

Abstract
We have developed a CRISPR-based method that uses catalytically active Cas9 and distinct sgRNA constructs to knock out and activate different genes in the same cell. These sgRNAs, with 14 15 bp target sequences and MS2 binding loops, can activate gene expression using an active Cas9 nuclease, without inducing DSBs. We use these ‘dead RNAs’ to perform orthogonal gene knockout and transcriptional activation in human cells.
The RNA-guided Cas9 nuclease from the microbial CRISPR (clustered regularly interspaced short palindromic repeats)-Cas system holds promise as a system that can be used for both gene knock out and activation 1,2. Once bound to its target DNA, the active sgRNA-Cas9 complex can induce a double stranded break (DSB) via its RuvC and HNH domains. By mutating these nuclease domains, Cas9 can be made catalytically inactive3,4 and repurposed for genetic perturbation beyond DNA editing. For example, this ‘dead’ Cas9 (dCas9) has been combined with protein domains that suppress or activate gene expression5-9 or change the epigenetic state of a target locus10.
The simplest CRISPR-based orthogonal gene control system would function with only catalytically active wild type Cas9, in contrast to previous work that used distinct Cas9 proteins from different organism2. Here we show that by reducing the length of the RNA targeting sequence to 14-15 nucleotides (nt), and by adding MS2 binding loops into the sgRNA backbone11, sgRNAs can guide catalytically active Cas9 to activate transcription without inducing DSBs (Fig. 1a). After systematically studying how the design of these ‘dead sgRNAs’ (dRNAs) influences transcriptional activation and indel formation, and characterizing off-target transcriptional perturbations, we show that this system can simultaneously knock out and upregulate target genes in melanoma cells. These data demonstrate that sgRNAs can be engineered to exert transcriptional control using active Cas9, and that a single active Cas9 protein can be used for orthogonal gene control in mammalian cells.
Figure 1.
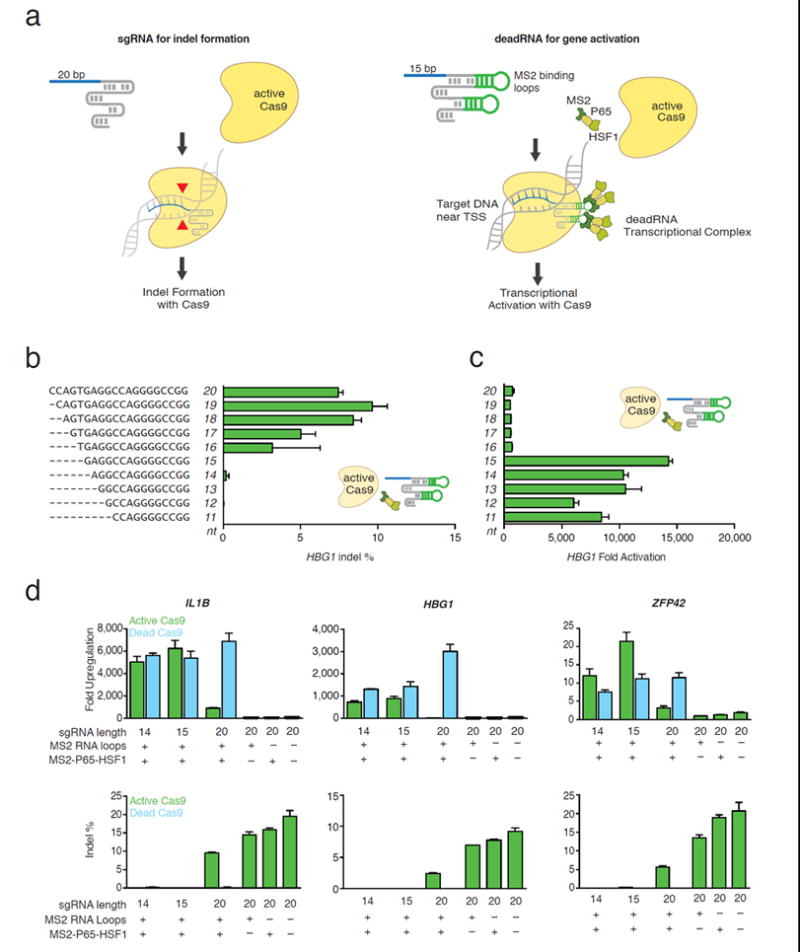
deadRNAs (dRNAs) can mediate robust gene activation using an active SpCas9. (a) dRNA-mediated gene activation. 15bp dRNA with MS2 loops on the scaffold can bind the MS2-P65-HSF1 (MPH) transcriptional activation complex and activate gene expression without inducing Cas9-mediated DNA indel formation. (b) The length of the RNA targeting sequence was varied from 11nt to 20nt. HBG1 indel frequencies were quantified with (c) HBG1 mRNA levels (normalized to GAPDH, and compared to cells transfected with GFP plasmid). No indel formation was observed when sgRNAs had less than 16bp of homology to target DNA. In all cases, guides were designed with MS2 binding loops in the tetraloops and stemloop two11, and were co-transfected with active Cas9 and the MPH transcriptional activation complex. (d) Three dRNAs targeting the promoter regions of IL1B, HBG1, and ZFP42 were tested for activation and indel formation. dRNAs with 14bp or 15bp of homology to target DNA did not induce detectable indel formation. dRNAs co- transfected with Cas9 and MPH activated transcription to a similar extent as 20nt sgRNA-MS2 co-transfected with dCas9 and MPH. (In all cases, mean +/- S.E.M. is plotted. N=2-3 replicates / group). Guide RNAs targeting IL1B, HBG1, and ZFP42 in Figure 1D were derived from the 20bp spacers AAAGGGGAAAAGAGTATTGG, GGCAAGGCTGGCCAACCCAT, and ACCCTGGCGGAGCTGATGGG, respectively
Changes in sgRNA structure and mismatches between the sgRNA targeting sequence and DNA can prevent Cas9- mediated DNA cleavage12-14. However, it is unknown whether these modified sgRNAs still allow binding of Cas9 to the DNA target. To test this, we designed sgRNAs with two structural characteristics. First, we added two aptamers that selectively bind dimerized MS2 bacteriophage coat proteins to the tetraloop and stem loop two of the sgRNA as previously described (sgRNA-MS2)11. Second, we shortened the length of the sgRNA guide sequence from 20nt to 11nt. We reasoned that these changes could result in a dRNA that would still enable binding of Cas9 while preventing nuclease activity (Fig. 1a).
We transfected eighty sgRNA-MS2s targeting four DNA sequences within 200 bp of the transcriptional start site of human hemoglobin 1 (HBG1) together with active Cas9 and the MS2-P65-HSF1 (MPH) activation complex. The MPH complex has previously been reported to mediate efficient target upregulation by binding to MS2 loops in the sgRNA11. We observed that guides from 20nt to 16nt resulted in indel formation, whereas shorter guides (11nt to 15nt) did not show detectable levels of indel formation in most cases (Fig. 1b). Notably, guides truncated to 11-15nt of complementarity to the target DNA were able to increase HBG1 mRNA expression by as much as 10,000 fold (Fig. 1c, Supplementary Fig. 1). We then investigated guides with mismatches on the 5’ end of the sgRNA analogous to our truncation experiments (Supplementary Fig. 2). In accordance with our results from truncated guides, we observed that guides with only 15bp complementarity to the target DNA were still able to mediate efficient activation in all four cases.
We proceeded to investigate the gene activation efficiency of 14 and 15nt dRNAs at three loci. In all cases the dRNAs, when co- transfected into HEK293FT cells with active Cas9 and the MPH complex, increased target mRNA expression of all three human genes (HBG1, Interleukin 1B (IL1B), and Zinc Finger Protease 42 (ZFP42)) without inducing significant indel formation (Fig. 1d). Notably, dRNA activation was comparable to the recently reported system using dCas9 in combination with a 20nt sgRNA-MS211. At all three loci 20nt sgRNAs cut target DNA and did not activate gene expression when combined with active Cas9. This was true for sgRNAs with and without the MS2 binding loops (Fig. 1d).
Biological studies utilizing activators will require specific target upregulation. Therefore, it is important to understand the specificity of Cas9 mediated gene activation. In addition, recent work has demonstrated that a single sgRNA can bind many sites in the genome, but the relationship between binding and transcriptional control is not clear15. Specificity may also change when the sgRNA is shortened to 15 bp. To assess the difference in specificity between 20nt sgRNA-MS2 and 15nt dRNAs we compared whole transcriptome mRNA levels in HEK293FT cells. Cells were co-transfected with dCas9, the MPH complex, and a 20nt activator sgRNA-MS2, or active Cas9, the MPH complex, and 15nt dRNA targeting the same sequence in the human HBG1/2 promoter. We previously determined that HBG1/2 upregulation induces limited downstream effects that could confound our analysis in HEK293FT cells. RNA-seq results showed that both the sgRNA/dCas9 and dRNA systems significantly activated HBG1/2 only, demonstrating that dRNAs can specifically upregulate target genes (Fig. 2a). We next performed off-target analysis on a second 15nt dRNA and 20nt sgRNA targeting the same HBG1/2 promoter. We found a significant number of perturbed transcripts for both the 15nt and 20nt guide RNAs (Fig. 2b)
Figure 2.
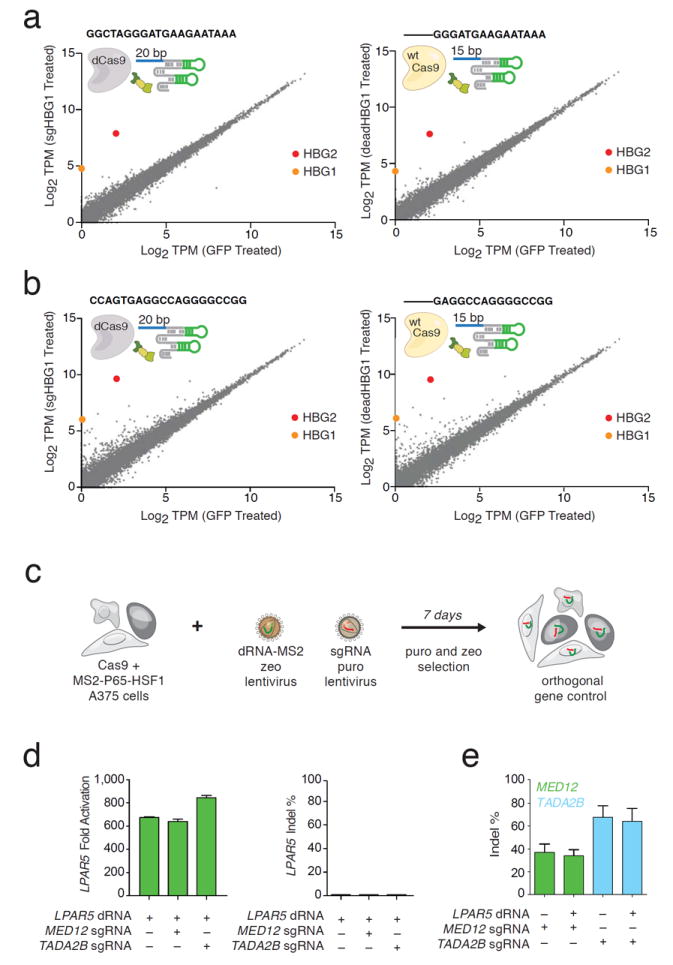
dRNAs can specifically upregulate gene expression, have a specificity profile similar to 20bp sgRNA activators, and can be used for orthogonal gene control in human cells. Sequences targeted to the HBG1/2 promoter were tested for off-target transcriptional activation using RNA-seq. 20nt sgRNAs with MS2 binding loops were co-transfected with dCas9 and the MPH activation complex. These were compared to dRNAs co-delivered with active Cas9 and the MPH activation complex. Both systems showed similar off-target profiles. (a) Zero significantly upregulated genes apart from HBG1/2 were observed for both the 20nt/dCas9 and dRNA/Cas9 treated cells. (b) A second guide showed 55 significantly upregulated genes apart from HBG1/2 for the 20nt/dCas9-treated cells, while 31 significantly upregulated genes were measured for dRNA-treated cells. (In all cases, N=3 replicates / group). (c) Orthogonal gene control in melanoma A375 cells expressing an active Cas9 and the MS2-P65-HSF1 fusion protein. Cells were transduced with lentivirus containing a dRNA targeting one gene and an sgRNA targeting a second gene. Selected cells were subsequently treated with BRAF- inhibitor PLX4720 and their survival was quantified. (d) Activation and indel % were measured for individually and orthogonally controlled genes. Left: LPAR5 transcriptional upregulation mediated by dRNA was robust in the presence and absence of sgRNAs targeting MED12 or TADA2B. Right: LPAR5 indel formation was <= 0.85% at the dRNA target site. (e) Robust indel formation was detected at DNA sites targeted by MED12 and TADA2B sgRNAs alone and when delivered together with a dRNA targeting LPAR5.
We expanded this analysis by studying whole transcriptome analysis on ten additional sgRNAs targeting the proximal promoter of HBG1/2 (Supplementary Fig. 3). Overall, four out of twelve 20bp guides exhibited high specificity (<3 significant genome-wide off-targets), confirming activators can be specific. Notably, a previously published algorithm that predicts off-target indels did not correlate with the number of non-targeted transcripts that were altered by each guide (R = 0.12, p = 0.7)13.
Finally, we aimed to test whether dRNAs in combination with sgRNAs could mediate orthogonal gene control (activation and knockout) using only active Cas9. We previously used CRISPR-Cas9 loss-of-function (LOF)16 and gain-of-function (GOF)11 screens to identify genetic modifiers that promote resistance of A375 melanoma cells to the BRAF inhibitor PLX-4720. We performed orthogonal gene regulation with hits selected from these screens. We first transduced and selected A375 cells with two lentiviral constructs encoding active Cas9 and the MPH complex, respectively (Fig. 2c). We then transduced these cells with lentiviral constructs encoding a dRNA targeting LPAR5 for activation and / or sgRNAs targeting MED12 or TADA2B for gene knockout. LPAR5 mRNA expression increased over 600-fold when cells were treated with dRNA targeting LPAR5, even when combined with sgRNAs targeting other genes. In all conditions, LPAR5 indels equal to 0.85% were detected (Fig. 2d). Indel rates of 0.6% and 0.05% at targeted loci were also measured after cells were treated with two additional dRNAs targeting EGFR and ITGA9, respectively (Supplementary Fig. 4). By contrast, the loci targeted by MED12 and TADA2B showed robust indel formation between 33-36% and 67.4-91.5%, respectively-even in the orthogonal conditions (Fig.2e).
dCas9 proteins have been engineered for diverse functions including gene suppression, gene activation, and epigenetic modification5-10. Here we have demonstrated that guide RNAs can be engineered to bind target DNA and successfully recruit a transcriptional activation complex, without inducing measurable indel formation using an active Cas9 nuclease. These dRNAs can be designed to specifically and potently upregulate gene expression (Fig. 1, 2). We anticipate that dRNAs may also be designed to perform other genetic functions, for example, epigenetic modification or mRNA suppression.
dRNAs can be used to simplify orthogonal gene control experiments, as nuclease-mediated gene knockout and transcriptional activation can be achieved in the same cell population with one Cas9 protein (Fig. 3). By activating LPAR5, a gene previously described to promote drug resistance when upregulated11, and knocking out known tumor suppressors16, we showed that orthogonal gene modifications can be performed in a biologically meaningful context (Fig. 3b-d). Streamlining orthogonal gene control is particularly important for in vivo experiments, since concurrently delivering two distinct Cas9 complexes to the same cell is challenging. Given the fact that mice have been engineered to constitutively and inducibly express Cas917,18, we anticipate that dRNAs can be utilized for in vivo orthogonal experiments, as the MPH activator complex combined with a dRNA and sgRNA fits within the 4.7kb packaging limit of AAV vectors.
Finally, our data align with previously reported work19 that show sgRNAs with seventeen or more nucleotides reliably and efficiently cut target DNA. It is notable that by reducing the guide length down to 15 nucleotides, we abrogate indel formation, but do not abrogate ‘functional binding’. Further evidence supporting this effect was recently published; active Cas9 fused to the transcriptional activator VPR and targeted to DNA by 14bp guide RNAs resulted in transcriptional activation20. This may point to a more fundamental mechanism, whereby the interactions between the protein, sgRNA, and DNA dictate whether the Cas9 protein cuts the target DNA.
Methods
Generation of sgRNA/dRNA and SpCas9 constructs
All DNA constructs used were delivered to cells as PCR products or plasmids. sgRNAs/dRNAs used in Figure 1 and Supplementary Figures 1 and 2 were designed to be expressed from a U6 promoter and delivered as a dsDNA PCR product. sgRNAs/dRNA used in Figures 2-4 were also expressed from U6 promoters but in a plasmid backbone (see supplementary text). The active Cas9, dCas9, and MPH constructs were cloned into plasmids and used an EF1-α promoter. All constructs are available from Addgene.
Transient transfection
HEK293FT cells (Life Technologies) were maintained in high-glucose DMEM with GlutaMax and sodium pyruvate (Life Technologies) supplemented with 10% heat-inactivated characterized HyClone fetal bovine serum (Thermo Scientific) and 1% penicillin/streptomycin (Life Technologies). For gene activation experiments, 20,000 HEK293FT cells/well were plated in 100 μL media in poly-D-lysine coated 96-well plates (BD BioSciences). 24 hours after plating, cells were transfected with a 1:1:1 mass ratio of:
sgRNA/dRNA PCR product/plasmid with gene-specific targeting sequence or an EGFP control plasmid
MS2-effector plasmid or pUC19.
dCas9 plasmid, activeCas9 plasmid, or pUC19.
The total plasmid transfected per well was 300 ng, and was transfected with Lipofectamine 2000 (Life Technologies) according to the manufacturer’s instructions. Culture medium was changed 4 hours after transfection.
Lentivirus Production
HEK293T cells were cultured as described above. 1 day prior to transfection, cells were seeded at ~40% confluency in a T25 flask. Cells were transfected when they reached 80-90% confluency. For each flask, 3.4 μg of plasmid containing the vector of interest, 1.7 μg of pMD2.G, and 2.6 μg of psPAX (Addgene) were transfected using 30_μL of Lipofectamine 2000 and 33 μL Plus Reagent (Life Technologies). Media was changed four hours after transfection. After 24 hours, 2mM (final conc.) caffeine was added to the media to boost viral titer. Virus supernatant was isolated 48 hours later, and filtered with a 0.45 μm PVDF filter (Millipore), and stored at −80 °C.
Lentiviral transduction
A375 cells (Sigma) were cultured in RPMI 1640 (Life Technologies) supplemented with 9% FBS (Seradigm) and 1% penicillin/streptomycin (Life Technologies). An A375 cell line was transduced with both an active SpCas9 lentivirus and an MPH lentivirus and selected for 7 days (with Blasticidin and Hygromycin, respectively). For the addition of sgRNAs/dRNAs, cells were transduced by spinfection in 12-well plates. 3 × 106 cells in 2 mL of media supplemented with 8 μg/mL polybrene (Sigma) were added to each well, supplemented with lentiviral supernatant at a multiplicity of infection (MOI) of 0.5 and centrifuged for 2h at 1000g. 24 hours later, cells were detached with TrypLE (Life Technologies) and counted. Cells were replated at low density in a T75 flask before a selection agent was added (Zeocin and/or Puromycin, all Life Technologies). Media was replaced 2 days later, and cells were passaged every other day starting four days after replating. The duration of selection was 4 days for Puromycin and 7 days for Zeocin, Lentiviral titers were determined by spinfecting cells with 6 different volumes of lentivirus ranging from 0 to 400 μL and counting the number of surviving cells after a complete selection.
RT-qPCR
48 hours after transfection, cells were lysed and reverse transcription were performed using a Cells-to-Ct kit (Life Technologies). RNA expression was quantified by quantitative PCR (qPCR) using TaqMan qPCR probes (Life Technologies) and Fast Advanced Master Mix (Life Technologies). RNA expression was normalized to GapDH, and compared to wells transfected with 300 ng of GFP plasmid. qPCR was carried out in 5 μL multiplexed reactions and 384- well format using the LightCycler 480 Instrument II.
Indel Analysis
48 hours after transient transfection with Lipofectamine 2000, and 7 days after lentiviral transduction, cells were lysed and genomic DNA was extracted from transfected cells using QuickExtract™ DNA Extraction Solution (Epicentre). Target regions for indel analysis were PCR amplified (which adds Illumina P5 adapters) and pooled into a library for next generation sequencing. Libraries were quantified using a Qubit 2.0 Fluorometer and sequenced using an Illumina MiSeq instrument using a 300 cycle v2 kit. Indels were analyzed computationally as previously described1. MiSeq reads were first filtered by requiring an average Phred quality (Q score) of at least 23. Reads were analyzed for potential indels by using the Python difflib package (based off of the Ratcliff and Obershelp algorithm) on a 30bp flanking region around the target site (80bp total). Reads were counted as indels if an insertion or deletion operation was detected. Using the negative control samples, maximum-likelihood estimation was used to calculate the true indel rate at each target site, as previously described2. DNA indel data were deposited into SRA with the accession code PRJNA296872.
RNAseq
Samples were prepped for RNA sequencing using a TruSeq Stranded mRNA Sample Prep Kit (Illumina) and deep- sequenced on Illumina sequencing machines. RSEM v1.273 was run on the sequencing reads to estimate expression levels using the following parameters: --no-bam-output --estimate-rspd --bowtie-chunkmbs 512 --paired. RSEM’s gene level expression estimates (transcript per million (TPM)) were transformed to log-space by taking log2(TPM+1). All genes detected were used to construct scatter plots comparing each condition to the control GFP condition, using the average across biological replicates (log2(mean(TPM)+1) value per gene).
To find differentially expressed genes, we performed Student’s t-test against cells treated with 300 ng of EGFP plasmid. The t-test was run on all genes that had expression levels above log2(TPM+1)>2.5 in at least two samples. This threshold was chosen as the minimal threshold for which the number of detected genes across all libraries was constant. Only genes that were significant (p-value pass 0.05 FDR correction) and had at least 1.5 fold change were reported. RNAseq data were deposited into SRA with the accession code PRJNA296872.
Supplementary Material
Acknowledgments
The authors thank Kaijie Zheng and Ian Slaymaker for their support and input. JED is supported by a Life Science Research Foundation post-doctoral fellowship of the Cystic Fibrosis Foundation. OOA is supported by a Friends of the McGovern Institute Fellowship. JSG is supported by a D.O.E. Computational Science Graduate Fellowship. FZ is supported by the NIMH (1DP1-MH100706), the Poitras, Vallee, Simons, Paul G. Allen, and New York Stem Cell Foundations, David R. Cheng, and Bob Metcalfe. The authors plan to make the reagents widely available to the academic community through Addgene and to provide software tools via the Zhang lab web site (www.genome-engineering.org).
Footnotes
Author Contributions
JED, OOA, FZ, and SK conceived this study and designed the experiments. JED, OOA, SK, JJ, and JSG performed experiments. JED, OOA, FZ, and SK wrote the manuscript with input from all authors.
Competing Financial Interests
JED, OOA, FZ, and SK have filed intellectual property related to material described in this publication.
References
- 1.Zalatan JG, et al. Engineering Complex Synthetic Transcriptional Programs with CRISPR RNA Scaffolds. Cell. 2015;160:339–350. doi: 10.1016/j.cell.2014.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Esvelt KM, et al. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nature methods. 2013;10:1116–1121. doi: 10.1038/nmeth.2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jinek M, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E2579–2586. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perez-Pinera P, et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. 2013;10:973–976. doi: 10.1038/nmeth.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mali P, et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nature biotechnology. 2013;31:833–838. doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maeder ML, et al. CRISPR RNA-guided activation of endogenous human genes. Nature methods. 2013;10:977–979. doi: 10.1038/nmeth.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Konermann S, et al. Optical control of mammalian endogenous transcription and epigenetic states. Nature. 2013;500:472–476. doi: 10.1038/nature12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gilbert LA, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hilton IB, et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nature biotechnology. 2015;33:510–517. doi: 10.1038/nbt.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Konermann S, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nishimasu H, et al. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156:935–949. doi: 10.1016/j.cell.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsu PD, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature biotechnology. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Briner AE, et al. Guide RNA functional modules direct Cas9 activity and orthogonality. Molecular cell. 2014;56:333–339. doi: 10.1016/j.molcel.2014.09.019. [DOI] [PubMed] [Google Scholar]
- 15.Wu X, et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nature biotechnology. 2014;32:670–676. doi: 10.1038/nbt.2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shalem O, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Platt RJ, et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159:440–455. doi: 10.1016/j.cell.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dow LE, et al. Inducible in vivo genome editing with CRISPR-Cas9. Nature biotechnology. 2015;33:390–394. doi: 10.1038/nbt.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nature biotechnology. 2014;32:279–284. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kiani S, et al. Cas9 gRNA engineering for genome editing, activation and repression. Nature methods. 2015 doi: 10.1038/nmeth.3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods References
- Ran FA, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature biotechnology. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.