IM7), CD25 (clone PC61), TCR-β (clone H57-597), Vβ5 (clone MR9-4), Vα2 (clone B20.1), and IL-1R1 (JAMA-147). Dead cells were excluded from sorting and analysis with propidium iodide or Live/Dead stain (Invitrogen). Anti-CD16/32 was used in all staining. Cells were acquired using a BD LSR II flow cytometer and were analyzed with FlowJo software (Tree Star). For cell sorting, BD Influx, FACS Aria (BD), or MoFlo XDP (Beckman Coulter) sorters were used, with a purity of sorted cells >95%. For TH2 polarization, naive T cells were stimulated with anti-CD3 (1 μg/mL) and anti-CD28 (10 μg/mL) and were cultured with IL-4 (10 ng/mL), IL-2 (5 ng/mL), and anti-IFNγ (10 μg/mL) for 10 d or as indicated in figures.
Airway Allergy and H. polygyrus Infection Models.
H. polygyrus.
Mice were infected with 200 H. polygyrus infective larvae (stage 3) by oral gavage. On day 14 and 15 mice were treated with an oral dose of pyrantel embonate (2 mg) to clear the helminth infection. On day 28 mice were given a challenge infection. TH2-dependent immunity to H. polygyrus determined 14 d after the secondary infection by counting luminal worms in the small intestine.
HDM/alum-induced airway allergy.
For HDM/alum-induced airway inflammation, mice were sensitized by i.p. injection with 10 μg HDM (Greer) with 2 mg of Imject Alum (Thermo Scientific) on day 0 and day 14 followed by intratracheal challenge with 10 μg of HDM in PBS on day 21 and day 24. The severity of airway inflammation and pathology was determined on day 25.
HDM-induced airway allergy, without alum.
Alternatively, mice were sensitized by intranasal (i.n.) delivery of 10 μg HDM (Greer) in PBS on day 0 followed by i.n. challenge with 10 μg of HDM in PBS on days 7–11, with airway allergy determined on day 14. Cytospins of bronchoalveolar lavage (BAL) recoveries were performed to determine cellular infiltrates. Pulmonary tissue was recovered in RNAlater (Thermo Fisher) for gene-expression analysis or in 10% neutral-buffered formalin for histopathology. Tissues were stained with Alcian blue Periodic acid-Schiff (AB-PAS) stain and H&E to detect mucus production and leukocyte infiltration, respectively. Local lymph nodes were recovered, and 2 × 105 cells were restimulated in 96-well plates with 10 μg of HDM to determine antigen-specific cytokine secretions. Serum was recovered for IgE analysis.
Western Blot for Trim24.
For immunoblotting, cells were lysed in 1× RIPA buffer [500 mM Tris⋅HCl (pH 7.5), 150 mM NaCl, 2 mM EDTA, 0.1% SDS, 0.5% deoxycholate, 1% Nonidet-P40] containing protein inhibitors per the manufacturer’s instructions (Roche), including 50 mM sodium flouride (NaF), 1 mM sodium orthovanadate, 100 nM Okadaic acid, and 2 mM sodium pyrophosphate tetrabasic in MilliQ water. Cell lysates were normalized to equal total protein content using the Pierce BCA Protein Assay Kit (Life Technologies) and were resolved on NuPAGE 4–12% Bis-Tris Gel (Life Technologies). Separated proteins were transferred onto Amersham Hybond-P PVDF membranes (GE Healthcare) via the semidry Trans-Blot Turbo system (Bio-Rad). Membranes were blocked with 0.1% TBS and Tween 20 (TBST) containing 5% milk (Sigma) and were incubated with primary Rabbit anti-TIF1α/TRIM24 (A300-815A; Bethyl) in 0.1% TBST containing 5% BSA and secondary antibodies (Rabbit IgG; GE Healthcare) in 0.1% TBST (Sigma) containing 5% milk (Sigma). Membranes were washed in 0.1% TBST, and specific bound antibodies were visualized by chemiluminescence (Immobilon; Merck Millipore).
RNA, qRT-PCR, Microarray, and in Silico Analysis.
RNA was isolated from tissues and cells using RNeasy mini spin columns according to the manufacturer’s instructions (Qiagen). For microarray analysis, cDNA was generated from 5 ng of total RNA using the WT-Ovation Pico (version 1) RNA Amplification System followed by double-stranded cDNA synthesis using the WT-Ovation Exon Module. cDNA quality was determined using an Agilent BioAnalyzer and through hybridization performance on Affymetrix GeneChip mouse Genome 430A 2.0 microarray (Affymetrix) by the Systems Biology Unit at The Francis Crick Institute. Microarray data were quantile-normalized and analyzed using GeneSpring software (Agilent). Differentially expressed genes were determined using ANOVA and t tests. Genes with false discovery rate-corrected P values <0.1 and fold-change values ≥1.5 were considered significant, as indicated in figure legends. Three to five biological replicates were used, as indicated. Data then were uploaded into IPA (Ingenuity Systems; www.ingenuity.com) for pathways and upstream analysis (34). For qRT-PCR, RNA was reverse-transcribed using miScript II RT Kit (Qiagen). Real-time RT-PCR was performed on an ABI Prism 7900HT Sequence Detection System (Applied Biosystems) with relative quantities of mRNA determined using SYBR Green PCR Master Mix (Applied Biosystems) and by the comparative threshold cycle method as described by Applied Biosystems for the ABI Prism 7700/7900HT Sequence Detection Systems. mRNA levels were normalized to hypoxanthine-guanine phosphoribosyltransferase (HPRT) or GAPDH and expressed as a relative increase or decrease compared with levels in controls or relative to HPRT, as indicated. The following primer pairs were used: Il4, forward: ACGAGGTCACAGGAGAAGGGA, reverse: AGCCCTACAGACGAGCTCACTC; Il5, forward: TGACAAGCAATGAGACGATGAGG, reverse: ACCCCCACGGACAGTTTGATTC; Il13, forward: CCTCTGACCCTTAAGGAGCTTAT, reverse: CGTTGCACAGGGGAGTCTT; Relma, forward: CCCTCCACTGTAACGAAGACTC, reverse: CACACCCAGTAGCAGTCATCC; Muc5ac, forward: CAGGACTCTCTGAAATCGTACCA, reverse: AAGGCTCGTACCACAGGGA; Gob5, forward: CATCGCCATAGACCACGACG, reverse: TTCCAGCTCTCGGGAATCAAA; Arg1, F-GGAAAGCCAATGAAGAGCTG, reverse: GCTTCCAACTGCCAGACTGT; Trim24, forward: TTACCAAACCCTAGAATGCAG, reverse: ACATTCTGGCTTGGTGAATATC.
ELISA.
Cytokines and IgE were measured by ELISA. Capture and biotinylated detection antibodies for IL-4, IL-5, IL-13, IFNγ, and IL-10 were from R&D Systems. Total IgE was measured in serum using Purified Rat Anti-Mouse IgE (R35-72; BD Pharmingen) at 2 μg/mL For measurement of HDM-specific IgE, IgG was adsorbed to protein G Sepharose beads before measurement of HDM-specific IgE on HDM-coated (10 μg/mL) plates, as previously described (75) followed by Biotin Rat Anti-Mouse IgE at 1 μg/mL (R35-118; BD Pharmingen) with IgE, k isotype standard (BD Pharmingen), and was detected with Streptavidin HRP at 1:000 (BD Pharmingen) and ABTS One Component HRP Microwell Substrate (SurModics). The concentration of analytes in the sample was determined from a serial-fold diluted standard curve with OD read at 405 nm in an ELISA reader (Tecan II Safire).
Statistical Analysis.
Datasets were compared by Mann–Whitney test using GraphPad Prism (V.5.0). Differences were considered significant at P ≤ 0.05.
Ethics Statement.
All animal experiments were carried out following United Kingdom Home Office regulations (project license 80/2506) and were approved by the Francis Crick Institute Ethical Review Panel.
Acknowledgments
We thank Abdul Sesay, Harsha Jani, and Leena Bhaw-Rosun in the Systems Biology Department for help with microarray experiments; Radma Mahmood and Radika Anand for help with histology; Graham Preece, Wayne Turnbull, Bhavik Patel, and Dr. Phil Hobson for assistance with flow cytometry; Trisha Norton, Keith Williams, Adebambo Adekoya, and the staffs of buildings B1, B2, and C for animal husbandry; Rose-Marie Vesin for technical assistance in Illkirch; and members of the M.S.W. laboratory for critical feedback, discussion, and reading of the manuscript. This work was supported by the Francis Crick Institute (Grant FCI01) which receives its core funding from Cancer Research UK, the UK Medical Research Council (MRC reference no. MC_UP_A253_1028), and the Wellcome Trust. Work in the I.D. laboratory was supported by grants from the CNRS, the INSERM, the Equipes Labellisées program of the Ligue Nationale contre le Cancer, and the ANR-10-LABEX-0030-INRT French state fund through the Agence Nationale de la Recherche (ANR) under the Future Investments Program labeled “ANR-10-IDEX-0002-02.”
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1522287113/-/DCSupplemental.
References
- 1.Olin JT, Wechsler ME. Asthma: Pathogenesis and novel drugs for treatment. BMJ. 2014;349:g5517. doi: 10.1136/bmj.g5517. [DOI] [PubMed] [Google Scholar]
- 2.Lloyd CM, Hessel EM. Functions of T cells in asthma: More than just T(H)2 cells. Nat Rev Immunol. 2010;10(12):838–848. doi: 10.1038/nri2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Busse WW, et al. Randomized, double-blind, placebo-controlled study of brodalumab, a human anti-IL-17 receptor monoclonal antibody, in moderate to severe asthma. Am J Respir Crit Care Med. 2013;188(11):1294–1302. doi: 10.1164/rccm.201212-2318OC. [DOI] [PubMed] [Google Scholar]
- 4.Corren J, et al. A randomized, controlled, phase 2 study of AMG 317, an IL-4Ralpha antagonist, in patients with asthma. Am J Respir Crit Care Med. 2010;181(8):788–796. doi: 10.1164/rccm.200909-1448OC. [DOI] [PubMed] [Google Scholar]
- 5.Ortega HG, et al. MENSA Investigators Mepolizumab treatment in patients with severe eosinophilic asthma. N Engl J Med. 2014;371(13):1198–1207. doi: 10.1056/NEJMoa1403290. [DOI] [PubMed] [Google Scholar]
- 6.Arron JR, Scheerens H, Matthews JG. Redefining approaches to asthma: Developing targeted biologic therapies. Adv Pharmacol. 2013;66:1–49. doi: 10.1016/B978-0-12-404717-4.00001-9. [DOI] [PubMed] [Google Scholar]
- 7.Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89(4):587–596. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- 8.Stritesky GL, et al. The transcription factor STAT3 is required for T helper 2 cell development. Immunity. 2011;34(1):39–49. doi: 10.1016/j.immuni.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity. 1996;4(3):313–319. doi: 10.1016/s1074-7613(00)80439-2. [DOI] [PubMed] [Google Scholar]
- 10.Ho IC, Hodge MR, Rooney JW, Glimcher LH. The proto-oncogene c-maf is responsible for tissue-specific expression of interleukin-4. Cell. 1996;85(7):973–983. doi: 10.1016/s0092-8674(00)81299-4. [DOI] [PubMed] [Google Scholar]
- 11.Okoye IS, Wilson MS. CD4+ T helper 2 cells--microbial triggers, differentiation requirements and effector functions. Immunology. 2011;134(4):368–377. doi: 10.1111/j.1365-2567.2011.03497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sims JE, Smith DE. The IL-1 family: Regulators of immunity. Nat Rev Immunol. 2010;10(2):89–102. doi: 10.1038/nri2691. [DOI] [PubMed] [Google Scholar]
- 13.Ben-Sasson SZ, et al. IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc Natl Acad Sci USA. 2009;106(17):7119–7124. doi: 10.1073/pnas.0902745106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Papazian D, Hansen S, Wurtzen PA. Airway responses towards allergens - from the airway epithelium to T cells. Clin Exp Allergy. 2015;45(8):1268–1287. doi: 10.1111/cea.12451. [DOI] [PubMed] [Google Scholar]
- 15.Kim W, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. 2011;44(2):325–340. doi: 10.1016/j.molcel.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wagner SA, et al. 2011. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol Cell Proteomics 10(10):M111.013284.
- 17.Danielsen JM, et al. 2011. Mass spectrometric analysis of lysine ubiquitylation reveals promiscuity at site level. Mol Cell Proteomics 10(3):M110.003590.
- 18.Brissoni B, et al. Intracellular trafficking of interleukin-1 receptor I requires Tollip. Curr Biol. 2006;16(22):2265–2270. doi: 10.1016/j.cub.2006.09.062. [DOI] [PubMed] [Google Scholar]
- 19.Teague TK, et al. Activation changes the spectrum but not the diversity of genes expressed by T cells. Proc Natl Acad Sci USA. 1999;96(22):12691–12696. doi: 10.1073/pnas.96.22.12691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ozato K, Shin DM, Chang TH, Morse HC., 3rd TRIM family proteins and their emerging roles in innate immunity. Nat Rev Immunol. 2008;8(11):849–860. doi: 10.1038/nri2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cammas F, Khetchoumian K, Chambon P, Losson R. TRIM involvement in transcriptional regulation. Adv Exp Med Biol. 2012;770:59–76. doi: 10.1007/978-1-4614-5398-7_5. [DOI] [PubMed] [Google Scholar]
- 22.Herquel B, Ouararhni K, Davidson I. The TIF1α-related TRIM cofactors couple chromatin modifications to transcriptional regulation, signaling and tumor suppression. Transcription. 2011;2(5):231–236. doi: 10.4161/trns.2.5.17725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou XF, et al. TRIM28 mediates chromatin modifications at the TCRα enhancer and regulates the development of T and natural killer T cells. Proc Natl Acad Sci USA. 2012;109(49):20083–20088. doi: 10.1073/pnas.1214704109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cambiaghi V, et al. TRIM proteins in cancer. Adv Exp Med Biol. 2012;770:77–91. doi: 10.1007/978-1-4614-5398-7_6. [DOI] [PubMed] [Google Scholar]
- 25.Versteeg GA, Benke S, García-Sastre A, Rajsbaum R. InTRIMsic immunity: Positive and negative regulation of immune signaling by tripartite motif proteins. Cytokine Growth Factor Rev. 2014;25(5):563–576. doi: 10.1016/j.cytogfr.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chikuma S, Suita N, Okazaki IM, Shibayama S, Honjo T. TRIM28 prevents autoinflammatory T cell development in vivo. Nat Immunol. 2012;13(6):596–603. doi: 10.1038/ni.2293. [DOI] [PubMed] [Google Scholar]
- 27.Jiang S, et al. TRIM24 suppresses development of spontaneous hepatic lipid accumulation and hepatocellular carcinoma in mice. J Hepatol. 2015;62(2):371–379. doi: 10.1016/j.jhep.2014.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khetchoumian K, et al. Loss of Trim24 (Tif1alpha) gene function confers oncogenic activity to retinoic acid receptor alpha. Nat Genet. 2007;39(12):1500–1506. doi: 10.1038/ng.2007.15. [DOI] [PubMed] [Google Scholar]
- 29.Herquel B, et al. Trim24-repressed VL30 retrotransposons regulate gene expression by producing noncoding RNA. Nat Struct Mol Biol. 2013;20(3):339–346. doi: 10.1038/nsmb.2496. [DOI] [PubMed] [Google Scholar]
- 30.Tisserand J, et al. Tripartite motif 24 (Trim24/Tif1α) tumor suppressor protein is a novel negative regulator of interferon (IFN)/signal transducers and activators of transcription (STAT) signaling pathway acting through retinoic acid receptor α (Rarα) inhibition. J Biol Chem. 2011;286(38):33369–33379. doi: 10.1074/jbc.M111.225680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khetchoumian K, et al. Trim24 (Tif1 alpha): An essential ‘brake’ for retinoic acid-induced transcription to prevent liver cancer. Cell Cycle. 2008;7(23):3647–3652. doi: 10.4161/cc.7.23.7123. [DOI] [PubMed] [Google Scholar]
- 32.Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol. 2015;16(1):45–56. doi: 10.1038/ni.3049. [DOI] [PubMed] [Google Scholar]
- 33.Okoye IS, et al. Transcriptomics identified a critical role for Th2 cell-intrinsic miR-155 in mediating allergy and antihelminth immunity. Proc Natl Acad Sci USA. 2014;111(30):E3081–E3090. doi: 10.1073/pnas.1406322111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krämer A, Green J, Pollard J, Jr, Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014;30(4):523–530. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reynolds LA, Filbey KJ, Maizels RM. Immunity to the model intestinal helminth parasite Heligmosomoides polygyrus. Semin Immunopathol. 2012;34(6):829–846. doi: 10.1007/s00281-012-0347-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Humphreys NE, Grencis RK. IL-1-dependent, IL-1R1-independent resistance to gastrointestinal nematodes. Eur J Immunol. 2009;39(4):1036–1045. doi: 10.1002/eji.200838938. [DOI] [PubMed] [Google Scholar]
- 37.Helmby H, Grencis RK. Interleukin 1 plays a major role in the development of Th2-mediated immunity. Eur J Immunol. 2004;34(12):3674–3681. doi: 10.1002/eji.200425452. [DOI] [PubMed] [Google Scholar]
- 38.Ritter M, et al. Functional relevance of NLRP3 inflammasome-mediated interleukin (IL)-1β during acute allergic airway inflammation. Clin Exp Immunol. 2014;178(2):212–223. doi: 10.1111/cei.12400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Broide DH, Campbell K, Gifford T, Sriramarao P. Inhibition of eosinophilic inflammation in allergen-challenged, IL-1 receptor type 1-deficient mice is associated with reduced eosinophil rolling and adhesion on vascular endothelium. Blood. 2000;95(1):263–269. [PubMed] [Google Scholar]
- 40.Nakae S, et al. IL-1 is required for allergen-specific Th2 cell activation and the development of airway hypersensitivity response. Int Immunol. 2003;15(4):483–490. doi: 10.1093/intimm/dxg054. [DOI] [PubMed] [Google Scholar]
- 41.Kopf M, et al. Disruption of the murine IL-4 gene blocks Th2 cytokine responses. Nature. 1993;362(6417):245–248. doi: 10.1038/362245a0. [DOI] [PubMed] [Google Scholar]
- 42.Torres-Padilla ME, Zernicka-Goetz M. Role of TIF1alpha as a modulator of embryonic transcription in the mouse zygote. J Cell Biol. 2006;174(3):329–338. doi: 10.1083/jcb.200603146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Niederreither K, Remboutsika E, Gansmuller A, Losson R, Dollé P. Expression of the transcriptional intermediary factor TIF1alpha during mouse development and in the reproductive organs. Mech Dev. 1999;88(1):111–117. doi: 10.1016/s0925-4773(99)00175-6. [DOI] [PubMed] [Google Scholar]
- 44.Liu X, et al. Overexpression of TRIM24 is associated with the onset and progress of human hepatocellular carcinoma. PLoS One. 2014;9(1):e85462. doi: 10.1371/journal.pone.0085462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miao ZF, et al. 2015. TRIM24 is upregulated in human gastric cancer and promotes gastric cancer cell growth and chemoresistance. Virchows Archive: European Journal of Pathology 466(5):525–532.
- 46.Li H, et al. Overexpression of TRIM24 correlates with tumor progression in non-small cell lung cancer. PLoS One. 2012;7(5):e37657. doi: 10.1371/journal.pone.0037657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Freilich RW, Woodbury ME, Ikezu T. Integrated expression profiles of mRNA and miRNA in polarized primary murine microglia. PLoS One. 2013;8(11):e79416. doi: 10.1371/journal.pone.0079416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Doisne JM, et al. iNKT cell development is orchestrated by different branches of TGF-beta signaling. J Exp Med. 2009;206(6):1365–1378. doi: 10.1084/jem.20090127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ravasi T, et al. An atlas of combinatorial transcriptional regulation in mouse and man. Cell. 2010;140(5):744–752. doi: 10.1016/j.cell.2010.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xue D, et al. Clinical significance and biological roles of TRIM24 in human bladder carcinoma. Tumour Biol. 2015;36(9):6849–6855. doi: 10.1007/s13277-015-3393-3. [DOI] [PubMed] [Google Scholar]
- 51.Cui Z, et al. TRIM24 overexpression is common in locally advanced head and neck squamous cell carcinoma and correlates with aggressive malignant phenotypes. PLoS One. 2013;8(5):e63887. doi: 10.1371/journal.pone.0063887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tsai WW, et al. TRIM24 links a non-canonical histone signature to breast cancer. Nature. 2010;468(7326):927–932. doi: 10.1038/nature09542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guiter C, et al. Constitutive STAT6 activation in primary mediastinal large B-cell lymphoma. Blood. 2004;104(2):543–549. doi: 10.1182/blood-2003-10-3545. [DOI] [PubMed] [Google Scholar]
- 54.Ni Z, et al. Selective activation of members of the signal transducers and activators of transcription family in prostate carcinoma. J Urol. 2002;167(4):1859–1862. [PubMed] [Google Scholar]
- 55.Skinnider BF, et al. Signal transducer and activator of transcription 6 is frequently activated in Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood. 2002;99(2):618–626. doi: 10.1182/blood.v99.2.618. [DOI] [PubMed] [Google Scholar]
- 56.Qin JZ, et al. Constitutive and interleukin-7- and interleukin-15-stimulated DNA binding of STAT and novel factors in cutaneous T cell lymphoma cells. J Invest Dermatol. 2001;117(3):583–589. doi: 10.1046/j.0022-202x.2001.01436.x. [DOI] [PubMed] [Google Scholar]
- 57.Pathiraja TN, et al. TRIM24 links glucose metabolism with transformation of human mammary epithelial cells. Oncogene. 2015;34(22):2836–2845. doi: 10.1038/onc.2014.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Michalek RD, et al. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186(6):3299–3303. doi: 10.4049/jimmunol.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thénot S, Henriquet C, Rochefort H, Cavaillès V. Differential interaction of nuclear receptors with the putative human transcriptional coactivator hTIF1. J Biol Chem. 1997;272(18):12062–12068. doi: 10.1074/jbc.272.18.12062. [DOI] [PubMed] [Google Scholar]
- 60.Bonds RS, Midoro-Horiuti T. Estrogen effects in allergy and asthma. Curr Opin Allergy Clin Immunol. 2013;13(1):92–99. doi: 10.1097/ACI.0b013e32835a6dd6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hall JA, Grainger JR, Spencer SP, Belkaid Y. The role of retinoic acid in tolerance and immunity. Immunity. 2011;35(1):13–22. doi: 10.1016/j.immuni.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bishop B, Lloyd CM. CC chemokine ligand 1 promotes recruitment of eosinophils but not Th2 cells during the development of allergic airways disease. J Immunol. 2003;170(9):4810–4817. doi: 10.4049/jimmunol.170.9.4810. [DOI] [PubMed] [Google Scholar]
- 63.Schuh JM, Blease K, Hogaboam CM. 2002. The role of CC chemokine receptor 5 (CCR5) and RANTES/CCL5 during chronic fungal asthma in mice. FASEB J 16(2):228–230.
- 64.Lajoie S, et al. 2014. IL-21 receptor signalling partially mediates Th2-mediated allergic airway responses. Clin Exp Allergy 44(7):976–985.
- 65.Müller KM, Jaunin F, Masouyé I, Saurat JH, Hauser C. Th2 cells mediate IL-4-dependent local tissue inflammation. J Immunol. 1993;150(12):5576–5584. [PubMed] [Google Scholar]
- 66.Xanthou G, et al. Osteopontin has a crucial role in allergic airway disease through regulation of dendritic cell subsets. Nat Med. 2007;13(5):570–578. doi: 10.1038/nm1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Corrigan CJ, et al. T-helper cell type 2 (Th2) memory T cell-potentiating cytokine IL-25 has the potential to promote angiogenesis in asthma. Proc Natl Acad Sci USA. 2011;108(4):1579–1584. doi: 10.1073/pnas.1014241108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tamachi T, et al. IL-25 enhances allergic airway inflammation by amplifying a TH2 cell-dependent pathway in mice. J Allergy Clin Immunol. 2006;118(3):606–614. doi: 10.1016/j.jaci.2006.04.051. [DOI] [PubMed] [Google Scholar]
- 69.Löhning M, et al. T1/ST2 is preferentially expressed on murine Th2 cells, independent of interleukin 4, interleukin 5, and interleukin 10, and important for Th2 effector function. Proc Natl Acad Sci USA. 1998;95(12):6930–6935. doi: 10.1073/pnas.95.12.6930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stylianou E, et al. Interleukin 1 induces NF-kappa B through its type I but not its type II receptor in lymphocytes. J Biol Chem. 1992;267(22):15836–15841. [PubMed] [Google Scholar]
- 71.Tomar D, Singh R. 2015. TRIM family proteins: Emerging class of RING E3 ligases as regulator of NF-kappaB pathway. Biol Cell 107(1):22–40.
- 72.Wang D, Li Y, Liu Y, Shi G. The use of biologic therapies in the treatment of rheumatoid arthritis. Curr Pharm Biotechnol. 2014;15(6):542–548. doi: 10.2174/138920101506140910150612. [DOI] [PubMed] [Google Scholar]
- 73.Palmer WS, et al. Structure-guided design of IACS-9571, a selective high-affinity dual TRIM24-BRPF1 bromodomain inhibitor. J Med Chem. 2015 doi: 10.1021/acs.jmedchem.5b00405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bennett J, et al. Discovery of a chemical tool inhibitor targeting the bromodomains of TRIM24 and BRPF. J Med Chem. 2015 doi: 10.1021/acs.jmedchem.5b00458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wilson MS, et al. Suppression of allergic airway inflammation by helminth-induced regulatory T cells. J Exp Med. 2005;202(9):1199–1212. doi: 10.1084/jem.20042572. [DOI] [PMC free article] [PubMed] [Google Scholar]