

Abstract
Hypothalamic fatty acid metabolism is involved in central nervous system controls of feeding and energy balance. Malonyl-CoA, an intermediate of fatty acid biosynthesis, is emerging as a significant player in these processes. Notably, hypothalamic malonyl-CoA has been implicated in leptin's feeding effect. Leptin treatment increases malonyl-CoA level in the hypothalamic arcuate nucleus (Arc), and this increase is required for leptin-induced decrease in food intake. However, the intracellular downstream mediators of malonyl-CoA's feeding effect have not been identified. A primary biochemical action of malonyl-CoA is the inhibition of the acyltransferase activity of carnitine palmitoyltransferase-1 (CPT-1). In the hypothalamus, the predominant isoform of CPT-1 that possesses the acyltransferase activity is CPT-1 liver type (CPT-1a). To address the role of CPT-1a in malonyl-CoA's anorectic action, we used a recombinant adenovirus expressing a mutant CPT-1a that is insensitive to malonyl-CoA inhibition. We show that Arc overexpression of the mutant CPT-1a blocked the malonyl-CoA-mediated inhibition of CPT-1 activity. However, the overexpression of this mutant did not affect the anorectic actions of leptin or central cerulenin for which an increase in Arc malonyl-CoA level is also required. Thus, CPT-1a does not appear to be involved in the malonyl-CoA's anorectic actions induced by leptin. Furthermore, long-chain fatty acyl-CoAs, substrates of CPT-1a, dissociate from malonyl-CoA's actions in the Arc under different feeding states. Together, our results suggest that Arc intracellular mechanisms of malonyl-CoA's anorectic actions induced by leptin are independent of CPT-1a. The data suggest that target(s), rather than CPT-1a, mediates malonyl-CoA action on feeding.
Keywords: carnitine palmitoyltransferase, food intake, hypothalamus
obesity is a major health problem and a major cause of insulin-resistance and diabetes. An imbalance between energy intake and energy expenditure can lead to overweight and contribute to the development of obesity and the metabolic syndrome. The hypothalamus plays a critical role in the central nervous system (CNS) control of feeding and energy balance (21, 28). A large body of evidence now shows that fatty acid metabolism participates in this action of the hypothalamus (1, 3, 5–7, 9, 11, 12, 15, 17, 18, 20, 23, 24). In this regard, malonyl-CoA, an intermediate in fatty acid biosynthesis, is emerging as a significant player in the hypothalamic control of feeding and body energy balance (5–7, 9, 11, 12, 15, 18). Recent data have linked malonyl-CoA signaling action to the hypothalamic intracellular pathways of leptin in the central regulation of energy balance (6, 9). In the hypothalamus, leptin treatment increases malonyl-CoA level via inhibiting AMP-activated protein kinase (AMPK) and activating acetyl-CoA carboxylase (ACC) (2, 6). Notably, the increase of hypothalamic malonyl-CoA induced by leptin takes place specifically in the arcuate nucleus (Arc) (6), a critical site in mediating leptin's central actions on feeding and energy balance (28). Despite these findings, the intracellular mechanisms by which malonyl-CoA impacts feeding remain unclear. It is known that malonyl-CoA inhibits the acyltransferase activity of carnitine palmitoyltransferase-1 (CPT-1) that converts long-chain fatty acyl-CoA (LCFA-CoA) to long-chain acylcarnitine (9, 23). CPT-1 has liver- and muscle-isoforms, with the hypothalamus mainly expressing the liver isoform (CPT-1a) (23). Pharmacological studies have demonstrated that intracerebroventricular administration of compound ST-1326, a specific CPT-1a inhibitor, inhibits Arc CPT-1a activity, increases cellular LCFA-CoA levels, and reduces food intake (23). Accumulation of hypothalamic LCFA-CoAs has been suggested to have anorectic effects as intracerebroventricular oleic acid (which can form LCFA-CoAs in cells) was shown to reduce food intake (24). Given that malonyl-CoA is a physiological inhibitor of CPT-1, inhibition of Arc CPT-1a and the ensuing increase of LCFA-CoAs have been proposed to mediate malonyl-CoA's anorectic signaling actions. However, a growing body of evidence now strongly challenges this hypothesis (16). For example, we previously demonstrated that exogenous leptin upregulates the malonyl-CoA level without affecting the level of LCFA-CoAs in the Arc (6). The result thus casts doubt on the role of CPT-1a as a mediator of malonyl-CoA's action in leptin feeding pathways. To clarify the role of CPT-1a in the Arc, we used a recombinant adenovirus expressing a mutant CPT-1a that is insensitive to malonyl-CoA inhibition (10). Using this mutant, we examined the feeding responses of the animals with a disruption of malonyl-CoA-mediated inhibition on CPT-1a acyltransferase activity.
MATERIALS AND METHODS
Animal preparations for feeding experiments.
Animal experiments were performed in accordance with the guidelines of the Canadian Council for Animal Care, and were approved by the University of Alberta Animal Policy and Welfare Committee. Male Sprague-Dawley rats (225–300 g) were purchased from Charles River Laboratories. The rats were housed in a controlled (12:12-h light-dark) environment, and were allowed ad libitum access to standard laboratory chow and water, unless otherwise noted. Before the feeding experiments were started, the rats were handled daily and subjected to mock injections to minimize the stress from the experimental manipulation. In the leptin study, leptin or vehicle was administered at 1 h before the dark onset. Food intakes at 3 h and 23 h (overnight) after the dark onset, were monitored. Overnight body weight changes (24 h after the injection) were also monitored. In the cerulenin study, at 5 h before the dark onset, food was removed. Then, cerulenin or vehicle was administered, and food was not provided until the dark onset. Food intakes were monitored at 3 h and 19 h (overnight) after dark onset. Overnight body weight changes (24 h after the injection) were also monitored.
Brain sample preparations.
At the designated time points, rats were euthanized by decapitation. Brains were rapidly dissected (within 1 min) and coronal brain sections were prepared using a cryostat or a brain matrix (Roboz Surgical Instrument, Gaithersburg, MD). Individual hypothalamic nuclei including Arc, ventromedial nucleus (VMN), lateral hypothalamic area (LHA), and paraventricular nucleus (PVN) were dissected according to the established protocol (6). The accuracy of the dissection was verified by comparing the characteristic neuropeptide mRNA levels as detailed previously (6).
Recombinant adenoviruses.
The adenoviruses were delivered into the Arc (1 × 107 pfu/μl; 0.4 μl per side) by bilateral stereotaxic injection (6). The coordinates were anterior-posterior: −2.8 mm; dorsal-ventral: −9.5 mm; and medial-lateral: ± 0.4 mm. The accuracy of the injections was confirmed by histological analysis as described previously (6). The adenovirus encoding malonyl-CoA decarboxylase (MCD) contains the full-length human MCD (hMCD) coding sequence (27). The adenovirus encoding the wild-type CPT-1a contains the nucleotide sequence (58-2700) including the entire coding region of rat CPT-1a, and the same sequence was used to generate the mutant CPT-1a with the 593-methionine residual mutated to serine residual (22). The feeding experiments with leptin and cerulenin were conducted in the second week following the initial delivery of the adenoviruses. Significantly high levels of protein expressions or enzyme activities were reliably detected after 1 wk and these high levels last until at least 14 days (2 wk) following the delivery of the viruses.
Cannulation surgery and intracerebroventricular injection.
Cannulas were implanted into either the lateral or third ventricle based on the established protocol (6). The accuracy of placement was confirmed by angiotensin-2 drinking test or histological analysis as previously described (6). After surgery, daily food intake and body weight were monitored. After body weights returned to the levels before surgery and the rats were fully habituated to the experimental manipulations, bolus injections of the chemicals (leptin and cerulenin) were administered intracerebroventricularly.
Chemicals.
Leptin was obtained from A. F. Parlow (National Hormone and Pituitary Program, National Institute of Diabetes and Digestive and Kidney Diseases). For intracerebroventricular injection, a dose of 15 μg of leptin dissolved in PBS was chosen based on our previous studies (6). Cerulenin was obtained from Sigma (St. Louis, MO) and 125 μg in 25% DMSO/75% PBS (vehicle) was used in intracerebroventricular injection as previously described (1).
Quantifications of malonyl-CoA, LCFA-CoAs, and long-chain acylcarnitines.
The CoA recycling assay was performed to measure the malonyl-CoA level, and HPLC was used to measure the levels of long-chain acyl-CoAs (consisting of palmitoyl-CoA, oleoyl-CoA, and stearoyl-CoA) as detailed elsewhere (6). To measure the levels of the long-chain acylcarnitines (LC-ACs), brain tissues were extracted with acetonitrile and 2-propanol (6). The extracts were dried under streams of nitrogen and were reconstituted in 300 μl acetonitrile and n-butanol (1:1). The samples were then filtered and loaded into HPLC coupled with mass spectrometer (13). The levels of LC-ACs (consisting of palmitoylcarnitine, oleoylcarnitine, and stearoylcarnitine) were quantitated as detailed previously (13).
MCD activity assay.
Adenovirus encoding hMCD or green fluorescent protein (null) was stereotaxically delivered into the Arc. Individual hypothalamic nuclei (Arc, VMN, LHA, and PVN) were dissected from coronal brain sections. The MCD activity assay was performed based on established protocol (6, 27).
Carnitine palmitoyltransferase-1 (CPT-1) activity assay.
The brain was removed from the skull within 40 s and was immediately sectioned using the brain matrix. The mediobasal hypothalamic area (MBH), LHA, and PVN were quickly dissected on ice, and the tissues were immediately homogenized in the cold lysis buffer (0.25 M sucrose, 5 mM Tris·HCl, and 1 mM EGTA, pH 7.4). The crude homogenate was centrifuged at 800 g for 10 min at 4°C. The resulting pellet was washed by resuspension in two volumes of the lysis buffer and was then centrifuged at 800 g. This step was repeated twice to maximize the yield of the mitochondrial fraction. The combined supernatant was centrifuged at 6,000 g for 15 min at 4°C. The resulting pellet (crude mitochondrial fraction) was gently resuspended in the lysis buffer and was used in the activity assay using a radiometric method (20).
Antibodies and Western blot analysis.
The CPT-1a antibody (Ab) was generated as described elsewhere (29). Actin (Santa Cruz Biotechnology, Santa Cruz, CA) was used as the loading control in the Western blot analyses. The procedures of protein electrophoresis, transfer, and Ab detection were performed based on standard Western blot analysis protocol (Invitrogen, California). Densitometry was performed using Scion Image software (Scion, Frederick, MD).
Statistical analysis.
Data are reported as means ± SE. Data consisting of two groups were analyzed by Student's t-test. Data consisting of three groups were analyzed by one-way ANOVA. These one-way ANOVAs, when they yielded significant overall effects, were further analyzed by the Newman-Keuls multiple comparison test for group comparisons. Data consisting of four or six groups were analyzed by two-way ANOVA. These two-way ANOVAs, when they yielded significant overall effects, were further analyzed by Bonferroni post tests for group comparisons. For all tests, P < 0.05 indicated significance.
RESULTS
M593S CPT-1a mutant is insensitive to malonyl-CoA inhibition.
In this study, we used a M593S CPT-1a mutant to address the role of CPT-1a in malonyl-CoA-mediated anorectic actions. The M593S mutation results in an impaired interaction between malonyl-CoA and the malonyl-CoA binding site in CPT-1a (22). We first verified malonyl-CoA insensitivity of this mutant using yeast cells that do not possess endogenous CPT-1 acyltransferase activity (22). We transfected yeast cells with the vector that expresses this mutant (CPT-1a mt) or the wild-type CPT-1a (CPT-1a wt), and then measured CPT-1 acyltransferase activity using the extracts from these cells. We found that malonyl-CoA inhibitory effect on the mutant was nearly abolished (Fig. 1A). Next, we evaluated CPT-1 acyltransferase activities in the hypothalamus. The adenovirus encoding the CPT-1a mt, the CPT-1a wt, or the null virus was delivered bilaterally into the Arc. Two weeks following delivering the viruses, rats were euthanized, and CPT-1a protein levels were quantified in individual hypothalamic nuclei (Arc, VMN, LHA, and PVN). Compared with the rats injected with the null adenovirus, CPT-1a (wt or mt) adenoviral infections induced increases in CPT-1a protein levels in the Arc, while the CPT-1a protein levels were not altered in the VMN, LHA, or PVN (Fig. 1B). Concomitant with the increased protein levels, CPT-1 acyltransferase activities were also increased selectively in the MBH encompassing the Arc (Fig. 1C). Together, these data demonstrate Arc-specific overexpressions of CPT-1a as well as activations of CPT-1 following the stereotaxic Arc delivery of the viruses. We then evaluated the response of CPT-1a to exogenous malonyl-CoA. We prepared crude mitochondrial extract from the MBH region of the animals with Arc overexpressing the CPT-1a wt or the CPT-1a mt. Exogenous malonyl-CoA (50 μM) (20) was then added to the extracts, and the CPT-1 activity assay was conducted. As expected, we observed that the CPT-1a mt was resistant to the inhibitory effect of malonyl-CoA (Fig. 1D).
Fig. 1.
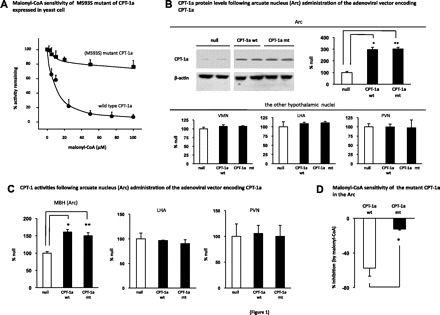
M593S mutant of carnitine palmitoyltransferase liver type (CPT-1a) is insensitive to malonyl-CoA inhibition. A: yeast extract (10 μg of protein) of wild-type CPT-1a (CPT-1a wt) or M593S mutant CPT-1a (CPT-1a mt) was incubated with increasing concentrations of malonyl-CoA, and the CPT-1 activities in the extract were measured (n = 4–5). B: adenovirus (Ade) expressing the control (null, n = 6), CPT-1a wt (n = 6), or M593S CPT-1a mt (n = 6) was administered into the arcuate nucleus (Arc). Two weeks following the administration of the viruses, the rats were euthanized. The individual hypothalamic nuclei tissues [Arc, ventromedial nucleus (VMN), lateral hypothalmic area (LHA), and paraventricular nucleus (PVN)] were dissected from the brain sections, and the CPT-1a protein levels were examined by Western blot analysis. Two representative blots from the Arc of each group are shown, and the ratios of the band intensity of CPT-1a to that of β-actin were quantitated (n = 6; *, **vs. null, P < 0.05). C: 2 wk following Arc administration of the Ades encoding the null, CPT-1a wt, and CPT-1 mt (n = 6), the CPT acyltransferase activities in the mediobasal hypothalmic area (MBH) encompassing the entire Arc and some VMN, LHA, and the PVN were measured. The CPT-1a activities of individual VMN tissues were not measured (*, **vs. null, P < 0.05). D: 2 wk following the delivery of the viruses (encoding CPT-1a wt and CPT-1a mt), the rats were euthanized. The mediobasal hypothalamus (MBH) area containing the Arc was dissected, and the crude mitochondrial fraction was prepared. Exogenous malonyl-CoA (50 μM) was added to the mitochondrial preparation, and CPT-1 activity assay was conducted (n = 8). %Activity inhibition by malonyl-CoA (compared with the assay without the addition of exogenous malonyl-CoA) are presented (*vs. CPT-1a wt, P < 0.05).
Increase in arc malonyl-CoA is required for leptin's anorectic actions.
Before addressing the role of CPT-1a in leptin's malonyl-CoA signaling pathway, we confirmed the importance of malonyl-CoA in the central control of feeding and in leptin's anorectic actions. The adenovirus encoding MCD (Ade-MCD) (27), which lowers malonyl-CoA level (9), was administered into the Arc of rats. Consistent with the previous finding (9), Arc delivery of the Ade-MCD increased daily food intakes and body weight gains, compared with the rats treated with the null virus (Fig. 2A). Delivery of the Ade-MCD induced an increase in MCD activity in the Arc (Fig. 2B), while the MCD activity was not altered in the VMN (Fig. 2B) or in the LHA and PVN (LHA: MCD, 87 ± 4.7% vs. null, 100 ± 13%; PVN: MCD, 105 ± 1% vs. null, 100 ± 10%). In addition, following the delivery of the Ade-MCD, the malonyl-CoA level in the Arc was reduced (Fig. 2C). We then administered (intracerebroventricularly) leptin to the rats with Arc-specific activation of MCD. As we demonstrated in the previous study (6), leptin treatment increased the malonyl-CoA level in the Arc (leptin: 220%, PBS: 100%; P < 0.05). We further showed that the MCD overexpression attenuated the level of the increase in malonyl-CoA and antagonized the anorectic actions by leptin treatment (Fig. 2D). These results confirm that the increase in the Arc malonyl-CoA level is a significant contributor to leptin's anorectic effects.
Fig. 2.
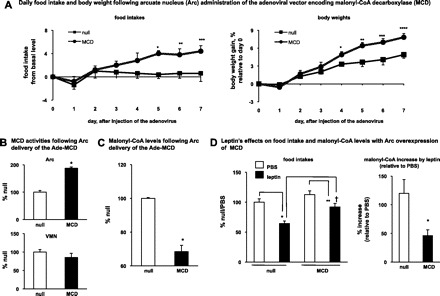
Increase of Arc malonyl-CoA is required in leptin's anorectic actions. A: adenovirus encoding the malonyl-CoA decarboxylase (MCD: n = 5) or enhanced green fluorescent protein (null, n = 5) was administered into the Arc on day 0. Daily food intakes and daily body weights were monitored. The daily food intake before the virus injection was used as the baseline level. The differences between the daily food intake from day 1 through day 7 and the basal level are presented. The daily body weight from day 1 through day 7 was compared with day 0 and the %body weight changes are presented. Food intakes: *, **vs. null, P < 0.05; ***vs. null, P = 0.05. Body weights: *, **, ***, ****vs. null, P < 0.05. B: after 1 wk following Arc delivery of the adenoviruses (MCD, n = 5; null, n = 5), the rats were euthanized. The MCD activities in individual hypothalamic nuclei (Arc, VMN, LHA, and PVN) were measured. The MCD activities from the Arc (n = 5) and the VMN (n = 5) are shown (*vs. null, P < 0.05). C: rats were subject to the similar procedures as described in B. Malonyl-CoA levels (n = 3–4) in the Arc were measured (*vs. null, P < 0.05). D: after 1 wk following the virus delivery, a bolus injection of leptin (15 μg, in PBS) was given intracerebroventricularly before the dark cycle. Then, the food intakes at 3 h after the dark onset were monitored (n = 6–9) (*vs. null/PBS, P < 0.05; **vs. MCD/PBS, P < 0.05; †vs. null/leptin, P < 0.05). Malonyl-CoA levels in the Arc were measured and %increases of malonyl-CoA level by leptin (as compared with PBS) are shown (n = 6–9; *P < 0.05, vs. null).
Blocking malonyl-CoA inhibition of CPT-1 acyltransferase activity does not affect leptin's anorectic actions.
We injected the adenovirus encoding CPT-1a wt (Ade-CPT-1a wt), the CPT-1a wt (Ade-CPT-1a mt) or the null (Ade-null) into the Arc of rats. Three to four days following the virus infections, daily food intakes and body weights returned to the preinjection levels. At least through the eighth day following the initial injection of the viruses, no significant body weight or feeding differences were found among these rats (data not shown). We then assessed the role of malonyl-CoA inhibition of CPT-1 in leptin's anorectic actions. During the second week following the initial delivery of the viruses (around the 11th day), leptin was injected to the rats. We found that Arc overexpression of CPT-1a mt (malonyl-CoA insensitive) did not affect leptin-induced feeding inhibition or weight loss (Fig. 3A). During this period (the 2nd week following the injection of the viruses), the adenoviral infections did not affect the daily (24 h) food intakes compared with the basal preinjection levels, and as in the first week described above, no significant differences of 24-h food intakes were found among all treated groups (basal level: 100 ± 3.2%, Ade-null: 103 ± 2.0%, Ade-CPT-1a wt: 97 ± 2.1%, Ade-CPT-1a mt: 95 ± 5.8%). Concomitant with producing similar anorectic effects, leptin induced similar increases in Arc malonyl-CoA levels in all treated groups (Fig. 3B). Since malonyl-CoA inhibition of CPT-1 acyltransferase activity is reversible (22), the CPT-1 activity assay using tissue extract is not informative in evaluating the in vivo effect of malonyl-CoA on CPT-1 activity. CPT-1 acyltransferase activity converts LCFA-CoAs to LC-ACs (13). We therefore assessed CPT-1a activity by measuring the levels of LC-ACs. As expected, activation of Arc CPT-1a increased LC-AC levels in the Arc (Fig. 3C). Leptin reduced LC-AC levels in the Arc that ectopically expresses the CPT-1a wt (Fig. 3C), indicating an inhibition of CPT-1 activity by leptin in these animals. In contrast, leptin did not affect LC-AC levels in the Arc overexpressing the CPT-1a mt (Fig. 3C), suggesting that leptin-induced accumulation of malonyl-CoA does not inhibit the CPT-1 activities in these animals. In parallel with the changes in the levels of LC-ACs, levels of total LCFA-CoAs (substrates for CPT-1a) were reduced by CPT-1a activation (Fig. 3D). Leptin induced the increase in LCFA-CoA levels in the Arc, ectopically expressing CPT-1a wt, while it did not affect LCFA-CoA levels in the Arc overexpressing the CPT-1a mt (Fig. 3D). It should be noted that these biochemical assays were performed at the time when the feeding experiment with leptin was conducted (i.e., in the 2nd week following viral injections). We also demonstrated (Fig. 1) that the effect of overexpressing the CPT-1a mt on antagonizing malonyl-CoA-mediated inhibition remained significant at a later time point (i.e., 2 wk following the viral injections). Thus, we demonstrated that during the period when the leptin-induced inhibition of CPT-1a were blocked, the leptin-induced feeding inhibition was not affected. Taken together, these data demonstrate that blocking malonyl-CoA inhibition of CPT-1a and the resulting increase in LCFA-CoA's level does not affect leptin's anorectic actions.
Fig. 3.
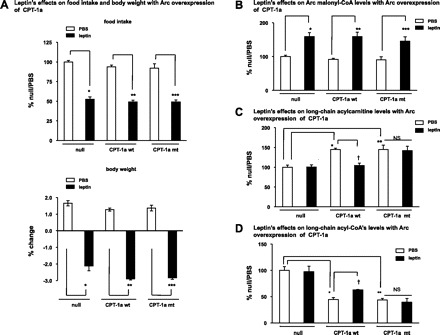
Blocking malonyl-CoA inhibition of CPT-1 acyltransferase activity does not affect the anorectic action of leptin. A: during the 2nd wk following the Arc administration of Ade-CPT-1a wt, Ade-CPT-1a mt, or Ade-null, a bolus injection of leptin (15 μg in PBS) was given intracerebroventricularly to rats before the dark cycle. Overnight food intake and body weight were monitored (n = 6). Values from the rat having a targeted Arc overexpression of CPT-1a and an increase of CPT acyltransferase activity were included in the data analysis (*vs. null/PBS, P < 0.05; **vs. CPT-1a wt/PBS, P < 0.05; ***vs. CPT-1a mt/PBS, P < 0.05). B–D: after 1 wk following the Arc delivery of the adenoviruses (CPT-1a wt, CPT-1a mt, and null), a bolus injection of leptin (15 μg in PBS) was given intracerebroventricularly to the rats. The Arc levels (at 3 h after icv injection) of the malonyl-CoA (B, n = 5–7), long-chain acylcarnitines (C, n = 5–6), and the long-chain acyl-CoA's (D, n = 5–8) were measured. NS, differences between CPT-1a mt/PBS and CPT-1a mt/leptin are not significant. B: *vs. null/PBS, P < 0.05; **vs. CPT-1a wt/PBS, P < 0.05; ***vs. CPT-1a mt/PBS, P < 0.05. C: *CPT-1a wt/PBS vs. null/PBS, P < 0.05; **CPT-1a mt/PBS vs. null/PBS, P < 0.05; †CPT-1a wt/leptin vs. CPT-1a wt/PBS, P < 0.05; NS, CPT-1a mt/leptin vs. CPT-1a mt/PBS, not significant. D: *CPT-1a wt/PBS vs. null/PBS, P < 0.05; **CPT-1a mt/PBS vs. null/PBS, P < 0.05; †CPT-1a wt/leptin vs. CPT-1a wt/PBS, P < 0.05; NS, CPT-1a mt/leptin vs. CPT-1a mt/PBS, not significant.
Blocking malonyl-CoA inhibition of CPT-1 acyltransferase activity does not affect the anorectic action of central cerulenin.
To assess the role of CPT-1a in the specific context of malonyl-CoA signaling actions, we examined the feeding response to central cerulenin that is an inhibitor of fatty acid synthase (FAS). FAS uses malonyl-CoA as a substrate and blocking FAS activity increases the malonyl-CoA level (15). We first show that overexpressing MCD attenuated the level of cerulenin-induced increase in Arc malonyl-CoA, and blocked the feeding inhibition induced by cerulenin treatment (Fig. 4A). These data demonstrate that an increase in Arc malonyl-CoA level is required for cerulenin's anorectic effects. As expected, Arc overexpression of CPT-1a mt did not affect the anorectic effects by cerulenin, and cerulenin treatment increased Arc malonyl-CoA to a similar level in all treated groups (Fig. 4B). It should be noted that the inhibition of FAS reduces the synthesis of LCFA/acyl-CoAs (14), which would contribute to lowering the LC-AC levels via mass action. As a result, we did not monitor the CPT-1 activity following cerulenin treatment. However, we found a similar level of increase (1.5- to 2-fold) in Arc malonyl-CoA by cerulenin compared with that seen following leptin treatment (compare Fig. 4B with Fig. 3B). We thus assume that cerulenin treatment produced the same effect on CPT-1a activity as leptin treatment did. Together, our data indicate that malonyl-CoA inhibition of Arc CPT-1a is not required for the anorectic effects of either leptin or cerulenin.
Fig. 4.
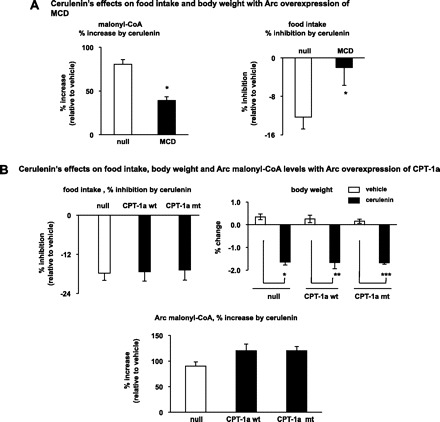
Blocking malonyl-CoA inhibition of CPT-1 acyltransferase activity does not affect the anorectic action of cerulenin. A: Ade-MCD or Ade-null was delivered into the Arc of rats. After 1 wk following Arc delivery of the adenoviruses, a bolus injection of cerulenin (125 μg, in 25% DMSO) was given intracerebroventricularly before the dark cycle. Then the malonyl-CoA levels in the Arc were measured. The %increase of the malonyl-CoA level by cerulenin (as compared with 25% DMSO) are shown (n = 6–7). Following intracerebroventricular injection of cerulenin, the food intakes at 3 h after the dark onset and the overnight (24 h) body weight changes were also monitored (n = 6–7). *P < 0.05, vs. null. B: Ade-CPT-1a wt, Ade-CPT-1a mt or Ade-null was delivered into the Arc of rats. After 1 wk following Arc delivery of the adenoviruses, a bolus injection of cerulenin (125 μg, in 25% DMSO) was given intracerebroventricully before the dark cycle and overnight food intake and overnight body weight were monitored (n = 8–10). Malonyl-CoA levels in the Arc were also measured and %increase of the malonyl-CoA level by cerulenin (as compared with 25% DMSO) are shown (n = 8–10; *vs. null/vehicle, P < 0.05; **vs. CPT-1a wt/vehicle, P < 0.05; ***vs. CPT-1a mt/vehicle, P < 0.05).
Changes in LCFA-acyl-CoA's level in the arc are dissociated from those in malonyl-CoA levels under fasting and refeeding conditions.
Under different nutritional states, such as fasting and refeeding, circulating leptin level changes in tight association with that of hypothalamic malonyl-CoA level (9, 14). If LCFA-CoAs act as downstream mediator of malonyl-CoA action in leptin's intracellular signaling pathways, the levels of LCFA-CoAs should also change in association with the level of malonyl-CoA. To assess this hypothesis, we examined the Arc levels of LCFA-CoAs and malonyl-CoA under fasting and refeeding conditions. Unexpectedly, we found that Arc LCFA-CoA levels were increased by fasting even though the malonyl-CoA level decreased (Fig. 5A), and these changes were reversed upon refeeding (Fig. 5A). Thus, in the Arc, the changes in LCFA-CoA levels appear to dissociate from those in malonyl-CoA's level under different feeding states. It is known that CPT-1-mediated fatty acid β-oxidation activity in the brain is trivial compared with the periphery (16). In determining the size of brain LCFA-CoA pool, other actions/pathways, such as exchange with the circulation, the action of acyl-CoA synthetase, and brain acyl-CoA hydrolase play more prominent roles (Fig. 5B and Ref. 16). Here, we showed that fasting elevated the fatty acid levels in the circulation, while refeeding brought down the elevated levels (Fig. 5C). Furthermore, the brain acyl-CoA hydrolase activity was lowered by fasting and elevated to prefasting level following refeeding (Fig. 5D). Although the physiological relevance of these changes is unclear, these metabolic events provide an interpretation for the observed dissociation of LCFA-CoAs from malonyl-CoA.
Fig. 5.
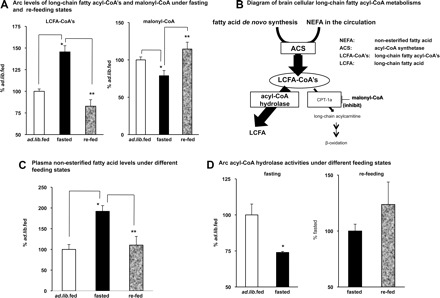
Changes of long-chain fatty acyl-CoAs (LCFA-CoAs) levels in the Arc are dissociated from the changes of malonyl-CoA levels under fasting and refeeding conditions. A: some rats were fasted for 48 h (fasted, n = 9), and the other rats were refed for 3 h after being fasted for 48 h (refed, n = 6). Arc levels of LCFA-CoAs and malonyl-CoA were measured. Ad libitum fed (n = 6) was used as the control. *Fasted vs. ad libitum fed, P < 0.05; **refed vs. fasted, P < 0.05. B: schematic diagram of the metabolism of brain cellular LCFA-CoAs is shown. Intracellular LCFA either synthesized de novo or transported from the circulation is esterified by acyl-CoA synthetase (ACS) to form LCFA-CoAs. Hydrolysis by acyl-CoA hydrolase and the CPT-1a-mediated mitochondrial β-oxidation are 2 pathways that lower the cellular LCFA-CoA levels. In the brain, acyl-CoA hydrolase action plays a major role, while the mitochondrial β-oxidation is a minor pathway, in controlling the cellular LCFA-CoA's levels. C: rats were subjected to the fasting and refeeding procedure as described in A. Plasma levels of free fatty acid were measured (n = 5). Ad libitum fed was used as the control. *Fasted vs. ad libitum fed, P < 0.05; **refed vs. fasted, P < 0.05. D: some rats were fasted for 1 overnight (fasted, n = 5) and the other rats were refed for 3 h after being fasted for 2 overnights (refed, n = 3). Arc acyl-CoA hydrolase activities were measured. Note: the acyl-CoA hydrolase activity levels between 24 h fasting and 48 h fasting are comparable (*vs. ad libitum fed, P < 0.05).
DISCUSSION
CPT-1a, a key enzyme in regulating mitochondrial fatty acid β-oxidation, has been proposed to be a candidate for mediating hypothalamic malonyl-CoA anorectic actions. In the CNS, as fatty acid β-oxidation activity is trivial, malonyl-CoA-mediated regulation of CPT-1 acyltransferase activity is not as significant in the brain as it is in the periphery (16). Indeed, in our present and previous studies (6), we found no change of the Arc levels of either LCFA-CoAs (substrates of CPT-1a) or LC-ACs (products of CPT-1a), upon leptin administration under normal conditions. These results suggest that leptin treatment does not affect CPT-1 acyltransferase activity in the Arc. Thus, Arc CPT-1a may not be implicated in leptin's central actions on feeding under normal conditions. In particular, our data indicate that CPT-1a is not a critical component of malonyl-CoA signaling mechanisms in leptin's anorectic actions. Some potential mechanisms underlie this conclusion. First, due to the inherent heterogeneity of CNS cells (28), malonyl-CoA metabolism and CPT-1a expression might not take place in the same cells (31). Thus, the change of the malonyl-CoA level in response to leptin may occur in a population of cells that does not express CPT-1a. Second, malonyl-CoA produced by the two isoforms of ACC (ACC-1 and ACC-2) has different effects on CPT-1a activity. Compared with ACC2, the ACC-1-associated malonyl-CoA does not significantly affect CPT-1a-mediated fatty acid β-oxidation (19). It should be noted that we have demonstrated leptin specifically activates ACC-1 to increase the malonyl-CoA level (6). It follows that malonyl-CoA may not inhibit Arc CPT-1a in leptin's anorectic actions. Finally, due to the inherent nature of low activity, the CPT-1a in the Arc may not be subject to the regulation by malonyl-CoA, particularly when malonyl-CoA is increased. Under physiological conditions, the already low activity of CPT-1a in the Arc may be resistant to a further inhibition by malonyl-CoA. Under artificial conditions, such as when CPT-1a wt is ectopically overexpressed, leptin treatment does inhibit CPT-1a activity and increase LCFA-CoA's levels. However, the blockades of these changes fail to affect leptin's anorectic actions. Therefore, in the Arc, malonyl-CoA inhibition of CPT-1a seems unlikely to play a key role in mediating leptin's feeding actions. Our data also show that leptin's effect on body weight is independent of malonyl-CoA-mediated CPT-1a inhibition. Together with the results of food intake, we speculate that leptin's action on body energy expenditure would also be independent of malonyl-CoA's inhibitory effect on CPT-1a. However, a definitive conclusion requires direct measurement of energy expenditure in the experimental paradigm.
There have been increasing concerns (16) with the hypothesis that inhibiting Arc CPT-1a with the ensuing increases of LCFA-CoAs can produce anorectic effects (23, 24). Our results provide further evidence to support the notion that these biochemical events are not implicated in leptin's anorectic actions. In addition, particularly under acute experimental conditions (24), intracellular LCFA-CoA levels may not go up following the treatment with LCFAs (16, 24). Moreover, a growing body of evidence now shows that accumulation of hypothalamic LCFA-CoAs is indeed associated with increase in food intake, and increases in adiposity and body weight (3, 4, 25). Notably, ghrelin, an orexigenic factor that potently stimulates feeding, increases hypothalamic LCFA-CoA levels while inhibiting hypothalamic ACC, which reduces the malonyl-CoA level (3). Observed increases in LCFA-CoA levels suggest that the action of LCFA-CoAs, at least in ghrelin hypothalamic pathways, can dissociate from that of malonyl-CoA. In line with this prediction, we demonstrate a dissociation of the levels of LCFA-CoAs from that of malonyl-CoA under fasting and refeeding conditions. It is also worth pointing out that Arc overexpression of acyl-CoA synthetase raising LCFA-CoA levels does not induce the expected anorexigenic actions (personal communication with Dr. Jason Dyck, University of Alberta, Edmonton, AB, Canada). Together, these data strongly challenge the proposed anorectic role of Arc LCFA-CoA, and they also argue against the view that LCFA-CoAs can act as effectors of Arc malonyl-CoA-mediated anorectic actions.
Further challenge to a role of CPT-1a and LCFA-CoAs as mediators of malonyl-CoA feeding action comes from the study using compound C89b, a CPT-1 activator (1). It was expected that C89b treatment, by activating CPT-1a and thus reducing LCFA-CoA levels, would stimulate food intake. Surprisingly, the study demonstrated that C89b induced the same feeding response, i.e., an inhibition, as the increase in Arc malonyl-CoA level (1). These C89b data also directly contradict the previous finding that CPT-1a inhibition (with ST1326) reduces feeding. Because opposing actions on the same targets produce similar feeding effects, these data further suggest that CPT-1a and LCFA-CoAs are not implicated in the hypothalamic control of feeding. In support of this prediction, our results are also unfavorable for a significant role of Arc CPT-1a per se in the CNS control of feeding and body weight. In our studies, we were unable to detect significant changes of either food intake or body weight gain following Arc-specific activation of CPT-1a. Furthermore, we demonstrated that CPT-1a activity (by measuring the levels of LC-ACs) in the Arc was not significantly altered under either fasting or refeeding condition (ad libitum fed: 100 ± 17%, fasted: 104 ± 14%, refed: 112 ± 10%). Taking these findings together, Arc CPT-1a is unlikely to play a direct and key role in the central controls of feeding and energy balance.
The recent discovery of the brain-specific CPT-1 isoform, CPT-1c (26), may provide some insights into differential roles of CPT-1 isoforms in the central control of feeding and body energy balance. CPT-1c is structurally similar to CPT-1a and CPT-1b, but does not have an appreciable CPT acyltransferase activity (26, 30). Notably, CPT-1c has been implicated in the hypothalamic control of energy balance (30). Given that CPT-1c exhibits a high amino acid sequence similarity to the other CPT-1 members, those CPT-1a regulators (i.e., ST1326 and C89b) may have affected CPT-1c with the same pharmacological action, thus resulting in the same feeding effect. Furthermore, we anticipate that CPT-1c is an alternative downstream mediator in malonyl-CoA's anorectic signaling action. Indeed, our studies have provided evidence that CPT-1c is a downstream mediator of the malonyl-CoA action in leptin Arc anorectic signaling pathways (addressed in another manuscript).
Perspectives and Significance
Our data strongly suggest that the intracellular downstream pathways mediating Arc malonyl-CoA's anorectic effects induced by leptin are independent of CPT-1a. Our study thus leaves open the possibility of other target(s) as mediators of Arc malonyl-CoA anorectic signaling actions.
GRANTS
These studies were funded by a grant from the Canadian Diabetes Association and a fellowship from Heart and Stroke Foundation of Canada (awarded to S. Gao). F. G. Hegardt, D. Serra, N. Casals, and P. Carrasco acknowledge grants from Ministry of Education and Science, Spain (Grant SAF2007-61926 to F. G. Hegardt), and from Instituto de Salud Carlos III (Grant CIBERobn CB06/03/0026 to F. G. Hegardt and research contract to P. Carrasco). G. D. Lopaschuk is a scientist of the Alberta Heritage Foundation for Medical Research.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
ACKNOWLEDGMENTS
We thank Amy Barr (University of Alberta, Alberta, Canada) for the preparation (amplification) of the recombinant adenoviruses. We thank Ken Strynadka and Thomas Panakkezhum (University of Alberta, Alberta, Canada) for the HPLC analysis of LCFA-CoAs.
REFERENCES
- 1. Aja S, Landree LE, Kleman AM, Medghalchi SM, Vadlamudi A, McFadden JM, Aplasca A, Hyun J, Plummer E, Daniels K, Kemm M, Townsend CA, Thupari JN, Kuhajda FP, Moran TH, Ronnett GV. Pharmacological stimulation of brain carnitine palmitoyl-transferase-1 decreases food intake and body weight. Am J Physiol Regul Integr Comp Physiol 294: R352–R361, 2008. [DOI] [PubMed] [Google Scholar]
- 2. Andersson U, Filipsson K, Abbott CR, Woods A, Smith K, Bloom SR, Carling D, Small CJ. AMP-activated protein kinase plays a role in the control of food intake. J Biol Chem 279: 12005–12008, 2004. [DOI] [PubMed] [Google Scholar]
- 3. Andrews ZB, Liu ZW, Walllingford N, Erion DM, Borok E, Friedman JM, Tschop MH, Shanabrough M, Cline G, Shulman GI, Coppola A, Gao XB, Horvath TL, Diano S. UCP2 mediates ghrelin's action on NPY/AgRP neurons by lowering free radicals. Nature 454: 846–851, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Benoit SC, Kemp CJ, Elias CF, Abplanalp W, Herman JP, Migrenne S, Lefevre AL, Cruciani-Guglielmacci C, Magnan C, Yu F, Niswender K, Irani BG, Holland WL, Clegg DJ. Palmitic acid mediates hypothalamic insulin resistance by altering PKC-θ subcellular localization in rodents. J Clin Invest 119: 2577–2589, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chakravarthy MV, Zhu Y, Lopez M, Yin L, Wozniak DF, Coleman T, Hu Z, Wolfgang M, Vidal-Puig A, Lane MD, Semenkovich CF. Brain fatty acid synthase activates PPARα to maintain energy homeostasis. J Clin Invest 117: 2539–2552, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gao S, Kinzig KP, Aja S, Scott KA, Keung W, Kelly S, Strynadka K, Chohnan S, Smith WW, Tamashiro KL, Ladenheim EE, Ronnett GV, Tu Y, Birnbaum MJ, Lopaschuk GD, Moran TH. Leptin activates hypothalamic acetyl-CoA carboxylase to inhibit food intake. Proc Natl Acad Sci USA 104: 17358–17363, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gao S, Lane MD. Effect of the anorectic fatty acid synthase inhibitor C75 on neuronal activity in the hypothalamus and brainstem. Proc Natl Acad Sci USA 100: 5628–5633, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guzman-Ruiz R, Somoza B, Gil-Ortega M, Merino B, Cano V, Attane C, Castan-Laurell I, Valet P, Fernandez-Alfonso MS, Ruiz-Gayo M. Sensitivity of cardiac carnitine palmitoyltransferase to malonyl-CoA is regulated by leptin: similarities with a model of endogenous hyperleptinemia. Endocrinology 151: 1010–1018. [DOI] [PubMed] [Google Scholar]
- 9. He W, Lam TK, Obici S, Rossetti L. Molecular disruption of hypothalamic nutrient sensing induces obesity. Nat Neurosci 9: 227–233, 2006. [DOI] [PubMed] [Google Scholar]
- 10. Herrero L, Rubi B, Sebastian D, Serra D, Asins G, Maechler P, Prentki M, Hegardt FG. Alteration of the malonyl-CoA/carnitine palmitoyltransferase I interaction in the beta-cell impairs glucose-induced insulin secretion. Diabetes 54: 462–471, 2005. [DOI] [PubMed] [Google Scholar]
- 11. Hu Z, Cha SH, Chohnan S, Lane MD. Hypothalamic malonyl-CoA as a mediator of feeding behavior. Proc Natl Acad Sci USA 100: 12624–12629, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hu Z, Dai Y, Prentki M, Chohnan S, Lane MD. A role for hypothalamic malonyl-CoA in the control of food intake. J Biol Chem 280: 39681–39683, 2005. [DOI] [PubMed] [Google Scholar]
- 13. Jauregui O, Sierra AY, Carrasco P, Gratacos E, Hegardt FG, Casals N. A new LC-ESI-MS/MS method to measure long-chain acylcarnitine levels in cultured cells. Anal Chim Acta 599: 1–6, 2007. [DOI] [PubMed] [Google Scholar]
- 14. Lane MD, Hu Z, Cha SH, Dai Y, Wolfgang M, Sidhaye A. Role of malonyl-CoA in the hypothalamic control of food intake and energy expenditure. Biochem Soc Trans 33: 1063–1067, 2005. [DOI] [PubMed] [Google Scholar]
- 15. Loftus TM, Jaworsky DE, Frehywot GL, Townsend CA, Ronnett GV, Lane MD, Kuhajda FP. Reduced food intake and body weight in mice treated with fatty acid synthase inhibitors. Science 288: 2379–2381, 2000. [DOI] [PubMed] [Google Scholar]
- 16. Lopaschuk GD, Ussher JR, Jaswal JS. Targeting intermediary metabolism in the hypothalamus as a mechanism to regulate appetite. Pharmacol Rev 62: 237–264, 2010. [DOI] [PubMed] [Google Scholar]
- 17. Lopez M, Lage R, Saha AK, Perez-Tilve D, Vazquez MJ, Varela L, Sangiao-Alvarellos S, Tovar S, Raghay K, Rodriguez-Cuenca S, Deoliveira RM, Castaneda T, Datta R, Dong JZ, Culler M, Sleeman MW, Alvarez CV, Gallego R, Lelliott CJ, Carling D, Tschop MH, Dieguez C, Vidal-Puig A. Hypothalamic fatty acid metabolism mediates the orexigenic action of ghrelin. Cell Metab 7: 389–399, 2008. [DOI] [PubMed] [Google Scholar]
- 18. Lopez M, Lelliott CJ, Tovar S, Kimber W, Gallego R, Virtue S, Blount M, Vazquez MJ, Finer N, Powles TJ, O'Rahilly S, Saha AK, Dieguez C, Vidal-Puig AJ. Tamoxifen-induced anorexia is associated with fatty acid synthase inhibition in the ventromedial nucleus of the hypothalamus and accumulation of malonyl-CoA. Diabetes 55: 1327–1336, 2006. [DOI] [PubMed] [Google Scholar]
- 19. Mao J, DeMayo FJ, Li H, Abu-Elheiga L, Gu Z, Shaikenov TE, Kordari P, Chirala SS, Heird WC, Wakil SJ. Liver-specific deletion of acetyl-CoA carboxylase 1 reduces hepatic triglyceride accumulation without affecting glucose homeostasis. Proc Natl Acad Sci USA 103: 8552–8557, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mera P, Bentebibel A, Lopez-Vinas E, Cordente AG, Gurunathan C, Sebastian D, Vazquez I, Herrero L, Ariza X, Gomez-Puertas P, Asins G, Serra D, Garcia J, Hegardt FG. C75 is converted to C75-CoA in the hypothalamus, where it inhibits carnitine palmitoyltransferase 1 and decreases food intake and body weight. Biochem Pharmacol 77: 1084–1095, 2009. [DOI] [PubMed] [Google Scholar]
- 21. Moran TH, Gao S. Looking for food in all the right places? Cell Metab 3: 233–234, 2006. [DOI] [PubMed] [Google Scholar]
- 22. Morillas M, Gomez-Puertas P, Bentebibel A, Selles E, Casals N, Valencia A, Hegardt FG, Asins G, Serra D. Identification of conserved amino acid residues in rat liver carnitine palmitoyltransferase I critical for malonyl-CoA inhibition. Mutation of methionine 593 abolishes malonyl-CoA inhibition. J Biol Chem 278: 9058–9063, 2003. [DOI] [PubMed] [Google Scholar]
- 23. Obici S, Feng Z, Arduini A, Conti R, Rossetti L. Inhibition of hypothalamic carnitine palmitoyltransferase-1 decreases food intake and glucose production. Nat Med 9: 756–761, 2003. [DOI] [PubMed] [Google Scholar]
- 24. Obici S, Feng Z, Morgan K, Stein D, Karkanias G, Rossetti L. Central administration of oleic acid inhibits glucose production and food intake. Diabetes 51: 271–275, 2002. [DOI] [PubMed] [Google Scholar]
- 25. Posey KA, Clegg DJ, Printz RL, Byun J, Morton GJ, Vivekanandan-Giri A, Pennathur S, Baskin DG, Heinecke JW, Woods SC, Schwartz MW, Niswender KD. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am J Physiol Endocrinol Metab 296: E1003–E1012, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Price N, van der Leij F, Jackson V, Corstorphine C, Thomson R, Sorensen A, Zammit V. A novel brain-expressed protein related to carnitine palmitoyltransferase I. Genomics 80: 433–442, 2002. [DOI] [PubMed] [Google Scholar]
- 27. Sambandam N, Steinmetz M, Chu A, Altarejos JY, Dyck JR, Lopaschuk GD. Malonyl-CoA decarboxylase (MCD) is differentially regulated in subcellular compartments by 5'AMP-activated protein kinase (AMPK). Studies using H9c2 cells overexpressing MCD and AMPK by adenoviral gene transfer technique. Eur J Biochem 271: 2831–2840, 2004. [DOI] [PubMed] [Google Scholar]
- 28. Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature 404: 661–671, 2000. [DOI] [PubMed] [Google Scholar]
- 29. Sebastian D, Herrero L, Serra D, Asins G, Hegardt FG. CPT I overexpression protects L6E9 muscle cells from fatty acid-induced insulin resistance. Am J Physiol Endocrinol Metab 292: E677–E686, 2007. [DOI] [PubMed] [Google Scholar]
- 30. Wolfgang MJ, Kurama T, Dai Y, Suwa A, Asaumi M, Matsumoto S, Cha SH, Shimokawa T, Lane MD. The brain-specific carnitine palmitoyltransferase-1c regulates energy homeostasis. Proc Natl Acad Sci USA 103: 7282–7287, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wolfgang MJ, Lane MD. Hypothalamic malonyl-CoA and CPT1c in the treatment of obesity. FEBS J 278: 552–558, 2011. [DOI] [PubMed] [Google Scholar]