

Abstract
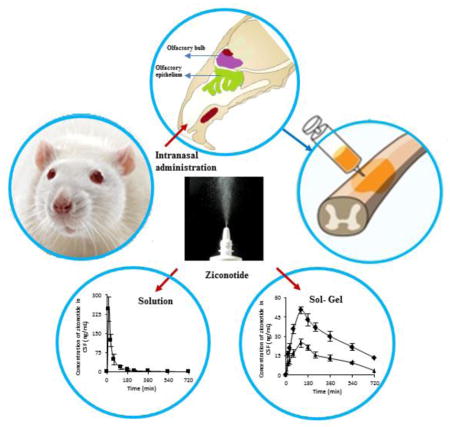
The purpose of the current study was to investigate the plausibility of delivery of ziconotide to the cerebrospinal fluid (CSF) via intranasal administration. Ziconotide was administered either in the form of solution or Kolliphor P 407 gels (KP 407) intranasally in Sprague-Dawley rats. The effect of incorporation of chitosan in the formulation was also investigated. Time course of drug in the CSF was investigated by collecting CSF from cisterna magna. Pharmacokinetics of ziconotide in CSF following intrathecal and intravenous (i.v) administration of ziconotide was investigated. Upon intrathecal administration the elimination rate constant of ziconotide in CSF was found to be 1.01 ± 0.34 h−1. The Cmax and Tmax of ziconotide in CSF following intravenous administration were found to be 37.78 ± 6.8 ng/mL and ~2 h respectively. The time required to attain maximum concentration (Tmax) in CSF was less upon intranasal administration (15 min) compared to i.v administration (120 min). Presence of chitosan enhanced the overall bioavailability of ziconotide from intranasal solution and gel formulations. The elimination rate constant of ziconotide in CSF following intranasal and intravenous administration of ziconotide solution was found to be 0.54 ± 0.08 h−1 and 0.42 ± 0.10 h−1 respectively. Whereas, intranasal administration of ziconotide in the form of in situ forming gel lowered the elimination rate significantly. These results suggest that intranasal administration could be a potential noninvasive and patient compliant method of delivering ziconotide to CSF to treat chronic pain.
Graphical Abstract
Introduction
Treatment of chronic or recurring pain is a major therapeutic challenge. Chronic pain is a widely prevalent universal problem and has been reported that more than 15% of the world’s population experience chronic pain. Reports support the fact that chronic pain significantly affects the quality of life of patients and obviously interferes with their participation in daily activities leading to reduced productivity [1, 2].
A variety of medications are generally administered via oral, transdermal and parenteral routes for the treatment of chronic neuropathic pain. Despite the availability of very potent drugs, the treatment of chronic pain remains challenging [3]. It has been found that systemic administration of drugs fail to achieve adequate pain relief in up to ~30% of patients [2]. The poor clinical responses to drugs are attributed mainly to poor bioavailability of drugs to the brain and cerebrospinal fluid (CSF) (due to the physiological barriers protecting the CNS) and short duration of activity of drugs due to rapid clearance of drug from these regions [4–6]. In addition, serious adverse effects restrict the use of analgesics systemically [7].
Ziconotide (ω-conotoxin) is a synthetic peptide (25 amino acid sequence) isolated from the magician’s cone snail, Conus magnus. It is used for the treatment of chronic pain and has a novel mechanism of action that involves potent and selective blockade of presynaptic neuronal N-type calcium channels in the spinal cord [8, 9]. Ziconotide when bound to the N-type calcium channel blocks the release of neurotransmitters from the primary afferent nerve terminals to the synaptic cleft [10, 11].
Ziconotide is found to be effective in patients who respond poorly to opioids and other analgesic drugs [12]. However, due to the peptide nature of drug and poor ability to cross the blood-CSF barrier, bioavailability to CSF following systemic administration is poor. Moreover, systemic administration of ziconotide is also known to cause profound side effects [13]. Therefore, recently this non-opioid drug was approved only for intrathecal infusion by FDA and European Medicines Agency.
Although, intrathecal administration of ziconotide is considered to be an effective route to target the spinal cord region, the procedural and device complications may be a major concern when delivering drugs via intrathecal route. Meningitis is a potential complication caused because of the contamination of the micro infusion device, pump pocket, or catheter tract. Meningitis occurred in 3% (40 cases) and 1% (1 case) of the ziconotide and placebo groups respectively during the ziconotide clinical trial. Of the 41 meningitis cases 38 were because of the use of external infusion systems [14, 15]. Complications like pump failure, program error, and incorrect refill may lead to overdose of ziconotide which might lead to exaggerated pharmacological effects like ataxia, spinal myoclonus or nystagmus. Failure in adopting proper titrations of ziconotide may also lead to cognitive and neuropsychiatric side effects [16].
Neurological deficits can be developed because of the procedure failure and inflammatory mass development catheter tip. Therefore, considering all the complications associated with existing therapy for intrathecal administration of ziconotide there is an urgent need to develop safer methods for delivery of ziconotide to the CSF.
Intranasal delivery is a noninvasive route that offers a direct pathway from nose to CSF via the olfactory apparatus [17–21]. Nose to CSF pathway could deliver drugs directly to the CSF bypassing the blood-CSF barrier. In addition, intranasal delivery would be patient compliant and allows frequent administration. Nasal formulations can be self-administered and do not require physician supervision during administration unlike in case of intrathecal or parenteral formulations [22]. Therefore, in this project the plausibility of delivering ziconotide to the CSF via intranasal route was investigated.
Intranasal administration of drugs to the CNS is limited because of the poor permeability of the mucosal epithelium to drugs. Tight junctions present in the epithelia are known to limit the permeability and bioavailability of the drug molecules. It was reported by Vaka and coworkers that chitosan leads to reversible permeabilization of olfactory mucosa leading to enhanced bioavailability of intranasally administered drugs [23]. The bioavailability of drugs is also limited by the rapid mucociliary clearance which could be resolved by administration of formulations that would increase the drug residence duration on the olfactory mucosa [24]. Therefore, in the current study Kolliphor P 407 (KP 407) was used in formulation of in situ gel to minimize the mucociliary clearance of drug
2. MATERIALS AND METHODS
2.1. Chemicals
Ziconotide acetate, chitosan (MW ~250 kDa, 75–80% deacetylation), Krebs-ringer bicarbonate (KRB) (premixed powder), acetone, hydrochloric acid were procured from Sigma chemicals (St. Louis, MO). Ziconotide acetate was radio labeled by PerkinElmer (Waltham, MA) using 125I. Sodium perchlorate, methanol, triflouro acetic acid and acetonitrile were obtained from Fischer Scientific (Atlanta, GA). Kolliphor P 407 (KP 407) was obtained from BASF (Florham Park, NJ).
2.2. Preparation of Olfactory mucosa
In vitro permeation studies were carried out across bovine olfactory mucosa (PelFreez Biologicals, Rogers, AR). Freshly excised frozen tissue was obtained from the supplier and used immediately. The tissue was thawed in KRB for a period of 30 min before carrying out the permeation studies.
2.3. Analytical Method
A). Quantification of unlabeled neat ziconotide
The HPLC system (Waters, 1525) consisting of a Phenomenex C-18 analytical column (4.6 mm × 150 mm, Luna 5.0 μ) and a variable wavelength detector (Waters, 2487). The mobile phase was made up of a gradient elution starting with 100% mobile phase A for 2 minutes, shifting to 70% mobile phase A:30% mobile phase B over 12 minutes, then to 30% mobile phase A:70% mobile phase B for next 10 min, and to 100% mobile phase A for approximately 6 minutes to re-equilibrate the column. The flow rate was 1.0 mL/min and the column effluent was monitored at 212 nm. Mobile phase A consisted of 98% 25 mmol/L sodium perchlorate, 2% methanol, and 0.05% trifluoroacetic acid; mobile phase B consisted of 49.95% acetonitrile, 49.95% water, and 0.1% trifluoroacetic acid [25]. The limit of detection for neat ziconotide was found to be 10 μg/mL.
b). Estimation of 125I ziconotide
The ziconotide samples labelled with 125I were injected into a HPLC column using the same conditions mentioned above. The fractions eluted from the column were collected at regular intervals to separate free iodine from the intact 125I ziconotide. The fractions collected every minute were subjected to radioactivity measurement using a Beckmann Gamma counter (Pasadena, CA) [26].
2.4. Stability of ziconotide
A). Stability of ziconotide in olfactory tissue homogenate
Bovine olfactory mucosa was homogenized in a glass vial using a high shear tissue homogenizer (Fisher Tissuemizer®) in KRB. To this, a known concentration of ziconotide was incorporated and incubated at 34°C for 6 h. Samples were collected at regular intervals, filtered and analyzed using HPLC.
b). Stability of ziconotide in CSF
CSF was collected from the cisternae magna of the Sprague-Dawley rats. The CSF was incorporated with a known concentration of ziconotide. The drug with CSF was incubated at 37°C for a period of 6 h and samples were collected intermittently at regular intervals. The collected samples were analyzed using HPLC.
2.5. Preparation of chitosan and 125I ziconotide solution
Chitosan solution was prepared by dissolving the required quantity of chitosan in 1% glacial acetic acid solution prepared in KRB (pH 5.5). The cold ziconotide and 125I ziconotide were dissolved separately in KRB. The ziconotide and chitosan solutions were mixed by vortexing. The solutions prepared without incorporation of chitosan served as control.
2.6. Experimental setup for In vitro permeation
In vitro permeation studies were carried out using vertical Franz diffusion apparatus (Logan Instruments, Somerset, NJ). The olfactory mucosa was sandwiched between the donor and receiver compartments such that the dorsal side of the tissue is facing the donor compartment and the ventral side facing the receiver compartment. Ag/AgCl electrodes (procured from Alfa Aesar, Ward Hill, MA) in the form of circular ring with a diameter of 0.5 mm were placed 2 mm away from the tissue in both donor and receiver compartments[22]. A load resistor RL (100 kΩ) was placed in series with olfactory mucosa after filling the donor and receiver compartments with 500 μL and 5 mL of KRB respectively. The voltage drop across the whole circuit (Vo) and across the mucosa (VE) were measured using a waveform generator and multimeter (Agilent Technologies, Santa Clara, CA) [27]. The resistance in kΩ cm2 was measured by applying a small voltage of about 100 mv at 10 Hz [23, 28].
Where RE is the olfactory mucosa resistance and RL is the load resistor in kΩ.
2.7. Effect of chitosan on permeation of ziconotide, in vitro
The effect of different concentrations of chitosan (0.1%, 0.25% and 0.5% w/v) on the permeation of ziconotide was investigated. Drug-chitosan solution was prepared as mentioned in section 2.5. Drug solution with chitosan (100 μL) was placed in the donor compartment and 5 ml KRB was filled in the receiver compartment. Control set of experiments were run without incorporation of chitosan in donor solution. The receiver compartment buffer was sampled at different time points, injected in to the HPLC to separate the free Iodine and the fractions of 125I-ziconotide were quantitated using a Beckmann gamma counter.
2.8. Preparation of in situ gel forming formulations of ziconotide
In situ gels were prepared by cold process. KRB was cooled to 4°C, appropriate amount of KP 407 was added and stirred overnight in the refrigerator. Drug-chitosan solution was prepared by dissolving the required quantity of chitosan in 1% glacial acetic acid solution in polymeric solution followed by addition of unlabeled neat ziconotide and 125I ziconotide. In case of control, unlabeled neat ziconotide and 125I ziconotide were dissolved in KP 407 solution without chitosan.
2.9. Rheological characterization of in-situ gel forming formulations
Rheological studies were carried out using a HR2 rheometer (TA Instruments, New Castle, DE) equipped with 25 mm parallel plate fixture. Temperature was controlled by using a Peltier stage connected to the rheometer. The effect of chitosan addition on gel strength, gelation temperature, and complex viscosity was determined using oscillatory shear rheology. Samples were equilibrated in the rheometer for 5 minutes before starting any experiments. Each sample was examined in triplicates.
a) Gelation temperature
Oscillatory rheology was used to determine the gelation temperature (Tsol/gel) of the in situ forming gels. The samples were subjected to a constant strain (0.5%) and frequency (0.5 Hz). The sample was heated from 10 °C to 37 °C at a rate of 3 °C and the elastic modulus (G′) as a function of temperature was measured. The phase transition from liquid to gel (Tsol/gel) was obtained by determining the intersection point of G′ and G″.
b) Complex viscosity
The complex viscosity as a function of temperature (10 °C to 37 °C) during the in situ gelation process was determined at a constant frequency of 4.75 rad/sec.
c) Gel strength
Elastic modulus (G′) is an indirect measure of the gel strength. The elastic modulus (G′) recorded during in situ gelation process, as described above, was considered.
2.10. In vitro release of ziconotide from the gels
In vitro release studies were carried out using vertical Franz diffusion cells (Logan Instruments Ltd, NJ). A spectra pore membrane with a cut of molecular weight 5kD, was used to carry out the release studies. The donor and receiver compartments were filled with 0.1 ml of the drug polymeric solution and 5 mL of KRB respectively. The temperature of the chamber was regulated at 34 ± 1°C by water circulation. The receiver compartment buffer was sampled at different time points and the amount of ziconotide was measured by HPLC and gamma counter.
2.11. In vitro permeation of ziconotide from gels
In vitro permeation studies were carried out as discussed in section 2.6. The donor compartments were replaced with 0.1 mL of polymeric solutions, allowed to equilibrate at 34 ± 1°C. Samples were collected from the receiver compartment at regular intervals of time and were estimated for ziconotide using HPLC and Gamma counter.
2.12. Extraction of ziconotide from plasma
Blank plasma obtained from untreated rats was spiked with known concentrations of ziconotide and the calibration curve was plotted using the analytical method as mentioned in section 2.3. The rat plasma samples containing ziconotide were extracted initially by vortexing 100 μL of plasma with 0.9 ml of acid acetone (Acetone:Water:HCl; 40:6:1). Proteins were then removed by centrifugation at 4°C for 15 min. The supernatant was injected into the HPLC for separation of free label 125I and intact 125I-ziconotide. Gamma counter was used to estimate the intact 125I-ziconotide [29].
2.13. Pharmacokinetic studies
Pharmacokinetic studies were performed in Sprague-Dawley rats (male, 250–300 g; Harlan Company, Indianapolis, IN, USA). The rats were categorized into six main groups (n=6). Each main group (I–III) was subdivided into seven sub-groups for seven time points with each sub-group having 6 rats. Whereas, the groups IV–VI, were subdivided into nine sub-groups corresponding to nine time points. The rats were anesthetized using ketamine (80 mg/kg) and xylazine (10 mg/kg) (i.p injection).
Ziconotide (100 μg/kg) solution without chitosan, with chitosan, Ziconotide KP 407 without chitosan (ZKP) and Ziconotide KP 407 with Chitosan (ZKP-0.25 Ch) was administered via intranasal route (i.e. administration of drug directly into the posterior segment of the nose using a microsyringe connected with a soft polymer capillary) to rats in groups III–VI respectively. Whereas, rats were administered with ziconotide (100 μg/kg) by i.v route in group II.
Intrathecally catheterized rats (Group I) were obtained from Charles River (Charles River, Wilmington MA). Ziconotide solution (100 μg/kg) was administered to carry out the pharmacokinetic studies of ziconotide in CSF and plasma following intrathecal administration. In groups I–III the time course of drug in the CSF and plasma was investigated by collecting CSF from cisterna magna and blood from retro orbital sinus at 15, 30, 60, 120, 180, 240 and 360 min. Whereas in groups IV–VI the time course of drug in CSF was obtained at 15, 30, 60, 120, 180, 240, 360, 540 and 720 min. Blood samples in groups IV–VI were collected at time points similar to groups I–III. Plasma was collected from blood samples by centrifuging at 1500 rpm for 15 min and ziconotide was extracted as mentioned in section 2.12. Both blood and CSF collected from each group at respective time points were analyzed using HPLC and gamma counter.
2.14. Aspiration of CSF
The rats were anesthetized using ketamine/xylazine and secured on a stereotaxic frame (Harvard Instruments, Holliston, MA, USA). An incision was made on the skin over the occipital bone and the first layer of the muscle was cut using a scalpel blade. A capillary tube was molded to a pointed edge on one end, whereas the other end with circular tip was connected to a 1 mL syringe with the aid of polyethylene tubing and was used for a cisternal puncture. Following i.v, intranasal or intrathecal administration of ziconotide formulations (25 μg in 100 μL) the CSF samples were collected at specified time intervals as mentioned in section 2.13 and quantified using HPLC and gamma counter.
2.15. Data analysis
The statistical analysis was carried out using GraphPad Prism 5 software. The t-test was selected as the test of significance, and p<0.05 was considered statistically significant.
2.16. Pharmacokinetic data analysis
The area under the CSF concentration-time curve (AUC) from the start of drug administration to the time of the last quantifiable concentration in CSF (AUC0-t) was calculated by using the trapezoidal rule. The slope and elimination rate constant (Ke) were calculated from the concentration-time curve on a semi logarithmic scale. The pharmacokinetic parameters between the groups were compared by t-test at a level of significance of p value<0.05.
3. Results and Discussion
3.1. Quantitation of ziconotide
The lowest concentration of neat unlabeled ziconotide that was detected by HPLC with the aid of UV detector at 212 nm was 10 μg/ml at a retention time of ~24.79 min. Ziconotide when radiolabeled with 125I and quantitated using gamma counter the limit of detection was 1.24 × 10−3 ng/mL. To quantitate the amount of ziconotide, the free 125I was separated from each biological sample using HPLC. The chromatogram representing the separation of free 125I and 125I bound to ziconotide is represented in figure 1. Mass spectral analysis was carried out for the fractions collected from 2–4 min using a Matrix-assisted laser desorption/ionization technique (MALDISYNAPT MS/HDMS). According to the mass spectra, peak of free Iodine was found at 126.87 m/z, which resembles the theoretical m/z value of Iodine value obtained from the Mass Lynx software and confirms the separation of the free Iodine (Figure 2).
Figure 1.

Chromatogram representing 125I and 125I ziconotide.
Figure 2.

Negative mode ESI-MS spectra of free Iodine acquired on a Waters synapt HDMS.
3.2. Stability of ziconotide in olfactory tissue homogenates and CSF
A large pool of enzymes is known to be present in the olfactory mucosa. These enzymes act as a protective agents and prevent accidental entry of toxins via nose to brain pathway [30]. The nasal mucosal surface has a variety of proteolytic enzymes which could result in degradation of peptide and protein drugs. Therefore, the stability of ziconotide in the homogenate of the mucosal tissue was investigated for a period of 6 h. Ziconotide was found to be stable for a period of 4 h and there was no significant loss of drug observed.
CSF is a product obtained from the filtrate of plasma and membrane secretions. It is a clear, colorless fluid with a composition of various ions, proteins, enzymes and other substances [31]. However, the ziconotide was found to be stable without any significant degradation in freshly spiked rat CSF which is attributable to unique structure of this peptide.
3.3. Kinetics of ziconotide in CSF - Intrathecal and Intravenous route of administration
Ziconotide was administered into the lumbar region of the spinal cord via intrathecal route. A concentration of 992.2 ± 76.43 ng/ml was attained within a period of 15 min following intrathecal administration. The elimination rate constant of ziconotide in CSF was found to be 1.01 ± 0.34 h−1. As mentioned earlier, the drug remained stable without significant degradation for 6 h in freshly aspirated rat CSF at 37°C. CSF is a dynamic system that generally has a high turnover rate. Generally, the clearance of drugs from the CSF is predominantly due to its high turnover rate. Some of the other drugs used in the treatment of pain management were also reported to have a short elimination half-life predominantly due to the rapid turnover of CSF. Vigdis and coworkers [32] reported that sufentanil when administered intrathecally in humans has a short half-life of 0.6 h and a low mean residence time of 0.9 h in CSF. Similarly, morphine was also found to have shorter half-life of ~1.2 h in CSF when administered intrathecally [33, 34]. Therefore, it is reasonable to speculate that the high fluid turnover rate of CSF (125 ml/h in rats) is the likely reason for rapid disappearance of ziconotide as well from the CSF.
When administered via intravenous route, the Cmax and Tmax of ziconotide in CSF were found to be 37.78 ± 6.8 ng/mL and ~2 h respectively. The AUC0-6 of ziconotide in CSF following i.v administration was 4.98 ± 0.97 min.μg/mL (100 μg/kg dose). The poor bioavailability of drug in CSF following intravenous administration is likely because of the poor ability of ziconotide to cross the blood-CSF barrier which is an interface formed by the epithelial cells of the choroid plexus [35, 36]. From the CSF concentration-time profile data following intravenous administration of drug shown in figure 3, it appears that blood-CSF barrier strongly limits the bioavailability of ziconotide into the CSF.
Figure 3.

Pharmacokinetics of ziconotide in the CSF following intrathecal (●) and intravenous (▲) administration.
3.4. In vitro permeation across the olfactory mucosa
In vitro permeation studies were carried out across the olfactory mucosa using Franz diffusion cells. The amount of ziconotide permeated across the olfactory mucosa from neat ziconotide solution (Control) following 4 h was found to be 0.65 ± 0.05 μg/cm2. Earlier studies have shown that chitosan acts as a permeation enhancer for both small (Cefotaxime) and macromolecules (NGF and BDNF) across the olfactory mucosa. Chitosan used in concentrations of 0.1, 0.25 and 0.5 % w/v enhanced the permeation of ziconotide across bovine olfactory mucosa by ~5, 12 and 14 fold at the end of 4 h respectively, over control (The permeability coefficients for control, 0.1, 0.25 and 0.5 % chitosan were 1.2 ± 0.06 × 10−4 cm/sec, 4.6 ± 0.3 × 10−4 cm/sec, 11.2 ± 0.5 × 10−4 cm/sec, 11.8 ± 0.3 × 10−4 cm/sec respectively) (Figure 4).
Figure 4.

In vitro permeation of ziconotide across the bovine olfactory mucosa in presence of control (◆), 0.1% chitosan (■), 0.25% chitosan (▲) and 0.5% chitosan (●).
Generally, with increase in concentration of the polymer, the viscosity increases reducing the diffusion coefficient and thus the permeation across mucosal membrane. In this case, it appears that the concentration dependent effect of chitosan on the membrane is predominant over the influence of viscosity. However, 0.25% w/v solution was used in vivo to investigate the pharmacokinetics of ziconotide following intranasal administration to minimize the potential discomfort to the animal due to interruption of respiratory pathway at higher polymer concentration.
3.5. Pharmacokinetics of ziconotide in CSF following administration of intranasal solution
The Cmax of ziconotide in the CSF following intranasal administration of ziconotide with chitosan was 7 fold higher than control (neat ziconotide) (33.55 ± 7.39 ng/mL) (Figure 5). But the amount of ziconotide in CSF following i.v administration (37.78 ± 6.8 ng/mL) was comparable to that of intranasal control group.
Figure 5.

Pharmacokinetic profile of ziconotide in CSF following intranasal administration of ziconotide with chitosan (■), without chitosan (◆) and intravenous (▲) administration of ziconotide.
The difference in Cmax attained following intranasal administration of neat ziconotide and ziconotide with chitosan is likely due to the ability of chitosan to open the tight junctions present in the olfactory epithelia. In addition, chitosan also acts as a mucoadhesive agent and would retain the drug for a longer period of time compared to the control.
In case of the group administered by ziconotide with chitosan, the bioavailability (AUC0-6) of ziconotide in CSF following intranasal administration was ~2 fold greater than the bioavailability (AUC0-6) following i.v administration (4.98 ± 0.98 min.μg/mL). Whereas, the bioavailability of neat ziconotide following intranasal administration (1.57 ± 0.44 min. μg/mL) was ~ 3 fold less compared to bioavailability (AUC0-6) following i.v administration (Table 1).
Table 1.
PK parameters of ziconotide in CSF following intranasal and intravenous administration.
| CSF | Intranasal ziconotide | Intranasal ziconotide with Chitosan | Intravenous ziconotide |
|---|---|---|---|
| AUC0-6 (min.μg/mL) | 1.57 ± 0.44 | 10.73 ± 2.92 | 4.98 ± 0.98 |
| Cmax (ng/mL) | 33.55 ± 7.39 | 244.34 ± 48.6 | 37.78 ± 6.8 |
| Tmax (min) | 15 | 15 | 120 |
| KE (h−1) | 0.54 ± 0.08 | 0.69 ± 0.17 | 0.43 ± 0.10 |
The time required to attain maximum concentration (Tmax) in CSF was less upon intranasal administration (15 min) compared to i.v administration (120 min). Similar observations have been reported in case of cephalexin. Sakane and co-workers [19] suggested that following intranasal administration of cephalexin, the drug CSF levels were higher in 15 min than at 30 min. It was reported that the rapid absorption of drug was attributed to the rapid distribution of the drug into the cervical lymph node via the perineural spaces of the olfactory neurons [19]. From figure 5 it is evident that the drug delivered via intranasal route in presence of chitosan attained a concentration of 244.34 ± 48.6 ng/mL (92 nM) in 15 min. The objective is to achieve this order of drug levels in the CSF in humans. Although we cannot extrapolate the rodent data to humans as there exists significant anatomical differences in the nose to brain pathway between the human and rats, the pharmacokinetic data in rats serve as proof of concept to escalate to the next step in humans.
The production or turnover rate of CSF is about 125 μl/h in rats [37]. This might contribute to considerable level of elimination of drug from the CSF. The elimination rate of ziconotide in CSF following intranasal and intravenous administration is 0.6 ± 0.17 h−1 and 0.42 ± 0.10 h−1 respectively. Although, the onset of action via intranasal route is rapid, the elimination of ziconotide from CSF is also considerably high because of its high turnover rate as discussed earlier in section 3.3 [8].
3.6. Rheological evaluation of ziconotide gels
The gelation of KP 407 occurs as a result of change in polymer desolvation and subsequent micellization. As the temperature was increased, the negative coefficient of the solubility effects micellization, expels the water from the core of micelles, leads to conformational changes in methyl group (PPO) orientation and results in gel formation [38–41]. Chen et al reported that KP 407 in concentrations that forms gel at physiological temperature, possess higher viscosity than normal nasal fluid, fast phase switching capability and withstand the antidilution effect [24]. Preliminary studies revealed that a minimum of 18% w/w KP 407 is required to obtain a gel at a temperature lower than 37 °C. As the concentration of the KP 407 increases, the gelation temperature decreases. The temperature of the nasal cavity is ~34 °C and a polymeric solution that forms gel close to the nasal temperature would be ideal for controlled release drug delivery applications. Further studies were carried out to determine a poloxamer concentration that gels in the range of 25 °C to 37 °C. The gelation temperatures of 18, 20 and 22% KP 407 were found to be ~31, 28 and 22 °C, respectively (Table 2 and Figure 6).
Table 2.
Gelation temperature, storage modulus and complex viscosity of 18, 20 and 22% KP 407.
| Poloxamer Concentration (%w/w) | Gelation Temperature (°C) | Storage Modulus (Pa) @ 34 °C | Complex Viscosity (Pa.s) @ 34 °C |
|---|---|---|---|
| 18% KP 407 | 31 | 5311 ± 25 | 1153 ± 41 |
| 20% KP 407 | 28 | 9366 ± 61 | 1976 ± 74 |
| 22% KP 407 | 22 | 14654 ± 110 | 3085 ± 94 |
| 20% KP 407-0.25% Chitosan | 34 | 7578 ± 94 | 1605.46 ± 38 |
Figure 6.

Determination of gelation temperature, storage and loss modulus of 18% (■,□), 20% (●,○) and 22% (▲,△) KP 407. Filled symbols represent storage modulus and empty symbols represent loss modulus.
The complex viscosity of 18, 20 and 22% KP 407 at 34 °C was found to be 1153 ± 41, 1976 ± 74 and 3085 ± 94 Pa.s, respectively.
Gel strength is an indirect measurement of storage modulus (G′). The gel strength of 18, 20 and 22% KP 407 at an angular frequency of 0.5 Hz and 34 °C were found to be 5311 ± 25, 9366 ± 61 and 14654 ± 110 Pa respectively (Figure 6). The gel strength increased with increase in the concentration of KP 407.
KP 407 with 20% w/w was considered for further studies as it has the gelation temperature close to the physiological temperature, considerable gel strength and a viscosity higher than nasal mucosa.
3.7 Effect of chitosan on gelation temperature, gel strength and viscosity
The effect of chitosan on gelation temperature, gel strength and viscosity were also examined. Chitosan when incorporated in a concentration of 0.25 % w/v in the KP407 polymeric solution, shifted the gelation temperature of 20% KP 407 from 30 °C to 34 °C. Addition of chitosan hinders gel formation and as a result gelation temperature increases to a higher temperature [42]. A similar change in temperature on addition of chitosan was also reported by Zaki et al [43]. In addition to increase in gelation temperature, the gel strength and the complex viscosity of the gel decreased due to incorporation of chitosan (Table 2). Although the viscosity of the formulation was lower than that observed for formulation without chitosan, it was still higher than the nasal mucosal viscosity which ranges between 0.02–500 Pa.s at a frequency of 0.01–100 1/sec [44, 45].
3.8. In vitro release and permeation studies of ziconotide gels
The percentage of ziconotide released from ZKP and ZKP-0.25Ch in 6 h was found to be 85.01 ± 0.86 and 69.22 ± 1.04, respectively.
Chitosan used in concentration of 0.25 % w/v enhanced the permeation of ziconotide across the bovine olfactory mucosa by ~5 fold at the end of 6 h compared to ziconotide alone (1.06 ± 0.09 μg/cm2). Therefore, ZKP and ZKP-0.25Ch were used to carry out the in vivo studies following intranasal administration.
3.9. Intranasal ziconotide gels
The Cmax of ziconotide in the CSF following intranasal administration of ZKP-0.25Ch was ~ 2 fold higher than ZKP (24.92 ± 2.91 ng/mL). Whereas, the amount of ziconotide in CSF following intranasal administration of ziconotide solution with chitosan was ~5 fold higher than ZKP-0.25Ch (50.21 ± 3.48 ng/mL) (Figure 7). The difference in Cmax attained following administration of solution and gel formulation could be attributed to enhanced viscosity of gel formulation compared to solution.
Figure 7.

Pharmacokinetic profile of ziconotide in CSF following intranasal administration of ziconotide KP 407 without chitosan (▲), with chitosan (◆) and ziconotide solution with chitosan (■).
The elimination rate of ziconotide in CSF following intranasal gel was reduced by ~7.5 fold compared to the elimination rate attained by intranasal solution (0.6 ± 0.17 h−1) as observed in sustained release dosage forms (Figure 7). It appears that the mucoadhesive formulation approach using in situ gel forming polymer is an excellent approach to retain drug in CSF longer by replenishing the CSF.
3.10 Pharmacokinetics of ziconotide in plasma
The AUC0-6 levels in the plasma following intrathecal administration of ziconotide solution, intranasal administration of ziconotide solution with and without chitosan were 53.15 ± 8.25 μg.min/mL, 54.84 ± 5.11 μg.min/mL and 27.88 ± 9.18 μg.min/mL, respectively. These levels were almost ~5–10 fold less compared to that of i.v administration (275.07 ± 23.44 μg.min/mL) (Figure 8). This data suggests that low systemic bioavailability following intranasal administration is likely to result in less systemic side effects compared to parenteral administration.
Figure 8.

Pharmacokinetic profile of ziconotide in plasma following intranasal administration of ziconotide KP 407 without chitosan (X), with chitosan (+), ziconotide solution with chitosan (■) without chitosan (◆), i.v (▲) and intrathecal (●) administration of ziconotide solution.
The AUC0-6 levels of ziconotide in plasma following intranasal administration of ZKP-0.25C and ZKP was found to be 37.89 ± 4.86 μg.min/mL and 31.77 ± 3.22 μg.min/mL (Figure 8). These levels are almost 7 and 8 fold less compared to that of i.v administration (275.07 ± 23.44 μg.min/mL).
This data suggests that low systemic bioavailability following intranasal administration is likely to result in less systemic side effects compared to parenteral administration.
4. Conclusion
These results indicate that intranasal route could be utilized as one of the potential routes for delivery of ziconotide to the CSF compared to intrathecal and intravenous route. Ziconotide solution when administered via intranasal route attained a faster Tmax compared to intravenous administration. Co-administration of ziconotide solution with chitosan via intranasal route enhanced the Cmax in CSF by ~5 folds when compared to its control, but had a faster elimination rate from CSF irrespective of the formulation.
Ziconotide when delivered using poloxamer as a vehicle not only attains therapeutic concentration rapidly but also prolonged the release of ziconotide and maintained therapeutic levels in CSF till 9 h compared to solution. Prolonged maintenance of ziconotide would potentially reduce the frequency of administration. The elimination rate of ziconotide from CSF was reduced by ~7.5 fold using gel formulations compared to intranasal solutions. Despite reducing the rate of elimination, intranasal route is also preferred over i.v and intrathecal route because of its noninvasiveness, opportunity for frequent administration, and potentially minimal side effects. The intranasal administration of ziconotide was also found to result in relatively low systemic bioavailability. These results suggest that intranasal delivery of ziconotide could be developed as a potential treatment approach for chronic pain.
Acknowledgments
This study was supported by a grant from National Institute of Neurological Disorders and Stroke (NINDS) Grant NS074362 and Institutional Development Award (IDeA) Grant Number P20GM104932 from the National Institute of General Medical Sciences (NIGMS). Both NINDS and NIGMS are components of the National Institutes of Health (NIH). The contents are solely the responsibility of the authors and do not necessarily represent the official view of NIGMS, NINDS or NIH. The authors would like to acknowledge supports from National Science Foundation (DMR-1352572, EPS-0903787) and thank Dr. Quentin Smith (Texas Tech University Health Science center, Amarillo, TX) for teaching the CSF collection technique. The authors would also like to thank Scott Rone health and safety officer at University of Mississippi for helping with all the required procedures for handling 125I.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Breivik H, Collett B, Ventafridda V, Cohen R, Gallacher D. Survey of chronic pain in Europe: prevalence, impact on daily life, and treatment. European journal of pain. 2006;10:287–287. doi: 10.1016/j.ejpain.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 2.Scholz J, Woolf CJ. Can we conquer pain? Nature neuroscience. 2002;5:1062–1067. doi: 10.1038/nn942. [DOI] [PubMed] [Google Scholar]
- 3.Staats PS, Yearwood T, Charapata SG, Presley RW, Wallace MS, Byas-Smith M, Fisher R, Bryce DA, Mangieri EA, Luther RR. Intrathecal ziconotide in the treatment of refractory pain in patients with cancer or AIDS: a randomized controlled trial. Jama. 2004;291:63–70. doi: 10.1001/jama.291.1.63. [DOI] [PubMed] [Google Scholar]
- 4.Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M. Synthetic therapeutic peptides: science and market. Drug discovery today. 2010;15:40–56. doi: 10.1016/j.drudis.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 5.Diao L, Meibohm B. Pharmacokinetics and pharmacokinetic–pharmacodynamic correlations of therapeutic peptides. Clinical pharmacokinetics. 2013;52:855–868. doi: 10.1007/s40262-013-0079-0. [DOI] [PubMed] [Google Scholar]
- 6.Thapa P, Espiritu MJ, Cabalteja CC, Bingham J-P. Conotoxins and their regulatory considerations. Regulatory Toxicology and Pharmacology. 2014;70:197–202. doi: 10.1016/j.yrtph.2014.06.027. [DOI] [PubMed] [Google Scholar]
- 7.W.H. Organization. Cancer pain relief: with a guide to opioid availability. World Health Organization; 1996. [Google Scholar]
- 8.Malmberg AB, Yaksh TL. Voltage-sensitive calcium channels in spinal nociceptive processing: blockade of N-and P-type channels inhibits formalin-induced nociception. The Journal of neuroscience. 1994;14:4882–4890. doi: 10.1523/JNEUROSCI.14-08-04882.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chaplan SR, Pogrel JW, Yaksh TL. Role of voltage-dependent calcium channel subtypes in experimental tactile allodynia. Journal of Pharmacology and Experimental Therapeutics. 1994;269:1117–1123. [PubMed] [Google Scholar]
- 10.Atanassoff PG, Hartmannsgruber MW, Thrasher J, Wermeling D, Longton W, Gaeta R, Singh T, Mayo M, McGuire D, Luther RR. Ziconotide, a new N-type calcium channel blocker, administered intrathecally for acute postoperative pain. Regional anesthesia and pain medicine. 2000;25:274–278. doi: 10.1016/s1098-7339(00)90010-5. [DOI] [PubMed] [Google Scholar]
- 11.Miljanich G. Ziconotide: neuronal calcium channel blocker for treating severe chronic pain. Current medicinal chemistry. 2004;11:3029–3040. doi: 10.2174/0929867043363884. [DOI] [PubMed] [Google Scholar]
- 12.Bowersox SS, Gadbois T, Singh T, Pettus M, Wang Y-X, Luther RR. Selective N-type neuronal voltage-sensitive calcium channel blocker, SNX-111, produces spinal antinociception in rat models of acute, persistent and neuropathic pain. Journal of Pharmacology and Experimental Therapeutics. 1996;279:1243–1249. [PubMed] [Google Scholar]
- 13.McGivern JG. Ziconotide: a review of its pharmacology and use in the treatment of pain. Neuropsychiatric disease and treatment. 2007;3:69. doi: 10.2147/nedt.2007.3.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.V.P.B.M.S.H.G.a.t.M.A. Panel. Ziconotide for Intrathecal Infusion (Prialt®) US Department of Veteran Affairs; 2006. [Google Scholar]
- 15.J Pharmaceuticals. Prialt Prescribing Information. 2013. [Google Scholar]
- 16.Schmidtko A, Lötsch J, Freynhagen R, Geisslinger G. Ziconotide for treatment of severe chronic pain. The Lancet. 2010;375:1569–1577. doi: 10.1016/S0140-6736(10)60354-6. [DOI] [PubMed] [Google Scholar]
- 17.Anand Kumar TC, David GF, Sankaranarayanan A, Puri V, Sundram KR. Pharmacokinetics of progesterone after its administration to ovariectomized rhesus monkeys by injection, infusion, or nasal spraying. Proceedings of the National Academy of Sciences. 1982;79:4185–4189. doi: 10.1073/pnas.79.13.4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumbale R, Frey W, Wilson S, Rahman Y. GM1 delivery to the CSF via the olfactory pathway. Drug Delivery. 1999;6:23–30. [Google Scholar]
- 19.Sakane T, Akizuki M, Yoshida M, Yamashita S, Nadai T, Hashida M, Sezaki H. Transport of cephalexin to the cerebrospinal fluid directly from the nasal cavity. Journal of Pharmacy and Pharmacology. 1991;43:449–451. doi: 10.1111/j.2042-7158.1991.tb03510.x. [DOI] [PubMed] [Google Scholar]
- 20.Chou K-J, Donovan MD. Lidocaine distribution into the CNS following nasal and arterial delivery: a comparison of local sampling and microdialysis techniques. International journal of pharmaceutics. 1998;171:53–61. [Google Scholar]
- 21.Hanson LR, Frey WH., II Strategies for intranasal delivery of therapeutics for the prevention and treatment of neuroAIDS. Journal of Neuroimmune Pharmacology. 2007;2:81–86. doi: 10.1007/s11481-006-9039-x. [DOI] [PubMed] [Google Scholar]
- 22.Manda P, Hargett JK, Kiran Vaka SR, Repka MA, Narasimha Murthy S. Delivery of cefotaxime to the brain via intranasal administration. Drug development and industrial pharmacy. 2011;37:1306–1310. doi: 10.3109/03639045.2011.571696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vaka SRK, Sammeta S, Day LB, Murthy SN. Delivery of nerve growth factor to brain via intranasal administration and enhancement of brain uptake. Journal of pharmaceutical sciences. 2009;98:3640–3646. doi: 10.1002/jps.21674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen E, Chen J, Cao S-l, Zhang Q-z, Jiang X-g. Preparation of nasal temperature-sensitive in situ gel of Radix Bupleuri and evaluation of the febrile response mechanism. Drug development and industrial pharmacy. 2010;36:490–496. doi: 10.3109/03639040903264371. [DOI] [PubMed] [Google Scholar]
- 25.Shields D, Montenegro R, Ragusa M. Chemical Stability of Admixtures Combining Ziconotide with Morphine or Hydromorphone During Simulated Intrathecal Administration. Neuromodulation: Technology at the Neural Interface. 2005;8:257–263. doi: 10.1111/j.1525-1403.2005.00034.x. [DOI] [PubMed] [Google Scholar]
- 26.Shields DE, Aclan J, Szatkowski A. Chemical stability of admixtures combining ziconotide with fentanyl or sufentanil during simulated intrathecal administration. Int J Pharm Compound. 2008;12:463–466. [PubMed] [Google Scholar]
- 27.Manda P, Sammeta SM, Repka MA, Murthy SN. Iontophoresis across the proximal nail fold to target drugs to the nail matrix. Journal of pharmaceutical sciences. 2012;101:2392–2397. doi: 10.1002/jps.23139. [DOI] [PubMed] [Google Scholar]
- 28.Manda P, Angamuthu M, Hiremath SR, Raman V, Murthy SN. Iontophoretic Drug Delivery for the Treatment of Scars. Journal of Pharmaceutical Sciences. 2014;103:1638–1642. doi: 10.1002/jps.23946. [DOI] [PubMed] [Google Scholar]
- 29.Newcomb R, Abbruscato TJ, Singh T, Nadasdi L, Davis TP, Miljanich G. Bioavailability of Ziconotide in brain: influx from blood, stability, and diffusion. Peptides. 2000;21:491–501. doi: 10.1016/s0196-9781(00)00175-3. [DOI] [PubMed] [Google Scholar]
- 30.Barnes L. Surgical pathology of the head and neck. CRC Press; 2000. [Google Scholar]
- 31.Di Terlizzi R, Platt S. The function, composition and analysis of cerebrospinal fluid in companion animals: Part I – Function and composition. The Veterinary Journal. 2006;172:422–431. doi: 10.1016/j.tvjl.2005.07.021. [DOI] [PubMed] [Google Scholar]
- 32.Hansdottir V, Hedner T, Woestenborghs R, Nordberg G. The CSF and plasma pharmacokinetics of sufentanil after intrathecal administration. Anesthesiology. 1991;74:264–269. doi: 10.1097/00000542-199102000-00012. [DOI] [PubMed] [Google Scholar]
- 33.Ionescu T, Drost R, Roelofs J, Winckers E, Taverne R, Van Maris A, van Rossum J. The pharmacokinetics of intradural morphine in major abdominal surgery. Clinical pharmacokinetics. 1988;14:178–186. doi: 10.2165/00003088-198814030-00006. [DOI] [PubMed] [Google Scholar]
- 34.Kotob H, Hand C, Moore R, Evans P, Wells J, Rubin A, McQuay H. Intrathecal morphine and heroin in humans: six-hour drug levels in spinal fluid and plasma. Anesthesia & Analgesia. 1986;65:718–722. [PubMed] [Google Scholar]
- 35.Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood–brain barrier. Neurobiology of disease. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 36.Brodie BB, Kurz H, Schanker LS. The importance of dissociation constant and lipid-solubility in influencing the passage of drugs into the cerebrospinal fluid. Journal of Pharmacology and Experimental Therapeutics. 1960;130:20–25. [PubMed] [Google Scholar]
- 37.Pardridge WM. Drug transport across the blood–brain barrier. Journal of Cerebral Blood Flow & Metabolism. 2012;32:1959–1972. doi: 10.1038/jcbfm.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Attwood D, Collett J, Tait C. The micellar properties of the poly (oxyethylene)-poly (oxypropylene) copolymer Pluronic F127 in water and electrolyte solution. International journal of pharmaceutics. 1985;26:25–33. [Google Scholar]
- 39.Gaisford S, Beezer AE, Mitchell JC, Bell PC, Fakorede F, Finnie JK, Williams SJ. Temperature induced aggregation in aqueous solution of a series of PEO–PPO–PEO copolymers. International journal of pharmaceutics. 1998;174:39–46. [Google Scholar]
- 40.Joshi R, Arora V, Desjardins JP, Robinson D, Himmelstein KJ, Iversen PL. In vivo properties of an in situ forming gel for parenteral delivery of macromolecular drugs. Pharm Res. 1998;15:1189–1195. doi: 10.1023/a:1011979505697. [DOI] [PubMed] [Google Scholar]
- 41.Rassing J, Attwood D. Ultrasonic velocity and light-scattering studies on the polyoxyethylene—polyoxypropylene copolymer Pluronic F127 in aqueous solution. International Journal of Pharmaceutics. 1982;13:47–55. [Google Scholar]
- 42.Choi H-G, Lee M-K, Kim M-H, Kim C-K. Effect of additives on the physicochemical properties of liquid suppository bases. International journal of pharmaceutics. 1999;190:13–19. doi: 10.1016/s0378-5173(99)00225-2. [DOI] [PubMed] [Google Scholar]
- 43.Zaki NM, Awad GA, Mortada ND, Abd ElHady SS. Enhanced bioavailability of metoclopramide HCl by intranasal administration of a mucoadhesivein situ gel with modulated rheological and mucociliary transport properties. European journal of pharmaceutical sciences. 2007;32:296–307. doi: 10.1016/j.ejps.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 44.Gwaltney JM, Hendley JO, Phillips CD, Bass CR, Mygind N, Winther B. Nose Blowing Propels Nasal Fluid into the Paranasal Sinuses. Clinical Infectious Diseases. 2000;30:387–391. doi: 10.1086/313661. [DOI] [PubMed] [Google Scholar]
- 45.Vasquez ES, Bowser J, Swiderski C, Walters KB, Kundu S. Rheological characterization of mammalian lung mucus. RSC Advances. 2014;4:34780–34783. [Google Scholar]